 Liang Wang1†
Liang Wang1† Menglong Chen1†
Menglong Chen1† Ruojie He1Yiming Sun2Juan Yang3Lulu Xiao4Jiqing Cao5Huili Zhang6Cheng Zhang1*
Ruojie He1Yiming Sun2Juan Yang3Lulu Xiao4Jiqing Cao5Huili Zhang6Cheng Zhang1*
- 1Department of Neurology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong, China
- 2Department of Health Care, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong, China
- 3Department of Neurology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China
- 4The Department of Tissue Typing Center, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, China
- 5Department of Neurology, Wuhan Central Hospital, Wuhan, Hubei, China
- 6Department of Neurology, Guangzhou First People’s Hospital, Guangzhou, Guangdong, China
Here, we investigated correlations between serum creatinine (SCRN) levels and clinical phenotypes of dystrophinopathy in young patients. Sixty-eight patients with dystrophinopathy at the Neuromuscular Clinic, The First Affiliated Hospital, Sun Yat-sen University, were selected for this study. The diagnosis of dystrophinopathy was based on clinical manifestation, biochemical changes, and molecular analysis. Some patients underwent muscle biopsies; SCRN levels were tested when patients were ≤3 years old, and reading frame changes were analyzed. Each patient was followed up, and motor function and clinical phenotype were assessed when the same patients were ≥4 years old. Our findings indicated that in young patients, lower SCRN levels were associated with increased disease severity (p < 0.01) and that SCRN levels were the highest in patients exhibiting mild Becker muscular dystrophy (BMD) (p < 0.001) and the lowest in patients with Duchenne muscular dystrophy (DMD) (p < 0.01) and were significantly higher in patients carrying in-frame mutations than in patients carrying out-of-frame mutations (p < 0.001). SCRN level cutoff values for identifying mild BMD [18 µmol/L; area under the curve (AUC): 0.947; p < 0.001] and DMD (17 µmol/L; AUC: 0.837; p < 0.001) were established. These results suggest that SCRN might be a valuable biomarker for distinguishing DMD from BMD in patients aged ≤3 years and could assist in the selection of appropriate treatment strategies.
Introduction
Dystrophinopathy is a recessive X-linked hereditary disease with an incidence of 1 in 3,500 newborn males, characterized by progressive muscle weakness, atrophy, and loss of ambulation (1). The disease can be divided into two clinical phenotypes according to different courses: Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). DMD exhibits rapid progression and loss of ambulation prior to age 12, whereas BMD exhibits relatively mild symptoms with slower progression. Following the successful cloning of DMD in 1985, the molecular pathogenesis of the disease was determined, and the importance of dystrophin in maintaining normal muscle function was discovered (2, 3). Different DMD mutations result in two primary forms of defective dystrophin: a truncated, non-functional protein associated with DMD and a partially functional protein associated with BMD (4).
Current treatment regimens for dystrophinopathy can delay the age at which loss of ambulation occurs and improve the patient’s life span, which are insufficient (5). As technology improves, new prospective treatments are proposed, such as stem cell therapy and gene therapy, including exon skipping and stop codon readthrough therapy (6–8). However, issues such as side effects and high costs remain unresolved (9). Therefore, they are not recommended for patients with mild BMD. As early treatment initiation leads to improved outcomes, identifying specific clinical phenotypes in young patients is essential for selecting appropriate treatments (10).
Prior to loss of ambulation, three main methods are used to distinguish clinical phenotypes: genotype–phenotype correlation, evaluation of dystrophin levels in the muscle tissues, and clinical manifestation. Genotype–phenotype correlations comprise the majority of the cases, with still some exceptions in both the dystrophies, but more commonly in cases with BMD (11, 12). Besides, genetic testing is expensive and time consuming, limiting its application in biomarker assessment. In addition, dystrophin expression does not always constitute an accurate marker of clinical phenotypes (13–15). Clinical manifestation is helpful to distinguish when patients exhibit obvious symptoms, which is therefore less useful in young asymptomatic or paucisymptomatic patients. While creatine kinase expression generally differs between patients with BMD and DMD, a high degree of interpatient and intrapatient variability makes accurate diagnosis difficult (16). Magnetic resonance imaging of the muscle tissue is another prospective method to distinguish the clinical phenotype; however, younger patients likely require sedation to ensure accurate results (17). Therefore, new biomarkers are needed to aid in distinguishing clinical phenotypes in young patients.
