 Hongfei Tai1
Hongfei Tai1 Liying Cui1,2*
Liying Cui1,2* Yuzhou Guan1Mingsheng Liu1Xiaoguang Li1Yan Huang1Jing Yuan1
Yuzhou Guan1Mingsheng Liu1Xiaoguang Li1Yan Huang1Jing Yuan1 Dongchao Shen1
Dongchao Shen1 Dawei Li1Feifei Zhai1
Dawei Li1Feifei Zhai1
- 1Department of Neurology, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 2Neuroscience Center, Chinese Academy of Medical Sciences, Beijing, China
Objective: To describe the characteristics of patients with amyotrophic lateral sclerosis (ALS) and myasthenia gravis (MG) overlap syndrome and explore the relationship between the two diseases.
Methods: We conducted a search of medical records at Peking Union Medical University Hospital from 1983 to 2015 for coexistence of ALS and MG and searched the PubMed database for all literature describing ALS and MG overlap syndrome published through December 2016. We analyzed the clinical and neurophysiological characteristics of patients by groups according to strict diagnostic criteria.
Results: We presented 2 patients in our database with combined ALS and MG, and together with 25 cases reported in the literature, the patients were divided into 4 groups: 12 patients with MG followed by ALS, 8 patients with ALS followed by MG, 5 ALS patients with false-positive anti-acetylcholine receptor, and the other 2 ALS patients with only myasthenia symptoms. Most patients had limb onset ALS, and myasthenia symptoms mainly affected ocular and bulbar muscles. Clinical and neurophysiological characteristics were summarized.
Conclusion: These findings support the conclusion that immunological mechanisms and alterations in the neuromuscular junction are related to ALS pathogenesis.
Introduction
Amyotrophic lateral sclerosis (ALS) and myasthenia gravis (MG) are different disorders affecting motor neurons and neuromuscular junctions, respectively. ALS is a progressive neurodegenerative disorder involving primarily motor neurons in the cerebral cortex, brain stem, and spinal cord, and it is characterized by muscle weakness and muscular atrophy (1). The pathogenesis of ALS is not well understood, resulting in a lack of appropriate therapy. By contrast, MG is a disorder of neuromuscular transmission, resulting from binding of autoantibodies to components of the neuromuscular junction, characterized by muscle weakness and fatigability (2). The most common antibody is against the acetylcholine receptor (AChR), and other targets, such as the muscle-specific kinase (MuSK) protein or lipoprotein-related protein 4 (LRP4), have been described (3). Treatment includes immune-regulating therapy and symptomatic therapy, resulting in good prognosis for most patients. Coexistence of ALS and MG has been described in a few reports. This coexistence is considered to be far beyond coincidence (4) and suggests a relationship between MG and ALS involving immunological mechanisms. In this study, we described two cases of our own and reviewed the cases previously published in English in detail.
Materials and Methods
Retrospective Study of Our Database
We searched all medical records at Peking Union Medical University Hospital (PUMCH) from January 1983 to December 2015 for International Classification of Diseases (ICD)-9 and ICD-10 diagnostic codes for coexistence of MG and ALS. We retrospectively re-evaluated the diagnoses based on clinical presentations and neurophysiological examinations, then followed up with patients by telephone.
Search of the Literature
PubMed, Embase, and Chinese biomedical databases were searched for papers published up to December 2016 that reported coexistence of ALS and MG. Search terms included (“amyotrophic lateral sclerosis” or “motor neuron disease” or “ALS” or “MND”) AND (“myasthenia gravis” or “myasthenia” or “MG”). Both text word and MeSH subject headings were used. The search strategy was supplemented by inspecting the reference lists of the included articles. Language was confined to English and Chinese. The papers were considered for inclusion if they described patients with coexistence of ALS and MG and provided diagnostic evidence. ALS was diagnosed according to the revised El Escorial World Federation of Neurology criteria (1). The diagnosis of MG was made based on clinical and accessory findings. Suggestive clinical features were fluctuating muscle weakness with exacerbation by exercise as well as improvement after cholinesterase inhibitor treatment. Laboratory tests supporting the diagnosis of MG were detection of specific autoantibody and a significant decrement in low-frequency repetitive nerve stimulation (RNS) test. Studies were excluded if they (1) reported coexistence of MG and other forms of neuronopathy, including spinal muscular atrophy and Kennedy’s disease (2); diagnosed either ALS or MG without sufficient evidence (3); and were epidemiological studies without sufficient patient details.
The clinical characteristics of each patient, including gender, nationality, interval between onset of the two diseases, clinical presentations, accessory exams, therapy, and prognosis, were extracted from the included studies. They were reviewed, and summarized and placed into groups according to strict diagnostic criteria.
Results
Case Presentation
Three cases were found from PUMCH medical records listed with diagnoses of both ALS and MG. Two of the cases were combined ALS and MG, and the other one was finally confirmed to be ALS that was misdiagnosed as MG at an early stage (not shown).
