 Zhengqing Zhao
Zhengqing Zhao Xiangxiang Zhao1†
Xiangxiang Zhao1† Sigrid C. Veasey
Sigrid C. Veasey- 1Department of Neurology, Changzheng Hospital, Second Military Medical University, Shanghai, China
- 2Center for Sleep and Circadian Neurobiology, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Approximately one-third of adolescents and adults in developed countries regularly experience insufficient sleep across the school and/or work week interspersed with weekend catch up sleep. This common practice of weekend recovery sleep reduces subjective sleepiness, yet recent studies demonstrate that one weekend of recovery sleep may not be sufficient in all persons to fully reverse all neurobehavioral impairments observed with chronic sleep loss, particularly vigilance. Moreover, recent studies in animal models demonstrate persistent injury to and loss of specific neuron types in response to chronic short sleep (CSS) with lasting effects on sleep/wake patterns. Here, we provide a comprehensive review of the effects of chronic sleep disruption on neurobehavioral performance and injury to neurons, astrocytes, microglia, and oligodendrocytes and discuss what is known and what is not yet established for reversibility of neural injury. Recent neurobehavioral findings in humans are integrated with animal model research examining long-term consequences of sleep loss on neurobehavioral performance, brain development, neurogenesis, neurodegeneration, and connectivity. While it is now clear that recovery of vigilance following short sleep requires longer than one weekend, less is known of the impact of CSS on cognitive function, mood, and brain health long term. From work performed in animal models, CSS in the young adult and short-term sleep loss in critical developmental windows can have lasting detrimental effects on neurobehavioral performance.
Chronic Short Sleep (CSS) in Humans is Commonly Observed
Chronic short sleep, defined as frequently obtaining ≤6 h sleep/24 h period, is highly prevalent in both adults and adolescents. In the United States, CSS is reported by 30% of employed adults (night and day shifts) or over 40 million individuals (1). The CSS definition cutoff of 6 h may underestimate the number of people regularly experiencing insufficient sleep. Specifically, an in-lab study of healthy young adult humans examining the cumulative effects of 4–10 h of sleep on vigilance performance determined with mathematical modeling that >8 h of sleep/night is needed to prevent decrements in vigilance (2). College students are also likely to experience CSS. A poll conducted among college students found that 25% of the students regularly obtained <6.5 h of sleep and 70% obtained <8 h/night (3). A poll obtained in the United States found that 97% of 12th grade students report less than the 9 h of sleep per night recommended for teenagers, and 75% reported regularly obtaining <8 h/night (4). There are intriguing geographical and/or cultural variations in reported sleep time. Within the United States, the region with the highest percentage of individuals reporting regularly obtaining insufficient sleep is within the mid-Atlantic Appalachian mountains (5). In South Korea, the mean reported sleep time for teenagers in one large study was 4.9 h/night (6). In contrast, Australian adolescents average 8.5–9.1 h/night and less commonly sleep in over weekends (7). Caffeine use, electronic devices in bedrooms, and school start times have all been shown to influence total sleep times for adolescents (8). Delaying school start time by 1 h in the United States increases sleep times in high school students and reduces both sleepiness and the incidence of student motor vehicle accidents (9–11), suggesting that circadian phase delay contributes at least in part to the CSS in American adolescents. In summary, high percentages of the workforce, college and high school students regularly experience CSS, where both cultural and biologic factors contribute to the high prevalence of CSS. While cardiovascular and metabolic effects of CSS have been substantiated (12–14), we are only beginning to explore the lasting neurobehavioral consequences of insufficient sleep. At present, we do not know whether the shorter sleep times have lasting effects on neurobehavioral performance and/or brain aging.
To try to understand needed sleep times, several recent studies have examined sleep times in adults living in pre-industrialized societies (15–17). One study compared two regionally proximal populations, one with and one without electricity, found that those with access to artificial light slept almost 1 h less (16). However, average sleep durations for three distinct groups of hunter-gatherers each on a different continent had total sleep times of 6–7 h (17), which is not different from average sleep times in developed countries. None of these studies, however, addressed the question how much sleep is needed for optimal performance. It is interesting that in controlled laboratory polysomnography studies in developed societies, sleep times in most individuals allowed 9 h time in bed were over 8 h (2, 18), suggesting that sleep needs may vary across developed and undeveloped societies, more than geographically. It is also possible that individuals are limited by wakefulness activities and duration, to a larger extent than sleep duration (2).
Protracted Recovery of Neurobehavioral Impairment Following CSS in Humans
Contrary to the widely appreciated subjective normalization of sleepiness after a weekend of recovery sleep, several studies have now shown clearly that shortened sleep across 1 week in healthy adults produces cumulative impairments in vigilance with incomplete recovery after three full nights of recovery sleep (2, 18, 19). Unfortunately in each of the studies examining recovery after partial sleep loss across the week, the recovery was not extended long enough to observe full recovery. Subjective improvement in sleepiness is greater than objective improvement, which may help explain why the pattern is repeated week after week in so many individuals (20). Incomplete recovery in vigilance raises the possibility of lasting injury or neuronal dysfunction from CSS. While vigilance remains impaired after three nights of recovery sleep, performance with other tasks sensitive to sleep loss, such as a visual-motor task, normalizes after the second night of recovery sleep (21). In adolescents, sleep architecture after seven nights of sleep restriction (5 h sleep) remained abnormal after three nights of recovery sleep, with greater sleep efficiency and a shorter latency to non-rapid eye movement (NREM) stage 2 sleep (22). Moreover, neurobehavioral performance in adolescents may be more vulnerable to sleep restriction than in adults. When adolescents are restricted to 4 h sleep per night for seven nights, processing speed remained slowed even after two full recovery nights of sleep (23). Collectively, these findings support: (1) recovery requiring >3 nights for specific neurobehavioral performances; (2) a differential susceptibility to CSS across various tasks, where vigilance is particularly sensitive to CSS; (3) a differential susceptibility for age in that adolescents may require longer to recover from CSS neurobehavioral impairments; and (4) recovery of circuits integral to behavioral state and/or vigilance is delayed following CSS.