Recent studies reported correlations between serum creatinine (SCRN) and dystrophinopathy phenotypes, with differences in the creatine metabolism pathway between patients with DMD and controls particularly striking (16, 18). SCRN, primarily produced in the muscle tissues, plays an important role in energy metabolism, and dysfunctional creatine metabolism has been reported in dystrophinopathy (19, 20). However, the correlation between SCRN levels and the clinical phenotypes in young patients aged ≤3 years with indistinguishable symptoms remains unknown. Therefore, this study investigated the relationships between SCRN levels and clinical dystrophinopathy phenotypes to determine the effectiveness of SCRN as a biomarker with applications in treatment strategy decision-making.
Materials and Methods
Study Participants
Sixty-eight patients with dystrophinopathy, admitted to the Neuromuscular Clinic at The First Affiliated Hospital, Sun Yat-sen University for regular visits, participated in this study. The diagnosis was confirmed by clinical manifestation, biochemical changes, and molecular analysis. A proportion of patients underwent a muscle biopsy, some of whom participated in our previous study (18, 21). All participants were male, and none was undergoing glucocorticoid treatment. Participants had a minimum age at their last checkup of 4.06 years, a maximum age of 11.04 years, and a median age of 4.92 years (interquartile range: 4.38–6.23 years). All participants were old enough to be adequately assessed. Clinical phenotypes were divided into mild BMD, severe BMD, and DMD according to the clinical manifestation. Patients with mild BMD exhibited completely normal gross motor functions, those with severe BMD exhibited higher degree of motor dysfunctions, and those with DMD exhibited severe motor dysfunctions. The typing criteria were adjusted according to age. For example, an 11-year-old patient who was unable to run or jump high, but who could stand from a squatting position or climb stairs slowly without using the railing, was classified as severe BMD, as this manifestation was milder than that observed in an 11-year-old patient with DMD.
Patients exhibiting the following confounding factors were excluded from the study: (a) increased SCRN levels owing to renal injury, diabetes mellitus, or consumption of certain drugs (22); (b) decreased SCRN levels owing to obvious malnutrition (determined either by clinical manifestation or decreased serum albumin levels), vegetarian diet, or advanced liver disease (23, 24).
Sample Processing
Blood samples were collected when the patients were ≤3 years old, at least 4 h after eating to decrease the influence of meat intake on SCRN levels, and were analyzed at our hospital (25). SCRN levels were then measured immediately. At the time of testing, patients had a minimum age of 4 months and a maximum age of 3.88 years (median: 3.16 years; interquartile range: 2.37–3.49 years).
Five milliliters of blood from each patient were centrifuged at 1,810 g for 5 min at 24°C, before sera were injected into individual tubes and analyzed using a Vitros 5.1FS chemistry analyzer (Johnson & Johnson Corporation, New Brunswick, NJ, USA). Concomitantly, 2 mL of blood from each patient was deposited in a separate tube for use in DMD mutation analysis. Multiplex ligation-dependent probe-amplification reactions were used to detect large sequence rearrangements, and next-generation sequencing, using an Illumina HiSeq 2000 system (Illumina Corporation, San Diego, CA, USA), was used to detect smaller scale mutations. The average sequencing depth was >200×.
Evaluation of Motor Function
As all participants exhibited normal upper limb motor functions, only lower limb motor function was evaluated, using the Vignos scale (18). Patients were required to walk, climb stairs under protection of their parents, and rise from chairs. Parents, with children failing to comply with instructions, were asked to provide details concerning conditions at home. Patients capable of walking and climbing stairs without assistance were scored as grade 1; walking and climbing stairs with the aid of the railing were scored as grade 2; walking and climbing stairs with the aid of the railing and requiring >25 s to complete eight standard steps were scored as grade 3; walking unassisted and rising from chairs, but who were unable to climb stairs were scored as grade 4; walking unassisted, but who were unable to rise from chairs or climb stairs were scored as grade 5; walking only with assistance or walking independently with long leg braces were scored as grade 6; walking in long leg braces, but requiring assistance for balance were scored as grade 7; standing in long leg braces, but unable to walk, even with assistance, were scored as grade 8; requiring a wheelchair were scored as grade 9; and confined to bed were scored as grade 10.
Assessment of Reading Frame Rule
Leiden DMD reading frame checker (http://www.dmd.nl/index.html) was used to determine whether patients with large genetic rearrangements due to mutations also carried frameshift mutations in DMD (11).