Case 1
One patient in his 40s presented with progressive left upper limb weakness with muscle atrophy for 8 months. Three months before consultation, fluctuating diplopia and ptosis appeared, along with paroxysmal palpitation, mild right hand tremor, irritability, chronic diarrhea, and a 5-kg weight loss. His right arm was also affected with mild weakness within the previous 3 weeks. On examination, bilateral lid paresis, ophthalmoplegia, and diplopia were noted, and the patient had a positive ptosis fatigue test. There was severe muscle atrophy in the left arm and both hands. The muscle strength of the patient’s upper limbs was 3/5 on MRC. Though manual muscle tests of the lower limbs were normal, fatigable weakness could be induced after exercise. The patient had brisk deep tendon reflexes in the lower limbs, bilaterally positive Hoffmann’s signs and Babinski’s signs. Electromyogram (EMG) revealed generalized motor neuron lesion with signs of both acute and chronic denervation. Significant decrement (up to 38%) was seen after RNS test on the left axillary nerve. A neostigmine test was positive. AChR antibodies (AChR-ab) were normal in concentration. A computed tomography (CT) scan of mediastinum showed thymic hyperplasia. The patient was also found to have hyperthyroidism and was prescribed Thyrozol. His ocular symptoms and fatigable weakness improved after treatment with intravenous immunoglobulin (IVIG) at a dose of 0.4 g/kg for 5 days, with sequential oral prednisone and pyridostigmine. However, limb weakness worsened irreversibly, and dysphagia, dysarthria, and dyspnea appeared during the following year.
Case 2
A patient in his 50s was diagnosed with ocular MG due to fluctuating bilateral ptosis and diplopia. The symptoms went into full remission after treatment with 60 mg QD pyridostigmine bromide. The patient was doing well without medication until 5 years later, when the ocular symptoms were aggravated, and he developed muscle weakness and wasting in bilateral lower limbs, progressively affecting all extremities and the bulbar region within 6 months. Pyridostigmine bromide (60 mg TID) could partially improve the ocular symptoms, but it could not be tolerated because of generalized fasciculation. Upon examination, he had bilateral ptosis, restricted eye movements and fasciculations in the tongue. Generalized brisk tendon reflexes, bilateral ankle clonus, Hoffmann’s sign, flexor plantar response, and spastic gait suggested upper motor neuronal damage. EMG showed fibrillation potentials and positive sharp waves together with signs of chronic denervation in all extremities, the paraspinal muscles, and the sternocleidomastoid. AChR-ab was 39.06 nmol/l (normal <0.4 nmol/l). RNS, thyroid function, and CT scan of the mediastinum were all normal. He was given 0.4 g/kg IVIG for 5 days, causing complete resolution of ocular symptoms, but the treatment had no effect on the general weakness. He was diagnosed with clinically probable ALS according to the revised El Escorial World Federation of Neurology criteria (1), and riluzole was prescribed. Respiratory function was eventually involved, and the patient died 28 months after ALS onset.
Characteristics of ALS and MG Overlap Syndrome
A total of 615 articles were identified. After removing duplicate articles, the 519 remaining articles were screened by title and abstract. A total of 23 full-text articles were assessed for eligibility. Four studies had an obscure diagnosis of either ALS or MG or lack of important information (5–8), five differentiated the two diseases but not their coexistence (9–13), two reported coexistence of MG and lower motor neuron syndrome (14, 15), and one described an ALS patient who developed myasthenia possibly due to the use of riluzole (16). Finally, 11 articles that reported cases diagnosed as coexistence of ALS and MG were identified (4, 17–26). A flow chart of the method of article retrieval is presented in Figure 1.
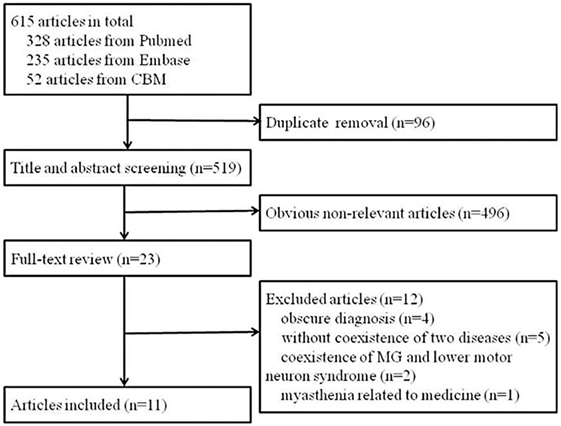
Figure 1. Flow chart of article retrieval.
Altogether, the characteristics of 25 cases reported in the literature and 2 cases of our own were reviewed. According to their clinical course and presentation, the cases could be roughly divided into four groups (Tables 1–3 for groups 1–3, respectively).