Animal Models of Sleep Loss
Various sleep disruption protocols have been implemented in animal research. Each model has its own set of benefits, limitations, and addressable sleep research questions as well as its own set of potentially confounding variables. Because of its simplicity and ease to administer, many chronic sleep loss studies have used permutations of the platform-over-water technique, in which animals must stay awake and maintain balance on a small platform to avoid falling into a water tray (24–28). The platforms can be sized to minimize primarily rapid eye movement sleep (REMS) or both non-rapid eye movement sleep (NREMS) and REMS. In earlier studies, animals were singly housed. This particular paradigm, however, causes significant stress for most rodents (28). More recently, chambers with multiple platforms have been utilized, that allow group housing (29). Nonetheless, animals cannot nest or sleep next to one another, and animals slept deprived by the platform method tend to lose weight, suggesting some influence on metabolism or stress (29). Several sleep deprivation apparatuses have been developed to enforce locomotion to maintain wakefulness. These devices have a sweeping disk, treadmill, or bar that prods a sleeping animal to move (30). This technique is also highly effective in inducing sleep fragmentation, but may not prevent all sleep (31). In fact, clever animals can learn to sleep upon the sweeping bar. The forced activity devices require single housing and animals will have microsleeps, between times when the bar or disk prods the animal (31). Mice also may be kept awake by gentle handling which allows mice to remain in their home cages with cage mates (32, 33). The gentle handling method is labor intensive, requiring a researcher to monitor animals continuously and provide a tactile stimulus when animals are still. An additional method effectively implemented for chronic sleep loss is environmental enrichment (34, 35). Researchers have used this technique to maintain continuous active wakefulness for 3–8 h/day across repeated days, modeling sleep patterns in shift workers or students pulling late study nights (35, 36). After the initial exposure, animals exposed to this model have normal corticosterone levels to suggest no major effects on stress (35). Moreover, animals exposed to enriched environment sleep loss do not lose weight across the paradigm. The animals catch up on some of the missed sleep across the undisturbed dark period, so that overall, the animals lose approximately 4 h of sleep in a 24-h period (35). This method of chronic sleep loss is labor intensive, requiring a researcher to be present at all times to monitor animals and add new bedding, toys, etc. when mice are still. This particular method is most appropriate when examining the effects of extended exploratory wakefulness, as mice can be effectively kept awake with minimal microsleeps for at least 8 h. In summary, the implemented sleep loss paradigms vary by stress induced, metabolic effects, locomotor effects, microsleeps, and intermittent sleep periods. Newer methods of sleep loss, including optogenetic and chemical genetic long-term stimulation of wake-activating neurons to induce extended wakefulness and sleep fragmentation, are being optimized for long-term effectiveness. Pharmacogenetics would allow non-invasive enforced wakefulness in group housed mice, while optogenetics is expected to offer a valuable window into the molecular responses in wake neurons to various levels and durations of activation of specific groups of neurons. Very few paradigms have been compared within a lab or across species to provide insight into species differences and sleep deprivation models differences in neural effects. Table 1 summarizes approaches, species, and neural findings for experiments.
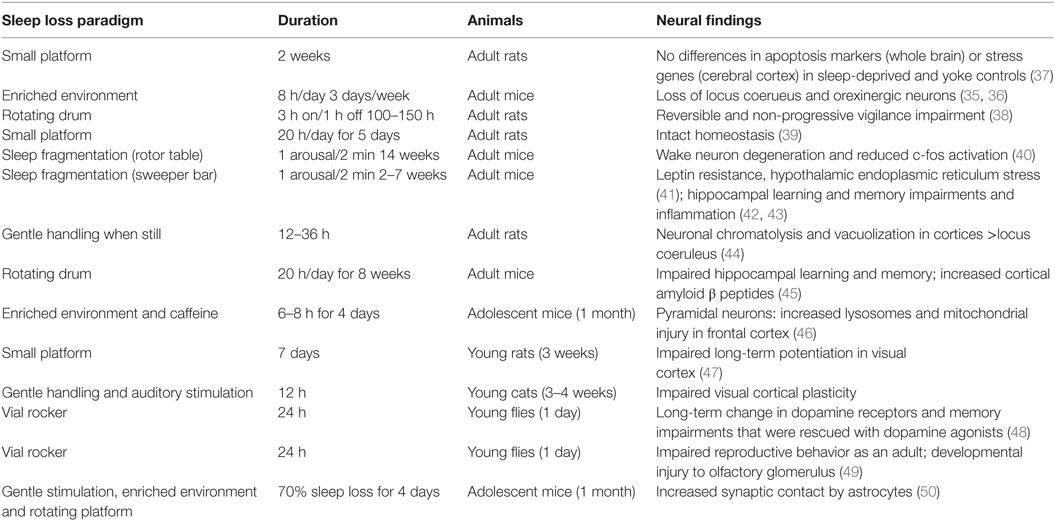
Table 1. Neural effects of sleep disruption across various experimental models and species.
Neural Findings in the Rechtschaffen Rat Studies of Sleep Deprivation
In landmark studies in the late 1970s and early 1980s, Allan Rechtschaffen and collaborators showed that continuous sleep deprivation in adult rats, using singly housed disk over water methodology, resulted in death of the animals within 2–4 weeks (51). While the cause of death was not evident in all cases, all rats lost tremendous amounts of weight and developed both malnutrition (protein wasting) and chronic skin ulcerations. Many of the animals succumbed to bacterial sepsis. Remarkably, rats on the brink of death from total sleep deprivation could experience what appeared to be a complete physiological recovery if allowed 2 weeks of uninterrupted recovery sleep (52). Despite the prominent peripheral physiological effects from total sleep deprivation, brains of rats exposed to prolonged sleep deprivation did not evidence obvious injury. Specifically, there was no increase in apoptosis or other neural markers of degeneration when examining 6–10 sections throughout the brain for all cell types (37). If animals exposed to this severe sleep loss do not incur obvious neurodegeneration, should we expect injury with the less severe chronic sleep restriction, as experienced by humans? Several important considerations are necessary to most accurately address this question. First, sleep-deprived animals in this platform paradigm were compared to yoked control animals that were also exposed to single housing on a platform over water (stressful for rodents) where some sleep loss and sleep fragmentation also occurred in these yoked controls. Thus, it is possible that less severe sleep loss in controls and severe sleep loss cause comparable injuries. Second, accurate determination of neurodegeneration requires performing neuronal stereology; other less robust measures may lead to inaccurate conclusions. Third, if only small subsets of specific neurons are injured, examining neurons collectively might obscure differences, and fourth, finding normal cell counts does not ensure normal brain function. The brain relies on connectivity and neuronal responsiveness to function well. For these reasons, it is possible that less severe chronic sleep loss could injure and even kill select populations of neurons in the brain, impair functionality and/or connectivity.