Statistical Analysis
Serum creatinine analysis included 68 data points. Statistical analysis was performed using SPSS version 20.0 (IBM Corp., Chicago, IL, USA), GraphPad PRISM version 7.01 (GraphPad Software, San Diego, CA, USA), and MedCalc version 16.8 (MedCalc Software, Ostend, Belgium). The distribution of data for each variable was assessed using the Shapiro–Wilk test (n ≤ 50), and normally distributed data were presented as mean ± SD, with others presented as medians and the 25th and 75th percentiles [median (interquartile range: P25–P75)]. In addition, 95% confidence interval was calculated. To analyze differences between normally distributed variables, Student’s t-test (two variables) or one-way analysis of variance (three variables) was used. To analyze differences between non-normally distributed variables, Mann–Whitney U test (two variables), or Kruskal–Wallis H test (three variables) was used, applying Bonferroni corrections to the differences between every potential pair of the three variables.
Receiver operating characteristic (ROC) curves were used to generate cutoff values, and the global performance of this test was assessed by determining the area under the curve (AUC). Differences in cutoff values between mild and non-mild BMD, and between DMD and non-DMD, were then analyzed. Cutoff values resulting in the largest Youden index (sensitivity + specificity − 1) were considered optimal. Considering the population of patients with childhood-onset dystrophinopathy, the prevalence of DMD was 12.57/100,000 boys, while the prevalence of BMD was 1.35/100,000 boys (26). Positive-predictive values (PVs+) and negative-predictive values were estimated according to this dystrophinopathy prevalence. All tests were two tailed. For the Shapiro–Wilk test, p < 0.1 was considered statistically significant. For all other tests, p < 0.05 was considered statistically significant.
Results
Clinical Data
Patient characteristics, including clinical features, were summarized in Table 1. The distribution of mutation types was similar to that reported in previous studies (11, 21, 27). Median ages at the time of SCRN testing of patients with mild BMD, severe BMD, and DMD were 3.27 years (interquartile range: 2.68–3.78 years; 95% confidence interval: 2.74–3.76 years), 3.24 years (interquartile range: 2.93–3.71 years; 95% confidence interval: 3.08–3.60 years), and 3.05 years (interquartile range: 2.00–3.43 years; 95% confidence interval: 2.46–3.30 years), respectively. Differences in the ages of the patients between the different groups were not significant (p = 0.082). The median SCRN level for all patients was 17.00 µmol/L (interquartile range: 14.00–20.00 µmol/L; 95% confidence interval: 16.00–18.00 µmol/L).
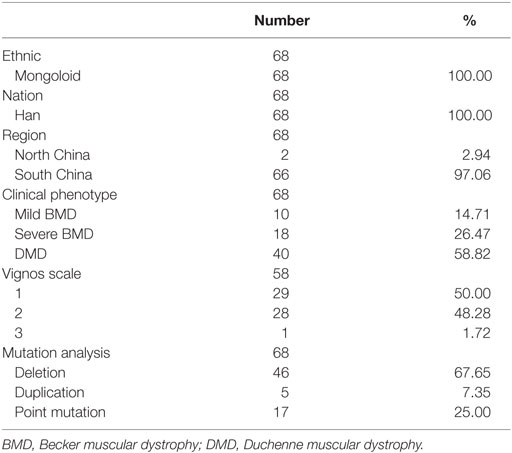
Table 1. The number and ethnic percentages of patients.
SCRN Levels Differ between Mild BMD, Severe BMD, and DMD
The negative correlation between SCRN level and Vignos score indicated that a more severe disease course corresponded to a lower SCRN level at an early age (p < 0.01, data not shown). The SCRN levels of patients at different ages, exhibiting different phenotypes, were analyzed. Among 2-year-old patients at the time of SCRN testing, levels differed significantly between patients with mild BMD, severe BMD, and DMD (p = 0.001). SCRN levels in patients with mild BMD (24.25 ± 3.40 µmol/L; 95% confidence interval: 18.83–29.67 µmol/L) were significantly higher than in those with either severe BMD (18.25 ± 3.86 µmol/L; 95% confidence interval: 12.10–24.40 µmol/L; p = 0.013) or DMD (15.30 ± 2.50 µmol/L; 95% confidence interval: 13.51–17.09 µmol/L; p < 0.001), whereas SCRN levels did not differ significantly between patients with severe BMD and DMD (p = 0.354). Among 3-year-old patients at the time of testing, SCRN levels also differed significantly between the three clinical phenotypes (p < 0.05). Analysis of SCRN levels in patients at different ages who exhibited the various clinical phenotypes indicated that SCRN levels did not differ significantly at different ages (p > 0.05). After merging data for similar clinical phenotypes at different ages, patients exhibiting mild BMD symptoms displayed the highest SCRN levels, while those with DMD displayed the lowest (p < 0.01; Figure 1), suggesting that higher SCRN levels at an early age correlated with a milder phenotype.