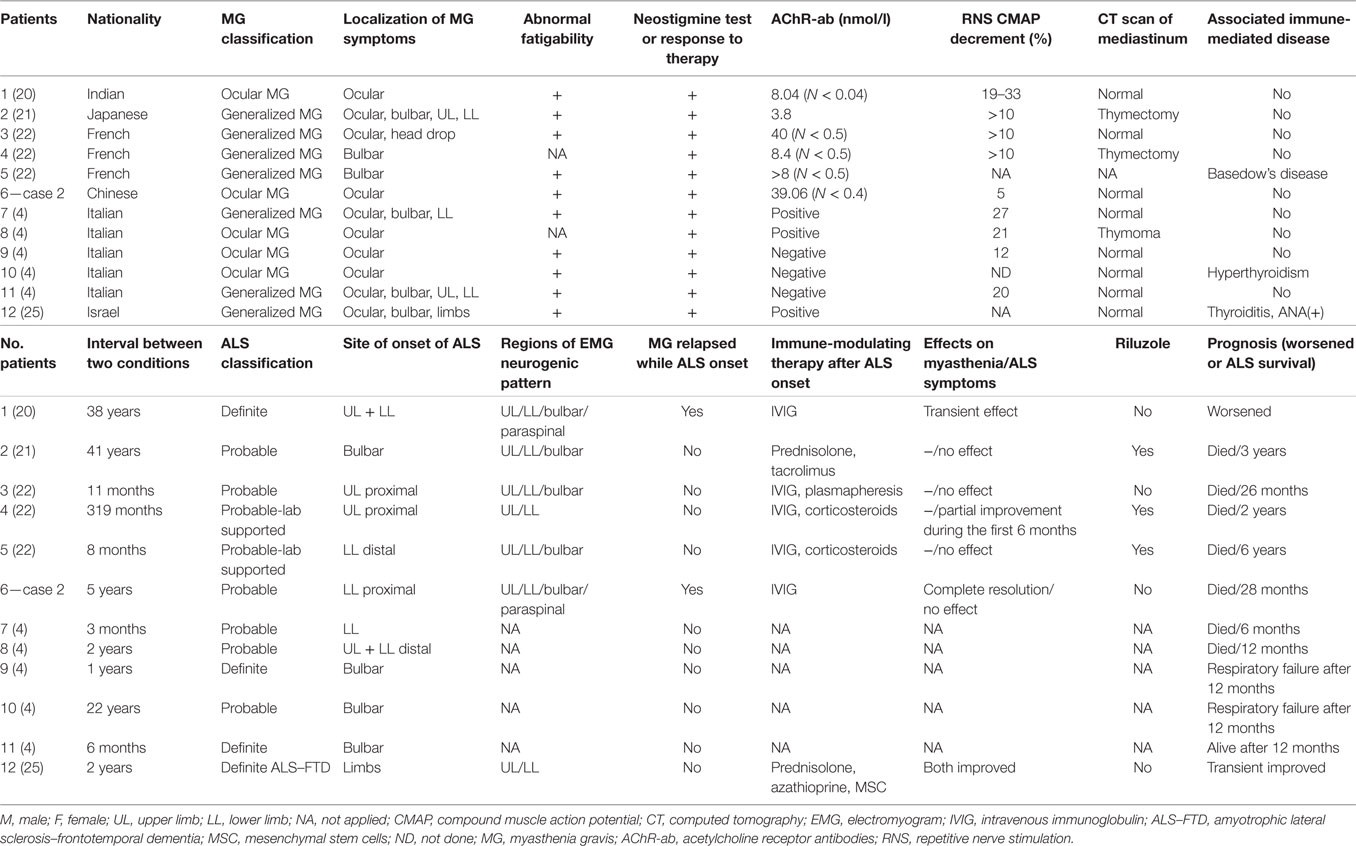
Table 1. Patients with MG as inaugural disease followed by ALS.
Group 1 included patients diagnosed with MG as the inaugural disease followed by ALS (details in Tables 1 and 4). There were 12 patients (male:female = 8:4) who had been diagnosed with MG 3 months to 41 years before they developed ALS. Although the onset of MG occurred at different ages (26–82 years old) and mainly affected ocular and bulbar muscles, they developed ALS at a mean age of 71 years old (55–83 years old) with limb or bulbar onset (limb:bulbar = 8:4). Comparing the localization of myasthenia symptoms and the site of onset of ALS for these patients, they were always in different regions except in three cases. Myasthenia symptoms in two patients relapsed or were aggravated concomitant with ALS symptoms, but they were sensitive to IVIG treatment. The mean survival time of seven cases was approximately 29 months.