Lasting Effects of CSS on Wake-Active Neurons
The incomplete cognitive recovery in vigilance observed in humans raises the possibility of lasting injury from CSS. Within the brain and brainstem are specific populations of neurons that are activated across wakefulness, quiescent only during sleep (53). One of these groups of neurons, the brainstem locus coeruleus neurons (LCn) is a collection of neurons essential for vigilance (54, 55). If sleep loss injures LCn from extended wakefulness, the injury should be greater for longer durations of wakefulness, and, indeed, LCn respond to short sleep loss (one period of 3 h of sleep loss) with an upregulation of antioxidant enzymes (35). With extended sleep loss (modeling 8 h of sleep loss on three consecutive days, followed by catch up sleep opportunities during the normal active period), there is no upregulation of antioxidant enzymes, and importantly, the neurons evidence increased reactive oxygen species (35). Sirtuins are deacetylase enzymes that play key roles in cellular homeostasis. The major mitochondrial sirtuin is sirtuin type 3 (SirT3) (56). Short-term sleep loss resulted in LC neuron upregulated SirT3, which then activates FOXO3a leading to antioxidant transcription (35). Importantly, in short-term sleep loss, SirT3 was essential for the adaptive upregulation of antioxidants and maintenance of redox homeostasis in LC neurons, in that mice lacking SirT3 did not upregulate antioxidants and consequently accrued superoxide in the LC across just 3 h of wake (35). The adaptive SirT3-dependent response, however, was short-lived. Upon extended sleep loss, 8 h sleep loss on three consecutive days, the SirT3 response failed, as evidenced by mitochondrial protein hyperacetylation (35); LC neurons did not upregulate antioxidant enzymes, and LC neurons succumbed to severe oxidative stress including the accumulation of lipofuscin, indicative of mitochondrial injury and senescence. Most importantly, extended wake resulted in a 25–30% loss in LC neurons, demonstrating for the first time that CSS can result in neuron loss (35). Recently, we extended the duration of CSS to 4 weeks in wild-type mice to ascertain whether longer CSS exposures increase injury further, to identify other groups of neurons might be injured and to examine reversibility (36). LC neuron loss was greater (40%) with 4 weeks CSS and did not recover with 1 month of rest, supporting irreversible loss of neurons rather than down regulation of noradrenergic specific markers (36). In addition to a loss of LC neurons, orexinergic neurons (another wake-activated group of neurons) were also lost in part (40%), while a sleep-active group of neurons, the melanin-concentrating hormone neurons, adjacent to orexinergic neurons in the hypothalamus, conferred resistance to CSS neuron loss (36). Importantly, CSS results in lasting sleep/wake impairments, in that despite 4 weeks recovery sleep, CSS mice had less sleep during the normal sleep period (lights-on) and less wake in the active period (dark period). This pattern is consistent with sleep patterns observed at later stages in life, supporting the concept that CSS may injure the brain, at least in part, by aging aspects of brain function. In support of aging of the brain, we examined sirtuins. SirT1 and SirT3 decline in the brain with aging, where reductions with age contribute to age-related degeneration (57–59). LC and orexinergic neurons evidenced reductions in SirT1 and SirT3 and had increased lipofuscin, and these age-dependent effects also persisted at least 1 month into recovery, suggesting heightened vulnerability for a long time after CSS (36). The injury to LCn may not only affect vigilance but may also affect brain health, including the temporal progression of Alzheimer’s disease (AD). Specifically, in mouse models of AD, LC neuron lesions increase plaque burden, cognitive impairments, and tau hyperphosphorylation, while replenishing norepinephrine lessens deficits (27, 29, 60–62). In summary, CSS induces degeneration in select populations of neurons, including LC and orexinergic neurons, reduces sirtuins in the remaining wake-activated neurons and imparts a lasting disruption in sleep/wake patterns, supporting the concept that chronic insufficient sleep has lasting effects on brain health and function.
Locus Coeruleus Response to Unihemispheric Sleep
A diverse collection of animal species demonstrate unihemispheric sleep, including aquatic mammals such as dolphin, seals, and sea lions, and many avian species. Fur seals can switch between bilateral hemispheric sleep when on land to unihemispheric sleep for extended periods when at sea (63). Presumably, unihemispheric sleep allows the animal motor activity to both keep the animal’s nose at the water’s surface and to maintain body temperature with muscle activity. Interestingly, during unihemispheric sleep, norepinephrine delivery to the cortex (from locus coeruleus) is reduced in both hemispheres (63). Similar responses are observed for two other wake monoaminergic neurotransmitters, histamine and serotonin. Thus, unihemispheric sleeping animals may have less demand on monoaminergic wake-active neurons across unihemispheric wakefulness. We do not know what happens to norepinephrine levels in the brain across sleep deprivation in unihemispheric sleepers, but it would be of great interest, as unihemispheric sleep deprivation leads to rebound only in the deprived hemisphere (64).
Chronic Sleep Restriction Effects on Vigilance in Animal Models
Chronic sleep restriction in rats has been performed to determine whether psychomotor vigilance is impaired after CSS in rats (38), as it is in humans. Unlike humans, the rats do not show progressive or cumulative declines in performance across sleep restriction days, and unlike mice and humans the recovery is complete after one 24-h recovery period. However, the model implemented here is distinct from the above extended wake studies in mice and in humans. For these studies, rats were placed in a rotating drum for enforced ambulation continuously for 3 h, and then, they are provided a rest period of 1 h to eat, drink, and have a rest opportunity. Thus, this is not a model of extended wakefulness for long periods of time, and like the short-term sleep loss studies above for LCn, it is possible that the 3 h duration increases antioxidants to protect the neurons. Remarkably, performance was most impaired on the second day of sleep restriction, and by the third or fourth day of sleep restriction, vigilance improved (38). In humans, it is not the amount of sleep lost but the duration of wakefulness that predicts impaired PVT performance (2). Leemburg et al. performed a rigorous study of chronic sleep restriction in which rats were allowed 4 h sleep at the beginning of the lights-on period followed by complete sleep deprivation for 20 h (39). In this paradigm, rats showed persistence in sleep homeostasis in multiple parameters including shortened sleep latency, increased sleep attempts, and increased slow-wave activity (39). In contrast, there was no evidence for allostasis to CSS (39). Vigilance tests and neuronal counts were not performed in these animals. Further studies are needed to determine whether rats confer resistance to CSS-induced vigilance impairments; whether brief nap opportunities across extended wake can mitigate wake neuron injury; and whether the pattern of intermittent brief naps enables adaptive responses that protect the brain across future longer sleep loss exposures.