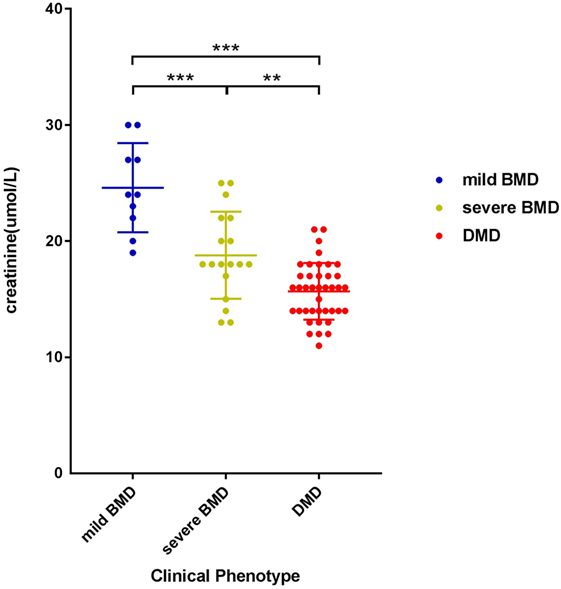
Figure 1. Scatter plot of serum creatinine levels versus phenotypes for dystrophinopathy patients. Differently colored dots signify different clinical phenotypes. Differences between different phenotypes were significant (**p < 0.01, ***p < 0.001).
Correlations between SCRN and Genotype
Among participants with large genetic rearrangements, 13 patients (25.49%) with in-frame mutations and 38 patients (74.51%) with out-of-frame mutations were found. Ages at the time of SCRN testing did not differ significantly between these two groups [3.12 years (interquartile range: 2.85–3.74 years; 95% confidence interval: 2.87–3.71 years) versus 3.17 years (interquartile range: 2.11–3.41 years; 95% confidence interval: 2.53–3.31 years), for patients with in-frame and out-of-frame mutations, respectively; p = 0.210]. However, SCRN levels were significantly higher in the in-frame group [23.38 ± 4.65 µmol/L (95% confidence interval: 20.58–26.19 µmol/L)] than in the out-of-frame group [16.00 µmol/L (interquartile range: 14.00–18.00 µmol/L; 95% confidence interval: 15.50–17.00 µmol/L); p < 0.001; Figure 2A]. SCRN data for seven patients carrying nonsense mutations are shown in Figure 2B. These data indicated that SCRN levels abide by the reading frame rule and that SCRN levels in exceptional cases might be consistent with phenotypes.
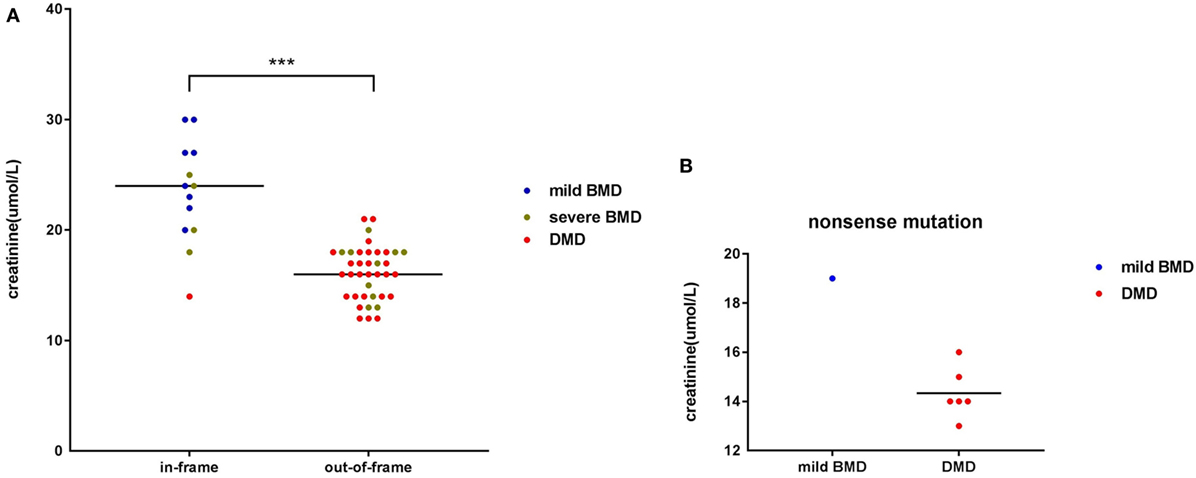
Figure 2. Scatter plot of serum creatinine (SCRN) levels versus different genotypes for dystrophinopathy patients. (A) Differently colored dots signify different clinical phenotypes. Differences in SCRN levels between groups exhibiting different changes to open reading frames were significant. (B) Differently colored dots signify different clinical phenotypes (***p < 0.001).