Group 2 contained patients diagnosed with ALS as the inaugural disease followed by MG (details in Tables 2 and 4), including eight patients (male:female = 6:2). There was a profound discrepancy between their ages at onset of ALS (34–89 years old). However, most patients developed MG within the following 18 months except one. Four patients developed the two diseases almost at the same time, and it was difficult to distinguish the order of onset. As in Group 1, most cases (5/7) were limb onset ALS, and myasthenia symptoms only affected ocular and bulbar muscles. In addition to the most common AChR-ab associated MG in six patients, the other two patients were positive for LRP4-ab (24). Myasthenia symptoms could be improved or relieved through immune-modulating therapy, and some recovered spontaneously. It seemed that ALS in younger patients progressed more slowly (22).
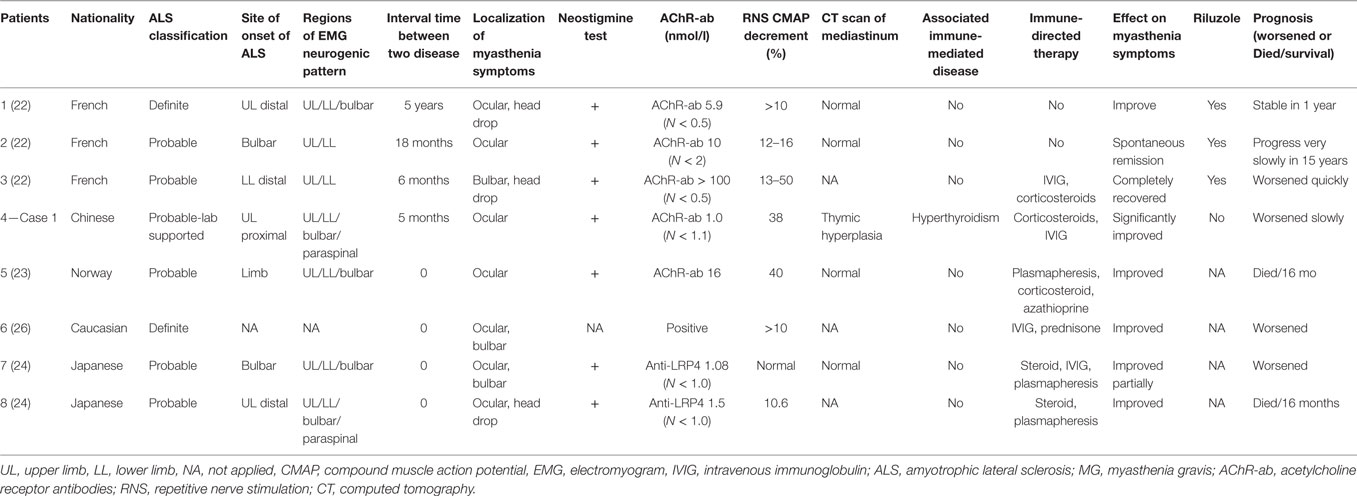
Table 2. Patients with ALS as inaugural disease followed by MG.
Group 3 contained five ALS patients (female:male = 3:2; age at onset of ALS: 37–88 years old) who were only positive for AChR-ab in sera (Table 3) but did not have any other characteristics of MG, including fluctuating symptoms, abnormal RNS test, or response to cholinesterase inhibitors. We characterize these patients as having ALS with false-positive AChR-ab as in previous papers (17).
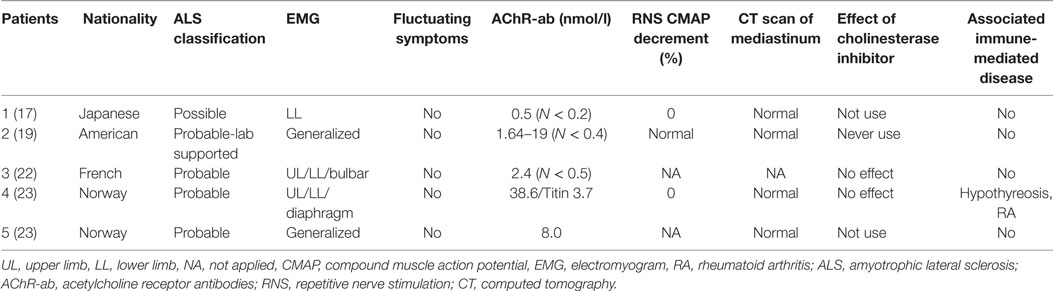
Table 3. ALS patients with false-positive AChR-ab.
Group 4: two clinically identified ALS patients presented with fluctuating ptosis and diplopia as initial symptoms followed by dysarthria, dysphagia, and limb muscle weakness, with normal concentration of sera AChR-ab and RNS test results. These patients were not responsive to cholinesterase inhibitors or immunosuppressive therapy (18). Evidence was considered insufficient to make the diagnosis of MG.