Similarities and Differences with Effects of Sleep Fragmentation vs CSS
Many individuals with sleep disorders, including sleep apnea and periodic limb movement disorder, regularly experience fragmentation of sleep. These individuals may have normal total sleep durations for 24 h, but periods of sleep are frequently disrupted. This frequent disruption has been modeled in adult mice and shown to induce lasting neuronal injury (65). As with CSS, there are several unique models of sleep fragmentation. One method implements a rotor shaker table (65). Mouse cages may be placed on the rotor platform that is then gently moved at intervals around the clock, and this has been shown to effectively fragment sleep, while allowing group housing and normal eating patterns (65). In a recent study, group housed adult male mice were exposed to this form of sleep fragmentation for 14 weeks continuously (40). The animals’ weights were similar across sleep fragmented and control mice. The effects of sleep fragmentation on sleep-wake activity appear distinct from CSS. While CSS blunted the diurnal rhythm of sleep, sleep fragmentation markedly reduced wakefulness within the first few hours of the dark period, the most active period. Like CSS, sleep fragmentation resulted in a loss of both LCn and orexinergic neurons, although effects on LC were more pronounced (50 vs 25%) (40). Sleep fragmentation also increased oxidative stress in LCn and reduced SirT3. One of the most intriguing findings was that TNF-α was upregulated LCn, not in microglia (40). Gozal and colleagues have examined the effects of chronic sleep fragmentation on hypothalamic function in mice (41). They used a singly housed cage with a sweeper bar that intermittently sweeps the entire cage prompting the mouse to wake and move. With this paradigm, his group has identified leptin resistance, which may contribute to increased obesity with sleep fragmentation (41). His groups has also found that chronic sleep fragmentation induces lasting spatial (hippocampal-dependent) learning and memory impairments that require augmentation of TNF-α signaling and activated of NADPH oxidase (42, 43). Whether the TNF-α and oxidative stress changes are upstream, downstream, or independent of sirtuin changes should now be determined to elucidate effective therapies to prevent cognitive impairments and neural injury with chronic sleep disruption.
Morphometric Effects of Chronic Sleep Loss
Several studies have now examined the ultrastructure of neurons in select brain regions in response to sleep loss. In an earlier study, the effect of 12, 24, and 36 h of total sleep deprivation on neuron ultrastructure was examined in four brain regions: the limbic cortex, the CA1 region of the hippocampus, the pontine reticular formation, and the locus coeruleus (44). At 12 h, all regions showed increased neurons with reparative changes (increased mitochondria, endoplasmic reticulum, lysosomes, chromatin, and nuclear invaginations). No degenerative changes were observed. At 24 h, greater percentages of neurons showed reparative changes in all four regions, and at 36 h, degenerative changes were observed in all regions. These degenerative changes included chromatolysis, somatodendritic vacuolization, and reductions in well-formed organelles. Intriguingly, layers III/IV of the limbic cortex and the CA1 region of the hippocampus showed greater percentages of injured neurons than the locus coeruleus. This work provides strong support that neurons are structurally altered differentially across subsets and by sleep loss duration. A recent study examined the effects of 2 months of sleep restriction to 4 h/day on the ultrastructure of cortical neurons (45). Although results were not quantified, the researchers concluded that there were more damaged mitochondria with missing or swollen cristae and swelling between the inner and outer membrane (45). Another recent study examined the effects of 4 days of sleep restriction on the ultrastructure of pyramidal neurons in the frontal cortex in young mice (4 weeks old) (46). CSS in this model was performed using a combination of approaches including an enriched environment and caffeine administration. Total daytime sleep time was reduced from 8.5 to 2 h for the 4 days. This study performed rigorous measures of neuronal mitochondria, finding larger mitochondria with CSS but no change in cristae or membranes to suggest injury (46). Because this study involved younger mice, a different sleep protocol and examined a different area than those above, it is not clear whether younger mice are less susceptible to CSS effects on neuronal mitochondria, whether the frontal cortex is less susceptible, or whether total sleep deprivation (longer extended wakefulness) provides a unique stress relative to CSS. CSS in these young rats does, however, increased lysosomes, consistent with above studies. In summary, both total and chronic partial sleep loss affect specific neuronal morphometrics, consistent with injury, and there is a sleep loss duration dose response for neuronal morphometric changes for severity of sleep loss. Both mitochondria and lysosomes seem particularly affected in most models.
Neural Consequences of Sleep Loss During Brain Development
Brain circuitry is most plastic during development. Interestingly, brain development occurs at a time when animals, including humans, experience large proportions of sleep time in rapid eye movement (REM) sleep. If sleep, particularly REMS, impacts plasticity, then effects should be most pronounced across the most active periods of brain development, and indeed the work of several research groups supports this prediction.
Monocular visual deprivation during the critical developmental window in rat pups dramatically enhances synaptic strength within the relevant visual circuitry, dramatically remodeling the visual cortex for life (66). In contrast, little effect is appreciated in the adult rat. Strikingly REMS deprivation within this critical developmental window profoundly affects the window of visual plasticity. Specifically, 1 week of REMS deprivation extends the critical window for visual plasticity (47). In addition to REMS influencing the temporal course of the critical window for plasticity, spontaneous NREMS immediately after monocular deprivation enhances plasticity in the kitten, while sleep loss across this same time frame prevents plasticity (ocular dominance) (67). While molecular mechanisms underlying the effects of sleep on visual plasticity during development are not known, the process requires neurotrophins and protein synthesis (68). In summary, both REM and NREM influence brain plasticity in development.
Persistence of neurobehavioral deficits following sleep loss during the critical developmental window comes from work with Drosophila melanogaster. When flies are deprived of sleep as newly eclosed flies (first day beyond the larval stage), young flies have memory impairments that persist at least 6 days (48). In contrast, adult flies, losing the same total amount of sleep had learning impairments that are readily reversed with a few hours of recovery sleep (48). These key findings were extended by Kayser and colleagues (49) who demonstrated that sleep within a critical developmental window for the fruit fly is essential for the structural development of olfactory neuronal circuits involved in courtship behavior, and that sleep loss at day 1 post-eclosure results in lasting diminution of a specific olfactory glomerulus. Intriguingly, as the particular glomerulus affected by sleep loss grows rapidly, the metabolic status or growth history may predict vulnerability of a particular circuit.