Determination of Cutoff Values
To determine an SCRN level cutoff that could be used to distinguish mild BMD, ROC curve analysis was performed (Figure 3A). The AUC was 0.947 (p < 0.001), and the SCRN level cutoff value giving the largest Youden index (81.03%) was 18 µmol/L; with PV+ of <36.2% as the prevalence of BMD, which includes both mild and severe cases, was used to estimate the prevalence of mild BMD. Currently, there are no accurate data concerning the prevalence of mild BMD specifically.
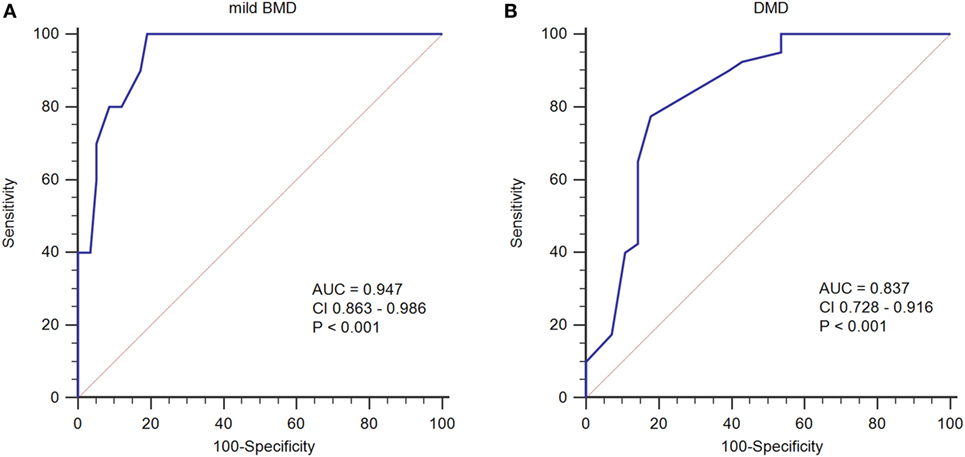
Figure 3. Receiver operating characteristic curve plots for serum creatinine (SCRN) levels. (A) SCRN levels for identifying patients with mild forms of Becker muscular dystrophy (BMD). (B) SCRN levels for identifying patients with Duchenne muscular dystrophy (DMD).
A similar approach was used to determine an SCRN level cutoff that could be used to distinguish DMD (Figure 3B). In this case, the AUC was 0.837 (p < 0.001), and the SCRN level cutoff value giving the largest Youden index (59.64%) was 17 µmol/L. Taken together, these results provided SCRN level cutoff values that could be used to distinguish between cases of mild BMD and DMD. Detailed information was provided in Table 2.
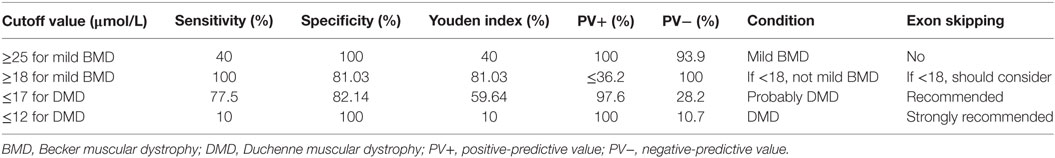
Table 2. Cutoff values and potential applications for exon skipping therapy.
Discussion
This study, based on previously reported results, investigated correlations between SCRN levels and dystrophinopathy (18). Sixty-eight patients, who were first sampled when they were young children with no obvious motor dysfunction, went on to develop some motor dysfunction by their last checkup. Our results indicated that SCRN level was associated with disease severity, clinical phenotype, and genotype.