Discussion
Herein, we presented two cases from our database and summarized the clinical characteristics of all the other cases that have been reported. Coexistence of ALS and MG is rare. However, Turner et al. (27) found that there were significantly more cases of ALS associated with a prior autoimmune disease, including MG. Association between two diseases may be driven by dysregulation of the immune system in both conditions. General and tissue-specific immune activation is seen in ALS (28). Deficiency or decrease of T-regulatory cells (Treg cells) (29, 30), upregulated atrophy-related atrogenes, and neuronal nitric oxide synthase abnormalities (31, 32) could be found in both MG and ALS. Immunoglobulin from ALS patients can affect neuromuscular junction functional characteristics (33), and the activity of AChRs also seems to play a role in the innervation and re-innervation of muscle fibers (28). Experimental evidence indicates that muscle and neuromuscular junctions may be sites of disease manifestation in a very early stage of ALS (34). According to the “dying-back” hypothesis, it is possible that neuromuscular transmission failure and postsynaptic membrane damage in MG patients may precede lower and upper motor neuron loss, thereby increasing the probability of developing ALS (28).
For patients who had ever been diagnosed with MG (as in Group 1), if their muscle weakness relapsed and/or progressed to more regions accompanied by upper motor neuron signs and was unresponsive to AChR inhibitors or immune-modulating therapy, it was not difficult to make an accurate diagnosis of ALS according to the EEC criteria (1). By comparing the localization of myasthenia symptoms and the site of onset of ALS in Group 1, we found that they were always at different regions except in three cases. In addition, muscle atrophy is a common sign of ALS but is relatively rare in MG patients. Therefore, when muscle weakness reappeared or was aggravated in affected muscles in different regions from prior MG or was accompanied by muscle wasting, reexamination of EMG should be recommended and a possible diagnosis of ALS should be taken into consideration. It has been reported that some bulbar onset MG with positive anti-AChR or anti-MuSK antibodies can mimic ALS (23, 35–37), so experimental immune-modulating therapy is necessary when it is too difficult to differentiate MG from ALS.
In the Italian case series, patients with ALS after MG had frequent bulbar onset of ALS symptoms (60%) (4) compared to 17% in a French study (22). The difference may come from bias due to a small number of cases. Taking all reported cases together, the proportion of bulbar onset ALS (36%, shown in Table 4) was consistent with epidemiological data of ordinary sporadic ALS (reported bulbar onset in 30–40% of ALS cases).
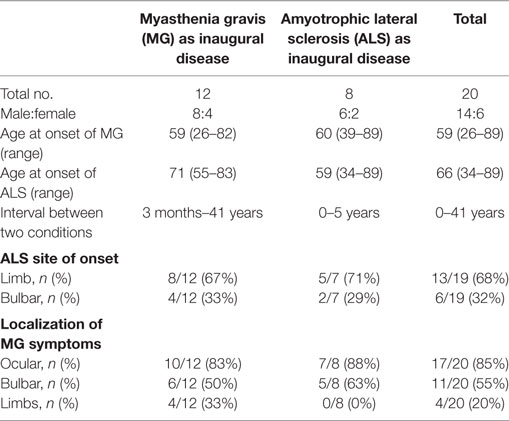
Table 4. Summary of patients in Group 1 and Group 2.
In Group 1, the mean age at onset of ALS was 71 years old, which is much older than that of patients in Group 2 (mean age of 59 years old, Table 4) or general sporadic ALS patients (reported mean age of 58–63 years old) (38). We speculate that immune-modulating therapy for prior MG at an early stage may have some protective or suppressive effect to delay the onset of motor neuron damage. Immune therapy trials have been unsuccessful in ALS (39, 40); however, timing is of great significance considering that ALS patients might be already in a late stage of the disease with irreparable motor neuron damage at the time of diagnosis. If we could identify ALS very early and initiate immune-modulating therapy at an asymptomatic stage, would it be effective? This possibility is supported by evidence that many molecular changes occur at the neuromuscular junctions at very early stages of ALS prior to symptom onset. By approaching therapeutic intervention from the three sides of the neuromuscular junction (the motor neuron, the muscle fiber and the terminal Schwann cells), the neuromuscular junction may be stimulated to remain in a healthy state for longer, and therefore, the onset of the disease might be more efficiently postponed (41). However, it should be noted that this is still speculation based on a clinical phenomenon, and the hypothesis needs to be verified by more well-established evidence.
As in Group 2, if ALS patients complain of fluctuating muscle weakness, concurrence of MG should be considered. RNS tests, assays of autoimmune antibodies related to the neuromuscular junction and experimental cholinesterase inhibitor treatment are useful methods for establishing diagnosis. As we can see in Group 2, the myasthenia symptoms mainly involved ocular and bulbar regions, presenting as ptosis, diplopia, dysphagia, dysarthria, and head drop, which might represent a phenotype prone to this association. Occurrence of ophthalmoplegia is rare in ALS; however, in those with advanced or bulbar onset ALS, it is likely that more extensive pathological changes in the brainstem give rise to supra-nuclear gaze palsy or slow saccades. When ophthalmoplegia occurs in prolonged or brainstem involved ALS without fluctuating phenomenon, fatigability, abnormal RNS and autoimmune antibodies, the diagnosis would not be myasthenia.