Effects of Sleep Loss on Neurogenesis
Across most of the adult lifespan, new neurons and glia are produced in specific brain regions. The production of new neurons (or neurogenesis) can be influenced by modifying health and lifestyle, including sleep time. Guzman-Marin et al. discovered that long-term sleep deprivation in adult rats (across 96 h) significantly suppressed the production of new progenitor cells in the hippocampus, relative to control animals and that this occurred without raising corticosterone levels (30). Moreover, adrenalectomized rats, lacking corticosterone, also demonstrate sleep deprivation suppression of neurogenesis (69). While the effects of sleep loss on neurogenesis are independent of corticosterone, sleep loss suppression of neurogenesis may require interleukin-1β (IL-1β). Specifically, mice lacking receptors for IL-1β do not reduce neurogenesis in response to sleep loss (70). Whether this is a direct or indirect effect (lifelong loss of a cytokine) has not been ascertained. In a follow-up experiment, the Guzman-Marin et al. group established that sleep loss specifically affected the generation of new mature neurons (71). A similar effect on neurogenesis is observed with sleep fragmentation where both 4 and 7 days of continuous sleep fragmentation suppressed neurogenesis, while 1 day did not (72). Neurogenesis is critical for optimal hippocampal learning and memory, and 3 days of sleep restriction (sleep loss during the first 6 h of the lights-on period) reduces the adult neurogenesis response to learning (73).
Intriguingly, an opposite effect of sleep loss on neurogenesis is observed for short-term sleep loss. Loss of one 12-h period of sleep actually increases both brain-derived neurotrophic factor (BDNF) and hippocampal neurogenesis and the increase in BDNF and neurogenesis are sustained for at least 1 month (74, 75). Junek et al. substantiated a time-dependent factor for sleep loss duration on hippocampal cell proliferation, finding no effect for 6 h sleep loss, while 12 h did indeed increase neurogenesis, while longer durations of sleep loss suppressed cell cycle cell proliferation (76).
Effects of Sleep Loss on Neuroinflammation
There are clear bidirectional influences between sleep and the immune system (77). Yet, little is known of how chronic sleep restriction affects the neuroinflammatory responses. Most of the studies have examined acute (short-term) sleep loss or chronic sleep fragmentation. Specifically, short-term sleep deprivation can suppress the peripheral immune responses to vaccination, and sleep disruption increases TNF-α, IL-1β, and interleukin-6 in specific brain regions and peripheral tissues (78–80). Total sleep loss for 24 h increases IL-1β in the hypothalamus and brainstem twofold (81). Mice exposed to chronic sleep fragmentation, but not reduced total sleep, develop increased TNF-α in cortical tissue (42). The increased TNF-α may contribute to sleepiness from sleep fragmentation, in that the sleepiness can be prevented in mice lacking TNF-α receptors (31). Acute sleep loss can also upregulate TNF-α within neurons (40). This may be in response to persistent neuronal activation, as whisker stimulation increases TNF-α in neurons in activated neurons in the barrel cortex (82, 83). Cytokine signaling in neurons can modify hypocretin signaling by reducing pre-pro-hypocretin and orexin receptor 2 mRNA (84). At the same time reducing central TNF-α signaling can reduce sleep, particularly slow-wave sleep (85), and IL-1β injected into the sleep-promoting preoptic area of the hypothalamus and wake-activating basal forebrain, increases the firing of sleep-active neurons and reduces the firing of wake-active neurons. Major questions prompted by the above findings include (1) What is the origin of sleep loss cytokine responses (neuronal or glial or other)? (2) How does neuronal activation increase intra-neuronal TNF-a and is this upregulation of consequence in the neuron? and (3) can suppression of cytokine responses hasten neurobehavioral recovery from sleep loss and/or prevent neuron loss?
Effects of Sleep Loss on Non-Neuronal Brain Cells
Once believed to be simply structural components of the brain, non-neuronal cells, including microglia, astrocytes, and oligodendrocytes play critical roles in synaptic maintenance, energy substrate delivery, and clearance of waste (86–89). Recent studies provide evidence that transcriptional regulation in both oligodendrocytes and astrocytes is influenced by behavioral state. Spontaneous sleep and wake differentially effect transcription profiles for enriched oligodendrocytes pooled from behaviorally monitored adult mice (90). Importantly, spontaneous wakefulness increases genes involved in cellular stress processes, including apoptosis genes (90). Intriguingly, optogenetic stimulation of astrocytes within the posterior hypothalamus at one specific frequency (10 Hz) increases both NREMS and REMS (91). How specifically this occurs and what messages are conveyed from astrocytes to neurons are not yet understood. Both spontaneous wake and enforced short-term wake reduce the number of oligodendrocyte progenitor cells (90), but whether total numbers and functionality of oligodendrocytes are compromised with extended wake has not been explored. Astrocytes demonstrate more pronounced responses to wake than oligodendrocytes with sevenfold more genes upregulated in wake than in sleep (50). Intriguingly, many of the genes upregulated are related to metabolism and/or energetics. Astrocytes release lactate as an energy substrate for neurons (92), and this is regulated in a wake-dependent fashion (93). In addition to changes in astrocyte genes related to metabolism, changes were observed for genes related to astrocyte process extension. The morphology of astrocytes has been examined with electron microscopy, showing that in response to CSS, astrocytic processes moves closer to synaptic clefts and this is not only wake-activated but activity specific so that regions of the brain activated by a given stimulus in waking show greater responses. However, because astrocytes were not marked specifically, it is possible that the processes examined included microglial processes. The collective neuronal and glial responses are summarized in Figure 1.