Our present findings suggest that patients with lower SCRN levels at young ages go on to develop more severe motor dysfunction and more severe disease phenotype as they age, indicating that SCRN level at a younger age is predictive of disease progression. Herein, we propose the following hypothesis to explain these observations. SCRN is associated with energy metabolism within muscle, and its levels are proportional to creatine and phosphocreatine degradation within muscle tissue (19). SCRN levels have been used to estimate muscle mass (28, 29); indeed, muscular dystrophy-associated reductions in muscle mass and creatine metabolism dysfunction are known to affect SCRN levels (20, 30, 31). Thus, SCRN levels in pediatric patients with dystrophinopathy are lower than in healthy children (16, 18). Patients with dystrophinopathy exhibit muscular lesions from the fetal stage, implying that the lesions in patients with slowly progressing BMD reflect a more moderate histopathology relative to that seen in patients with DMD (32); patients with DMD possess more severe muscular lesions than patients with BMD, even if they are asymptomatic during early childhood, resulting in reduced muscle mass and more severe creatine metabolism dysfunction as DMD becomes apparent. This hypothesis might explain the observed differences in SCRN levels between different clinical phenotypes.
Previously, we proposed a correlation between SCRN level and genotype (18). In this study, we demonstrated that SCRN levels might reflect the type of genetic mutation found in patients with dystrophinopathy. Particularly, SCRN levels of two cases with exceptional genotype–phenotype correlations, a DMD case with in-frame mutation and mild BMD case with a nonsense mutation, were consisted with clinical phenotype. While out-of-frame and nonsense mutations are thought to result in relatively severe phenotypes associated with DMD, there are some exceptions where special mechanisms, such as alternative splicing, occur (33–35). In the two exceptional cases noted above, genotype modifications may have occurred by unusual mechanisms, and the observed SCRN levels may be indicative of phenotypic differences related to these modifications. Therefore, it is possible that SCRN levels supplement the deficiencies of genotyping in predicting clinical phenotypes and act as a useful biomarker. Given that SCRN levels can be analyzed quickly, easily, and cost effectively in virtually all hospitals, SCRN measurements could be taken before deciding on a treatment strategy without significantly delaying treatment. The SCRN level cutoff values that we report here as being indicative of specific disease phenotypes could be useful in practical clinical applications, aiding clinicians when choosing treatment strategies. Table 2 shows cutoff values that could be used prior to the initiation of exon skipping treatment.
There were some limitations to this study that should be considered when interpreting the data. First, despite excluding confounding factors that could influence SCRN levels, such as age, ethnicity, diet, and renal injury, other potential confounders including fluid status could not be excluded (25). In the minority of older patients, some large shifts in SCRN level were observed (data not shown) for reasons that remain unclear. Second, the SCRN level distribution curves for the different clinical phenotypes contained some overlapping, making it difficult to discern clinical differences using SCRN levels exclusively. Third, our study included few patients aged <2 years, because these patients are generally almost asymptomatic and so not easily identifiable. However, we believe that the results of this study can be still applied to the whole patient population (age ≤3) because the SCRN level distribution was similar across patients of different ages. Finally, the follow-up duration was not ideal, and longer observational periods are necessary to make definitive conclusions.
In summary, SCRN levels in patients with dystrophinopathy aged ≤3 years constitute a potential biomarker for distinguishing clinical phenotypes that overcomes some of the disadvantages associated with existing methods. Furthermore, the use of SCRN as a biomarker can potentially aid clinicians when determining treatment strategies for patients with dystrophinopathy.
Ethics Statement
This study was carried out in accordance with the recommendations of “guidelines for clinical study, ICE for Clinical Research and Animal Trials of the First Affiliated Hospital of Sun Yat-sen University.” The protocol was approved by the “ICE for Clinical Research and Animal Trials of the First Affiliated Hospital of Sun Yat-sen University.” This study was approved to waive the informed parental consents by “ICE for Clinical Research and Animal Trials of the First Affiliated Hospital of Sun Yat-sen University,” because it was impossible to get written informed parental consents from every participant, and this study did not present personal information and was not harmful to any participant.