Acetylcholine receptor antibodies are considered to be highly specific for the diagnosis of MG, and these autoantibodies could also be found in 5% of ALS patients without clinical evidence of MG (42). One study traced the fluctuation of AChR-ab titers during the progression of ALS, and it seemed to increase during the periods when the patient’s ALS became more severe and decrease during periods when the patient was clinically more stable (17). The mechanism by which AChR-ab are produced in ALS cases is unknown. It may be related to the morphological alterations of neuromuscular junctions that are present at an early stage (43) due to autoimmune reaction against degenerative AChR at neuromuscular junctions. There were three patients with negative AChR-ab in Group 2, and two of them were positive for LRP4 antibodies. A recent study noted that LRP4 antibodies could be detected in 24 out of 104 ALS patients (23%) whose plasma was negative for AChR antibodies (44). Since LRP4 antibodies are found more frequently in ALS patients than in MG patients, they may have a direct pathogenic activity in the denervation process of ALS. The first patient we described was AChR-ab and MUsK-ab negative, but we did not test for LRP4 antibodies. It is worth noting that a compound muscle action potential decrease in an RNS test is not a specific phenomenon of MG, as it can also be found in 53% of typical ALS patients (45, 46). In ALS and MG overlap patients, an RNS test could also be negative, as for the last two patients in Table 2 (24). All patients in Group 2 were responsive to cholinesterase inhibitors, but patients in Groups 3 and 4 were not. Considering that fluctuating symptoms could be overlooked due to a marked weakness in ALS and that the specificity of the RNS test may be decreased, we recommend that in the context of ALS, the diagnosis of concomitant MG syndrome should thus be discussed only in the presence of suggestive clinical features (fluctuating symptoms and fatigability especially involved in ocular and/or bulbar regions) or autoimmune antibodies related to neuromuscular junction in addition to a positive response to cholinesterase inhibitors.
Conclusion
The coexistence of ALS and MG is rare and requires thoughtful interpretation of clinical manifestations. These findings indicate a relationship between the two diseases and support the hypothesis that immunological mechanisms and alterations in the neuromuscular junction are related to ALS pathogenesis. Immune-modulating therapy at an early stage before onset of ALS symptoms might have protective effects on postponing motor neuron degeneration.
Ethics Statement
This study was approved by the Ethics Committee of Clinical Research of Peking Union Medical College Hospital (Beijing, China), with written informed consent from all subjects whose medical records were reviewed.
Author Contributions
HT: conception of the work, data acquisition and analysis, literature search, and writing of the first draft. LC: conception and organization of the work, manuscript review, and critique. YG, ML, XL, YH, JY, DS, DL, and FZ: clinical evaluation of patients, review, and critique.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work was supported by CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-12M-1-004).
References
1. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord (2000) 1(5):293–9. doi: 10.1080/146608200300079536
2. Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology (2016) 87(4):419–25. doi:10.1212/WNL.0000000000002790
3. Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B. Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun (2014) 4(8–49):143–8. doi:10.1016/j.jaut.2014.01.003
4. de Pasqua S, Cavallieri F, D’Angelo R, Salvi F, Fini N, D’Alessandro R, et al. Amyotrophic lateral sclerosis and myasthenia gravis: association or chance occurrence? Neurol Sci (2017) 38(3):441–4. doi:10.1007/s10072-016-2787-3
5. Noseworthy JH, Rae-Grant AD, Brown WF. An unusual subacute progressive motor neuronopathy with myasthenia-like features. Can J Neurol Sci (1988) 15(3):304–9. doi:10.1017/S0317167100027797
6. Mulder DW, Lambert EH, Eaton LM. Myasthenic syndrome in patients with amyotrophic lateral sclerosis. Neurology (1959) 9:627–31. doi:10.1212/WNL.9.10.627
7. Pandian JD, Mathuranath PS, Suresh PA, Radhakrishnan K. Myasthenia gravis, motor neuron syndrome and thymoma. Neurol India (1998) 46(3):232–4.
8. Piccirillo G, Trojsi F, Monsurrò MR, Habetswallner F, De Martino BM, Tedeschi G. Intravenous immunoglobulins efficacy in a case of ALS with myasthenic symptoms. Eur J Neurol (2016) 23:647. doi:10.1111/ene.13094
9. Carpenter RJ III. The primary head and neck manifestations of myasthenia gravis and amyotrophic lateral sclerosis: a diagnostic aid. Conn Med (1980) 44(5):284–6.