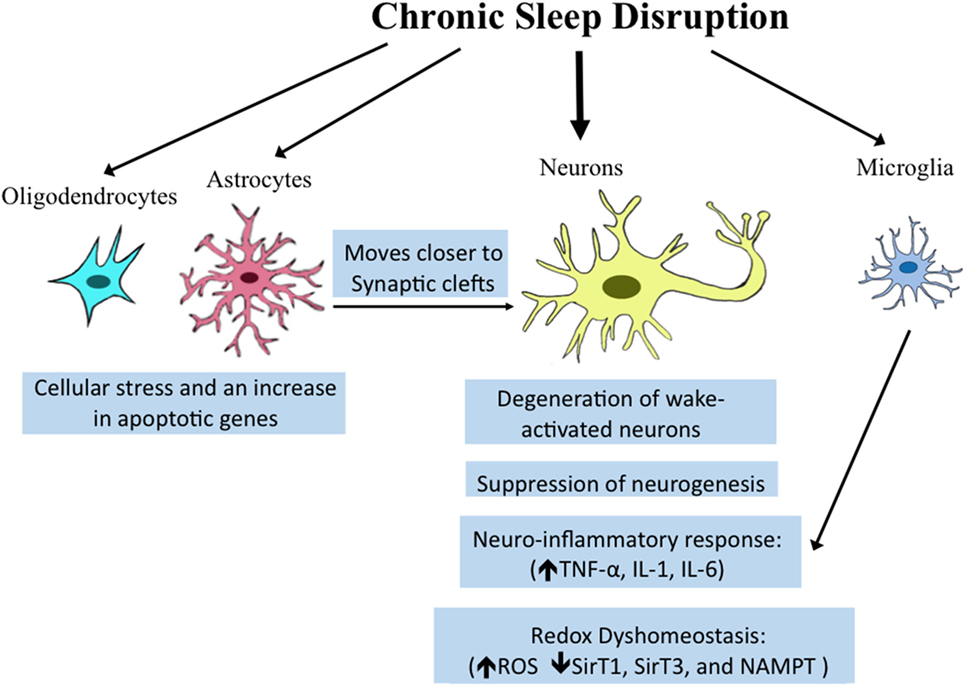
Figure 1. Overview of reported effects of sleep/wake disturbances on neural cells. Chronic sleep disruption which includes chronic total and partial sleep deprivation, as well as sleep fragmentation, can influence diverse brain cell types. Established molecular and morphologic responses within specific cell types are highlighted in blue boxes below the various cell types. Microglia share a pro-inflammatory response, also observed in neurons in resonse to sleep loss. The effects of sleep disruption on astrocytes on synaptic clefts may influence neuronal synapse function.
Effects of Sleep Loss on Hippocampal Function and Connectivity
An elegant body of research demonstrates a critical role for sleep in learning in the immediate period after contextual training. This hippocampal-dependent form of learning can be attenuated by sleep loss in a window of 5–6 h after training, and this effect from sleep loss on diminishing recall is not secondary to learning other material while wake (interference learning) (32, 94, 95). This vital role played by sleep has been systematically elucidated largely by the work of the Ted Abel lab. Short-term sleep loss in the window after training reduces cAMP in hippocampal neurons, leading to reduced protein kinase A and less phosphorylation of CREB for its activation (94) Activation of CREB is essential for the memory consolidation (96). This in turn prevents mTORC1-dependent protein translation for memory consolidation (97).
Less is known of how CSS affects hippocampal function and health. A recent study in humans examined hippocampal and thalamic gray matter volume by MRI before and after 72 h total sleep deprivation (98). The group found reduced volume in the thalamus but not in the hippocampus. There is, however, a differential susceptibility to impaired hippocampal function upon sleep loss, while hippocampal structure can predict the extent of impairment in hippocampal function following sleep loss (99). Whether sleep loss changes in synaptic densities or neuronal function occur in humans remains an open question, but this has been examined in wild-type adult male mice (100). Intriguingly, the loss of spines was regionally specific (in CA1 and not CA3) and dependent upon upregulation of cofilin, a protein that degrades actin filaments, structural elements of dendritic spines. This intriguing finding suggests that short-term sleep loss impairs memory, in part by degrading synapses and supports the concept that specific synapses are either lost or reinforced in sleep (101–104). In summary, short-term sleep loss reduces dendritic spines and therefore connectivity in specific regions of the hippocampus, and loss of spines predicts learning impairment for hippocampal-dependent learning and memory. Whether CSS acts in a similar way and whether effects are reversible with recovery sleep should be explored.
Concluding Remarks
The importance of sleep within an acute window in learning and memory is now firmly established, as is the need for adequate sleep long term for peripheral metabolic homeostasis and cardiovascular health. The concept that chronic sleep loss may have lasting or even protracted recovery effects on brain function has only recently begun to be substantiated, where it is now evident that chronic sleep loss can have profound effects on brain health and function, including neuronal survival. CSS that occurs within the temporal window of brain development imparts lasting disturbances in behaviors critical to the perpetuation of a species. Select populations of wake-activated neurons are highly susceptible to degeneration in conditions of CSS in young adult mice, where extended wakefulness results in mitochondrial metabolic and inflammatory stress, in part, by reducing sirtuins and increasing cytokine responses, respectively, in vulnerable neurons and/or brain regions. Neurons are not the only brain cells affected by sleep loss. Significant changes occur in oligodendrocytes, astrocytes and microglia. Molecular pathways involved are beginning to be understood, providing novel pharmacotherapeutic avenues to protect the brain across sleep loss. Collectively, this body of research on CSS emphasizes the need to develop therapies to prevent neural injury in the common practice of CSS.
Author Contributions
SV, ZZ, and XZ all identified relevant literature, read the body of work related to the topic, discussed format, wrote sections, and edited others. ZZ made the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work was supported in part by NIH grants HL123331, HL 124576, and AG054104 to SV and National Natural Science Foundation of China (Nos. 81100991 and 81671314). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
1. Liu Y, Wheaton AG, Chapman DP, Cunningham TJ, Hua L, Croft JB. Short sleep duration among workers – United States, 2010. MMWR Morb Mortal Wkly Rep (2012) 61:281–5.
2. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep (2003) 26:117–26. doi: 10.1093/sleep/26.2.117
3. Lund HG, Reider BD, Whiting AB, Prichard JR. Sleep patterns and predictors of disturbed sleep in a large population of college students. J Adolesc Health (2010) 46:124–32. doi:10.1016/j.jadohealth.2009.06.016
4. National Sleep Foundation. 2006 Teens and Sleep. Sleep in America Polls. (2006). Available from: http://www.sleepfoundation.org/article/sleep-america-polls/2006-teens-and-sleep
5. Grandner MA, Smith TE, Jackson N, Jackson T, Burgard S, Branas C. Geographic distribution of insufficient sleep across the United States: a county-level hotspot analysis. Sleep Health (2015) 1:158–65. doi:10.1016/j.sleh.2015.06.003
6. Yang CK, Kim JK, Patel SR, Lee JH. Age-related changes in sleep/wake patterns among Korean teenagers. Pediatrics (2005) 115:250–6. doi:10.1542/peds.2004-0815G
7. Olds T, Maher C, Blunden S, Matricciani L. Normative data on the sleep habits of Australian children and adolescents. Sleep (2010) 33:1381–8. doi:10.1093/sleep/33.10.1381
8. Owens J; Adolescent Sleep Working Group; Committee on Adolescence. Insufficient sleep in adolescents and young adults: an update on causes and consequences. Pediatrics (2014) 134:e921–32. doi:10.1542/peds.2014-1696
9. Gariepy G, Janssen I, Sentenac M, Elgar FJ. School start time and sleep in Canadian adolescents. J Sleep Res (2017) 26(2):195–201. doi:10.1111/jsr.12475
10. Paruthi S, Brooks LJ, D’Ambrosio C, Hall WA, Kotagal S, Lloyd RM, et al. Consensus statement of the American Academy of Sleep Medicine on the recommended amount of sleep for healthy children: methodology and discussion. J Clin Sleep Med (2016) 12:1549–61. doi:10.5664/jcsm.6288
11. Wheaton AG, Chapman DP, Croft JB. School start times, sleep, behavioral, health, and academic outcomes: a review of the literature. J Sch Health (2016) 86:363–81. doi:10.1111/josh.12388
12. Lusardi P, Zoppi A, Preti P, Pesce RM, Piazza E, Fogari R. Effects of insufficient sleep on blood pressure in hypertensive patients: a 24-h study. Am J Hypertens (1999) 12:63–8. doi:10.1016/S0895-7061(98)00200-3
13. Spaeth AM, Dinges DF, Goel N. Effects of experimental sleep restriction on weight gain, caloric intake, and meal timing in healthy adults. Sleep (2013) 36:981–90. doi:10.5665/sleep.2792
14. Kaneita Y, Uchiyama M, Yoshiike N, Ohida T. Associations of usual sleep duration with serum lipid and lipoprotein levels. Sleep (2008) 31:645–52. doi:10.1093/sleep/31.5.645
15. Moreno CR, Vasconcelos S, Marqueze EC, Lowden A, Middleton B, Fischer FM, et al. Sleep patterns in Amazon rubber tappers with and without electric light at home. Sci Rep (2015) 5:14074. doi:10.1038/srep14074
16. de la Iglesia HO, Fernández-Duque E, Golombek DA, Lanza N, Duffy JF, Czeisler CA, et al. Access to electric light is associated with shorter sleep duration in a traditionally hunter-gatherer community. J Biol Rhythms (2015) 30:342–50. doi:10.1177/0748730415590702
17. Yetish G, Kaplan H, Gurven M, Wood B, Pontzer H, Manger PR, et al. Natural sleep and its seasonal variations in three pre-industrial societies. Curr Biol (2015) 25:2862–8. doi:10.1016/j.cub.2015.09.046
18. Belenky G, Wesensten NJ, Thorne DR, Thomas ML, Sing HC, Redmond DP, et al. Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. J Sleep Res (2003) 12:1–12. doi:10.1046/j.1365-2869.2003.00337.x
19. Pejovic S, Basta M, Vgontzas AN, Kritikou I, Shaffer ML, Tsaoussoglou M, et al. The effects of recovery sleep after one workweek of mild sleep restriction on interleukin-6 and cortisol secretion and daytime sleepiness and performance. Am J Physiol Endocrinol Metab (2013) 305(7):E890–6. doi:10.1152/ajpendo.00301.2013
20. Simpson NS, Diolombi M, Scott-Sutherland J, Yang H, Bhatt V, Gautam S, et al. Repeating patterns of sleep restriction and recovery: do we get used to it? Brain Behav Immun (2016) 58:142–51. doi:10.1016/j.bbi.2016.06.001
21. Rabat A, Gomez-Merino D, Roca-Paixao L, Bougard C, Van Beers P, Dispersyn G, et al. Differential kinetics in alteration and recovery of cognitive processes from a chronic sleep restriction in young healthy men. Front Behav Neurosci (2016) 10:95. doi:10.3389/fnbeh.2016.00095
22. Ong JL, Lo JC, Gooley JJ, Chee MW. EEG changes across multiple nights of sleep restriction and recovery in adolescents: the need for sleep study. Sleep (2016) 39:1233–40. doi:10.5665/sleep.5840
23. Lo JC, Ong JL, Leong RL, Gooley JJ, Chee MW. Cognitive performance, sleepiness, and mood in partially sleep deprived adolescents: the need for sleep study. Sleep (2016) 39:687–98. doi:10.5665/sleep.5552
24. Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science (2009) 326:1005–7. doi:10.1126/science.1180962
25. Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med (2012) 4:150ra122. doi:10.1126/scitranslmed.3004291
26. Jouvet D, Vimont P, Delorme F, Jouvet M. [Study of selective deprivation of the paradoxal sleep phase in the cat]. C R Seances Soc Biol Fil (1964) 158:756–9.
27. Coenen AM, Van Hulzen ZJ. Paradoxical sleep deprivation in animal studies: some methodological considerations. Prog Brain Res (1980) 53:325–30. doi:10.1016/S0079-6123(08)60072-2
28. Bergmann BM, Kushida CA, Everson CA, Gilliland MA, Obermeyer W, Rechtschaffen A. Sleep deprivation in the rat: II. Methodology. Sleep (1989) 12:5–12. doi:10.1093/sleep/12.1.5
29. Medeiros R, Lenneberg-Hoshino C, Hoshino K, Tufik S. Neuroethologic differences in sleep deprivation induced by the single- and multiple-platform methods. Braz J Med Biol Res (1998) 31:675–80. doi:10.1590/S0100-879X1998000500012
30. Guzman-Marin R, Suntsova N, Stewart DR, Gong H, Szymusiak R, McGinty D. Sleep deprivation reduces proliferation of cells in the dentate gyrus of the hippocampus in rats. J Physiol (2003) 549:563–71. doi:10.1113/jphysiol.2003.041665
31. Kaushal N, Ramesh V, Gozal D. TNF-alpha and temporal changes in sleep architecture in mice exposed to sleep fragmentation. PLoS One (2012) 7:e45610. doi:10.1371/journal.pone.0045610
32. Graves LA, Heller EA, Pack AI, Abel T. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem (2003) 10:168–76. doi:10.1101/lm.48803
33. Veasey SC, Valladares O, Fenik P, Kapfhamer D, Sanford L, Benington J, et al. An automated system for recording and analysis of sleep in mice. Sleep (2000) 23:1025–40.