Author Contributions
WL and CM designed the study, analyzed the data, and drafted the manuscript. HR, SY, YJ, CJ, and ZH assisted in clinical data collection. XL and ZC assisted in data analysis and in drafting the manuscript. WL and CM contributed equally.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, RJ, and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors would like to acknowledge Feng Pingning for his technical assistance.
Funding
This study was supported by grants from the National Key Clinical Department, National Key Discipline, Guangdong Provincial Key Laboratory for Diagnosis and Treatment of Major Neurological Diseases (grant no. 2014B030301035), the Natural Science Foundation of China (grant nos. 81471280 and 81271401), and the Guangdong Province Science and Technology Plan (grant nos. 2014A020212130 and 1561000153/201508020012).
Abbreviations
SCRN, serum creatinine; DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; AUC, area under the curve; ROCs, receiver operating characteristics; PV+, positive-predictive value; PV−, negative-predictive value.
References
1. Jarmin S, Kymalainen H, Popplewell L, Dickson G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin Biol Ther (2014) 14:209–30. doi: 10.1517/14712598.2014.866087
2. Ray PN, Belfall B, Duff C, Logan C, Kean V, Thompson MW, et al. Cloning of the breakpoint of an X;21 translocation associated with Duchenne muscular dystrophy. Nature (1985) 318:672–5. doi:10.1038/318672a0
3. Monaco AP, Bertelson CJ, Middlesworth W, Colletti CA, Aldridge J, Fischbeck KH, et al. Detection of deletions spanning the Duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature (1985) 316:842–5. doi:10.1038/316842a0
4. Flanigan KM. Duchenne and Becker muscular dystrophies. Neurol Clin (2014) 32:671–88. doi:10.1016/j.ncl.2014.05.002
5. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol (2010) 9:77–93. doi:10.1016/s1474-4422(09)70271-6
6. Yokota T, Duddy W, Echigoya Y, Kolski H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: too many mutations, too few patients? Expert Opin Biol Ther (2012) 12:1141–52. doi:10.1517/14712598.2012.693469
7. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics (1988) 2:90–5. doi:10.1016/0888-7543(88)90113-9
8. Koo T, Wood MJ. Clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy. Hum Gene Ther (2013) 24:479–88. doi:10.1089/hum.2012.234
9. Yokota T, Duddy W, Partridge T. Optimizing exon skipping therapies for DMD. Acta Myol (2007) 26:179–84.
10. Wu B, Cloer C, Lu P, Milazi S, Shaban M, Shah SN, et al. Exon skipping restores dystrophin expression, but fails to prevent disease progression in later stage dystrophic dko mice. Gene Ther (2014) 21:785–93. doi:10.1038/gt.2014.53
11. Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve (2006) 34:135–44. doi:10.1002/mus.20586
12. Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum Mutat (2008) 29:728–37. doi:10.1002/humu.20722
13. Patel K, Leevers S, Abbs S, Hart KA, Heckmatt JZ, Bobrow M, et al. Absence of dystrophin in Becker muscular dystrophy. Lancet (1989) 1:47. doi:10.1016/S0140-6736(89)91705-4
14. van den Bergen JC, Wokke BH, Janson AA, van Duinen SG, Hulsker MA, Ginjaar HB, et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry (2014) 85:747–53. doi:10.1136/jnnp-2013-306350
15. Zatz M, Pavanello RC, Lazar M, Yamamoto GL, Lourenco NC, Cerqueira A, et al. Milder course in Duchenne patients with nonsense mutations and no muscle dystrophin. Neuromuscul Disord (2014) 24:986–9. doi:10.1016/j.nmd.2014.06.003
16. Boca SM, Nishida M, Harris M, Rao S, Cheema AK, Gill K, et al. Discovery of metabolic biomarkers for Duchenne muscular dystrophy within a natural history study. PLoS One (2016) 11:e0153461. doi:10.1371/journal.pone.015346110.5061/dryad.qg6q2
17. Wokke BH, Van Den Bergen JC, Hooijmans MT, Verschuuren JJ, Niks EH, Kan HE. T2 relaxation times are increased in skeletal muscle of DMD but not BMD patients. Muscle Nerve (2016) 53:38–43. doi:10.1002/mus.24679
18. Zhang H, Zhu Y, Sun Y, Liang Y, Li Y, Zhang Y, et al. Serum creatinine level: a supplemental index to distinguish Duchenne muscular dystrophy from Becker muscular dystrophy. Dis Markers (2015) 2015:141856. doi:10.1155/2015/141856
20. Fitch CD, Sinton DW. A study of creatine metabolism in diseases causing muscle wasting. J Clin Invest (1964) 43:444–52. doi:10.1172/JCI104929
21. Yang J, Li SY, Li YQ, Cao JQ, Feng SW, Wang YY, et al. MLPA-based genotype-phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet (2013) 14:29. doi:10.1186/1471-2350-14-29
22. Perrone RD, Madias NE, Levey AS. Serum creatinine as an index of renal function new insights into old concepts. Clin Chem (1992) 38:1933–53.