10. Fiacchino F, Scaioli V, Antozzi C, Giannini A. Upper motor neuron dysfunction reduces the EMG evidence of myasthenia gravis. J Neurosurg Anesthesiol (1995) 7(3):178–82. doi:10.1097/00008506-199507000-00004
11. Garfinkle TJ, Kimmelman CP. Neurologic disorders: amyotrophic lateral sclerosis, myasthenia gravis, multiple sclerosis, and poliomyelitis. Am J Otolaryngol (1982) 3(3):204–12. doi:10.1016/S0196-0709(82)80056-2
12. Wong WW, Lane RJ. Transient myasthenia gravis in an elderly woman. J Neurol Neurosurg Psychiatry (2004) 75(9):1363. doi:10.1136/jnnp.2003.034124
13. Nanri K, Harukawa H, Niiyama K, Ishiko T, Saito H, Taguchi T, et al. Three cases of motor neuron disease resembling progressive amyotrophy with examination findings suggestive of myasthenia gravis. J Tokyo Med Univ (2006) 64(5):483–8.
14. Sostarko M, Brzovic Z, Vranjes D. Motor neurone disease associated with several immunological disorders: a case report. J Neurol Sci (1994) 124(Suppl):70–1. doi:10.1016/0022-510X(94)90183-X
15. Steiner I, Goldstein L, Hellmann MA, Lotan I. Prior damage to lower motor neuron triggering myasthenia gravis. Muscle Nerve (2016) 54(1):167–9. doi:10.1002/mus.25026
16. Restivo DA, Bianconi C, Ravenni R, De Grandis D. ALS and myasthenia: an unusual association in a patient treated with riluzole. Muscle Nerve (2000) 23(2):294–5. doi:10.1002/(SICI)1097-4598(200002)23:2<294::AID-MUS25>3.0.CO;2-G
17. Okuyama Y, Mizuno T, Inoue H, Kimoto K. Amyotrophic lateral sclerosis with anti-acetylcholine receptor antibody. Intern med (1997) 36(4):312–5. doi:10.2169/internalmedicine.36.312
18. Pinto S, de Carvalho M. Amyotrophic lateral sclerosis patients and ocular ptosis. Clin Neurol Neurosurg (2008) 110(2):168–70. doi:10.1016/j.clineuro.2007.08.022
19. Mehanna R, Patton EL Jr, Phan CL, Harati Y. Amyotrophic lateral sclerosis with positive anti-acetylcholine receptor antibodies. Case report and review of the literature. J Clin Neuromuscul Dis (2012) 14(2):82–5. doi:10.1097/CND.0b013e31824db163
20. Naik KR, Saroja AO, Mahajan M. Unusual occurrence of amyotrophic lateral sclerosis in myasthenia gravis. Amyotroph Lateral Scler (2012) 13(5):477–8. doi:10.3109/17482968.2012.711836
21. Yamashita S, Fujimoto A, Mori Y, Hirahara T, Mori A, Hirano T, et al. Coexistence of amyotrophic lateral sclerosis and myasthenia gravis. J Neuromuscul Dis (2014) 1(1):111–5.
22. Del Mar Amador M, Vandenberghe N, Berhoune N, Camdessanche JP, Gronier S, Delmont E, et al. Unusual association of amyotrophic lateral sclerosis and myasthenia gravis: a dysregulation of the adaptive immune system? Neuromuscul Disord (2016) 26(6):342–6. doi:10.1016/j.nmd.2016.03.004
23. Gotaas HT, Skeie GO, Gilhus NE. Myasthenia gravis and amyotrophic lateral sclerosis: a pathogenic overlap. Neuromuscul Disord (2016) 26(6):337–41. doi:10.1016/j.nmd.2016.03.003
24. Takahashi H, Noto YI, Makita N, Kushimura-Okada Y, Ishii R, Tanaka A, et al. Myasthenic symptoms in anti-low-density lipoprotein receptor-related protein 4 antibody-seropositive amyotrophic lateral sclerosis: two case reports. BMC Neurol (2016) 16(1):229. doi:10.1186/s12883-016-0758-1
25. Petrou P, Argov A, Lennon VA, Gotkine M, Kassis I, Vaknin-Dembinsky A, et al. Rare combination of myasthenia and motor neuronopathy, responsive to MSC-NTF stem cell therapy. Muscle Nerve (2014) 49(3):455–7. doi:10.1002/mus.24143
26. Vincent H, McNeal T. Double trouble: a case of myasthenia gravis and amyotrophic lateral sclerosis. J Investig Med (2011) 59(2):374–5. doi:10.231/JIM.0b013e31820bab4c
27. Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology (2013) 81(14):1222–5. doi:10.1212/WNL.0b013e3182a6cc13
28. Malaspina A, Puentes F, Amor S. Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol (2015) 27(3):117–29. doi:10.1093/intimm/dxu099
29. Rentzos M, Evangelopoulos E, Sereti E, Zouvelou V, Marmara S, Alexakis T, et al. Alterations of T cell subsets in ALS: a systemic immune activation? Acta Neurol Scand (2012) 125(4):260–4. doi:10.1111/j.1600-0404.2011.01528.x
30. Alahgholi-Hajibehzad M, Kasapoglu P, Jafari R, Rezaei N. The role of T regulatory cells in immunopathogenesis of myasthenia gravis: implications for therapeutics. Expert Rev Clin Immunol (2015) 11(7):859–70. doi:10.1586/1744666X.2015.1047345
31. Meinen S, Lin S, Ruegg MA, Punga AR. Fatigue and muscle atrophy in a mouse model of myasthenia gravis is paralleled by loss of sarcolemmal nNOS. PLoS One (2012) 7(8):e44148. doi:10.1371/journal.pone.0044148
32. Suzuki N, Mizuno H, Warita H, Takeda S, Itoyama Y, Aoki M. Neuronal NOS is dislocated during muscle atrophy in amyotrophic lateral sclerosis. J Neurol Sci (2010) 294(1–2):95–101. doi:10.1016/j.jns.2010.03.022
33. Appel SH, Engelhardt JI, Garcia J, Stefani E. Immunoglobulins from animal models of motor neuron disease and from human amyotrophic lateral sclerosis patients passively transfer physiological abnormalities to the neuromuscular junction. Proc Natl Acad Sci U S A (1991) 88(2):647–51. doi:10.1073/pnas.88.2.647
34. Krakora D, Macrander C, Suzuki M. Neuromuscular junction protection for the potential treatment of amyotrophic lateral sclerosis. Neurol Res Int (2012) 2012:379657. doi:10.1155/2012/379657
35. Basiri K, Ansari B, Okhovat AA. Life-threatening misdiagnosis of bulbar onset myasthenia gravis as a motor neuron disease: how much can one rely on exaggerated deep tendon reflexes. Adv Biomed Res (2015) 4:58. doi:10.4103/2277-9175.151874
36. Furuta N, Ishizawa K, Shibata M, Tsukagoshi S, Nagamine S, Makioka K, et al. Anti-MuSK antibody-positive myasthenia gravis mimicking amyotrophic lateral sclerosis. Intern Med (2015) 54(19):2497–501. doi:10.2169/internalmedicine.54.4645
37. Huijbers MG, Niks EH, Klooster R, de Visser M, Kuks JB, Veldink JH, et al. Myasthenia gravis with muscle specific kinase antibodies mimicking amyotrophic lateral sclerosis. Neuromuscul Disord (2016) 26(6):350–3. doi:10.1016/j.nmd.2016.04.004
38. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet (2011) 377(9769):942–55. doi:10.1016/S0140-6736(10)61156-7
39. Meucci N, Nobile-Orazio E, Scarlato G. Intravenous immunoglobulin therapy in amyotrophic lateral sclerosis. J Neurol (1996) 243(2):117–20. doi:10.1007/BF02444000
40. Werdelin L, Boysen G, Jensen TS, Mogensen P. Immunosuppressive treatment of patients with amyotrophic lateral sclerosis. Acta Neurol Scand (1990) 82(2):132–4. doi:10.1111/j.1600-0404.1990.tb01602.x
41. Moloney EB, de Winter F, Verhaagen J. ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci (2014) 8:252. doi:10.3389/fnins.2014.00252
42. Ashizawa T. False positive anti-acetylcholine receptor antibodies in motorneurone disease. Lancet (1986) 1(8492):1272. doi:10.1016/S0140-6736(86)91408-X
43. Bruneteau G, Bauche S, Gonzalez de Aguilar JL, Brochier G, Mandjee N, Tanguy ML, et al. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol (2015) 2(4):362–72. doi:10.1002/acn3.179
44. Tzartos JS, Zisimopoulou P, Rentzos M, Karandreas N, Zouvelou V, Evangelakou P, et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol (2014) 1(2):80–7. doi:10.1002/acn3.26
45. Killian JM, Wilfong AA, Burnett L, Appel SH, Boland D. Decremental motor responses to repetitive nerve stimulation in ALS. Muscle Nerve (1994) 17(7):747–54. doi:10.1002/mus.880170708
Keywords: amyotrophic lateral sclerosis, myasthenia gravis, overlap, clinical characteristics, immunological mechanism
Citation: Tai H, Cui L, Guan Y, Liu M, Li X, Huang Y, Yuan J, Shen D, Li D and Zhai F (2017) Amyotrophic Lateral Sclerosis and Myasthenia Gravis Overlap Syndrome: A Review of Two Cases and the Associated Literature. Front. Neurol. 8:218. doi: 10.3389/fneur.2017.00218
Received: 13 January 2017; Accepted: 04 May 2017;
Published: 22 May 2017
Edited by:
Mark P. Burns, Georgetown University, USAReviewed by:
Ian Paul Johnson, University of Adelaide, AustraliaPatrizia Longone, Fondazione Santa Lucia, Italy
Copyright: © 2017 Tai, Cui, Guan, Liu, Li, Huang, Yuan, Shen, Li and Zhai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liying Cui, cHVtY2hjdWlseUB5YWhvby5jb20=