23. Thongprayoon C, Cheungpasitporn W, Kashani K. Serum creatinine level, a surrogate of muscle mass, predicts mortality in critically ill patients. J Thorac Dis (2016) 8:E305–11. doi:10.21037/jtd.2016.03.62
24. MacAulay J, Thompson K, Kiberd BA, Barnes DC, Peltekian KM. Serum creatinine in patients with advanced liver disease is of limited value for identification of moderate renal dysfunction are the equations for estimating renal function better. Can J Gastroenterol (2006) 20:521–6. doi:10.1155/2006/858053
25. Nair S, O’Brien SV, Hayden K, Pandya B, Lisboa PJ, Hardy KJ, et al. Effect of a cooked meat meal on serum creatinine and estimated glomerular filtration rate in diabetes-related kidney disease. Diabetes Care (2014) 37:483–7. doi:10.2337/dc13-1770
26. Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord (2014) 24:482–91. doi:10.1016/j.nmd.2014.03.008
27. Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, Baena M, Verdura E, Nascimento A, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS One (2015) 10:e0135189. doi:10.1371/journal.pone.0135189
28. Schutte JE, Longhurst JC, Gaffney FA, Bastian BC, Blomqvist CG. Total plasma creatinine: an accurate measure of total striated muscle mass. J Appl Physiol Respir Environ Exerc Physiol (1981) 51:762–6.
29. Kim SW, Jung HW, Kim CH, Kim KI, Chin HJ, Lee H. A new equation to estimate muscle mass from creatinine and cystatin C. PLoS One (2016) 11:e0148495. doi:10.1371/journal.pone.0148495
30. Fitch CD, Oates JD, Dinning JS. The metabolism of creatine-1-C14 by mice with hereditary muscular dystrophy. J Clin Invest (1961) 40:850–6. doi:10.1172/JCI104319
31. Franciotta D, Zanardi MC, Albertotti L, Orcesi S, Berardinelli A, Pichiecchio A, et al. Measurement of skeletal muscle mass in Duchenne muscular dystrophy: use of 24-h creatinine excretion. Acta Diabetol (2003) 40:S290–2. doi:10.1007/s00592-003-0089-8
32. Heckel S, Favre R, Flori J, Koenig M, Mandel J, Gasser B, et al. In utero fetal muscle biopsy a precious aid for the prenatal diagnosis of Duchenne muscular dystrophy. Fetal Diagn Ther (1999) 14:127–32. doi:10.1159/000020905
33. Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Howard MT, Sampson JB, et al. Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat (2011) 32:299–308. doi:10.1002/humu.21426
34. Greer K, Mizzi K, Rice E, Kuster L, Barrero RA, Bellgard MI, et al. Pseudoexon activation increases phenotype severity in a Becker muscular dystrophy patient. Mol Genet Genomic Med (2015) 3:320–6. doi:10.1002/mgg3.144
Keywords: Becker muscular dystrophy, Duchenne muscular dystrophy, dystrophinopathy, serum creatinine, molecular markers
Citation: Wang L, Chen M, He R, Sun Y, Yang J, Xiao L, Cao J, Zhang H and Zhang C (2017) Serum Creatinine Distinguishes Duchenne Muscular Dystrophy from Becker Muscular Dystrophy in Patients Aged ≤3 Years: A Retrospective Study. Front. Neurol. 8:196. doi: 10.3389/fneur.2017.00196
Received: 23 January 2017; Accepted: 21 April 2017;
Published: 08 May 2017
Edited by:
Eugene R. Schnitzler, Loyola University Chicago, USAReviewed by:
Kumar Sannagowdara, Medical College of Wisconsin, USASatinder Aneja, Lady Hardinge Medical College, India
Ryan Jacobson, Loyola University Chicago, USA
Copyright: © 2017 Wang, Chen, He, Sun, Yang, Xiao, Cao, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Zhang, Y2hlbmd6aGFuZzEwMEBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work.