34. Gompf HS, Mathai C, Fuller PM, Wood DA, Pedersen NP, Saper CB, et al. Locus ceruleus and anterior cingulate cortex sustain wakefulness in a novel environment. J Neurosci (2010) 30:14543–51. doi:10.1523/JNEUROSCI.3037-10.2010
35. Zhang J, Zhu Y, Zhan G, Fenik P, Panossian L, Wang MM, et al. Extended wakefulness: compromised metabolics in and degeneration of locus ceruleus neurons. J Neurosci (2014) 34(12):4418–31. doi:10.1523/JNEUROSCI.5025-12.2014
36. Zhu Y, Fenik P, Zhan G, Somach R, Xin R, Veasey S. Intermittent short sleep results in lasting sleep wake disturbances and degeneration of locus coeruleus and orexinergic neurons. Sleep (2016) 39:1601–11. doi:10.5665/sleep.6030
37. Cirelli C, Shaw PJ, Rechtschaffen A, Tononi G. No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Res (1999) 840:184–93. doi:10.1016/S0006-8993(99)01768-0
38. Deurveilher S, Bush JE, Rusak B, Eskes GA, Semba K. Psychomotor vigilance task performance during and following chronic sleep restriction in rats. Sleep (2015) 38:515–28. doi:10.5665/sleep.4562
39. Leemburg S, Vyazovskiy VV, Olcese U, Bassetti CL, Tononi G, Cirelli C. Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc Natl Acad Sci U S A (2010) 107:15939–44. doi:10.1073/pnas.1002570107
40. Zhu Y, Fenik P, Zhan G, Xin R, Veasey SC. Degeneration in arousal neurons in chronic sleep disruption modeling sleep apnea. Front Neurol (2015) 6:109. doi:10.3389/fneur.2015.00109
41. Hakim F, Wang Y, Carreras A, Hirotsu C, Zhang J, Peris E, et al. Chronic sleep fragmentation during the sleep period induces hypothalamic endoplasmic reticulum stress and PTP1b-mediated leptin resistance in male mice. Sleep (2015) 38:31–40. doi:10.5665/sleep.4320
42. Ramesh V, Nair D, Zhang SX, Hakim F, Kaushal N, Kayali F, et al. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-alpha pathway. J Neuroinflammation (2012) 9:91. doi:10.1186/1742-2094-9-91
43. Nair D, Zhang SX, Ramesh V, Hakim F, Kaushal N, Wang Y, et al. Sleep fragmentation induces cognitive deficits via NADPH oxidase-dependent pathways in mouse. Am J Respir Crit Care Med (2011) 184(11):1305–12. doi:10.1164/rccm.201107-1173OC
44. Abushov BM. Morphofunctional analysis of the effects of total sleep deprivation on the CNS in rats. Neurosci Behav Physiol (2010) 40:403–9. doi:10.1007/s11055-010-9271-y
45. Zhao H, Wu H, He J, Zhuang J, Liu Z, Yang Y, et al. Frontal cortical mitochondrial dysfunction and mitochondria-related beta-amyloid accumulation by chronic sleep restriction in mice. Neuroreport (2016) 27:916–22. doi:10.1097/WNR.0000000000000631
46. de Vivo L, Nelson AB, Bellesi M, Noguti J, Tononi G, Cirelli C. Loss of sleep affects the ultrastructure of pyramidal neurons in the adolescent mouse frontal cortex. Sleep (2016) 39:861–74. doi:10.5665/sleep.5644
47. Shaffery JP, Sinton CM, Bissette G, Roffwarg HP, Marks GA. Rapid eye movement sleep deprivation modifies expression of long-term potentiation in visual cortex of immature rats. Neuroscience (2002) 110:431–43. doi:10.1016/S0306-4522(01)00589-9
48. Seugnet L, Suzuki Y, Donlea JM, Gottschalk L, Shaw PJ. Sleep deprivation during early-adult development results in long-lasting learning deficits in adult Drosophila. Sleep (2011) 34:137–46. doi:10.1093/sleep/34.2.137
49. Kayser MS, Yue Z, Sehgal A. A critical period of sleep for development of courtship circuitry and behavior in Drosophila. Science (2014) 344:269–74. doi:10.1126/science.1250553
50. Bellesi M, de Vivo L, Tononi G, Cirelli C. Effects of sleep and wake on astrocytes: clues from molecular and ultrastructural studies. BMC Biol (2015) 13:66. doi:10.1186/s12915-015-0176-7
51. Everson CA, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: III. Total sleep deprivation. Sleep (1989) 12:13–21. doi:10.1093/sleep/12.1.13
52. Everson CA, Gilliland MA, Kushida CA, Pilcher JJ, Fang VS, Refetoff S, et al. Sleep deprivation in the rat: IX. Recovery. Sleep (1989) 12:60–7.
53. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron (2010) 68:1023–42. doi:10.1016/j.neuron.2010.11.032
54. Aston-Jones G, Rajkowski J, Kubiak P, Alexinsky T. Locus coeruleus neurons in monkey are selectively activated by attended cues in a vigilance task. J Neurosci (1994) 14:4467–80.
55. Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci (2009) 10:211–23. doi:10.1038/nrn2573
56. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature (2010) 464:121–5. doi:10.1038/nature08778
57. Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev (2000) 14:1021–6. doi:10.1101/gad.14.9.1021
58. Haigis MC, Guarente LP. Mammalian sirtuins – emerging roles in physiology, aging, and calorie restriction. Genes Dev (2006) 20:2913–21. doi:10.1101/gad.1467506
59. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell (2010) 143:802–12. doi:10.1016/j.cell.2010.10.002
60. Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci (2006) 26:1343–54. doi:10.1523/JNEUROSCI.4236-05.2006
61. Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, et al. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiol Aging (2007) 28:1206–14. doi:10.1016/j.neurobiolaging.2006.06.003
62. Kalinin S, Polak PE, Lin SX, Sakharkar AJ, Pandey SC, Feinstein DL. The noradrenaline precursor l-DOPS reduces pathology in a mouse model of Alzheimer’s disease. Neurobiol Aging (2012) 33:1651–63. doi:10.1016/j.neurobiolaging.2011.04.012
63. Lyamin OI, Lapierre JL, Kosenko PO, Kodama T, Bhagwandin A, Korneva SM, et al. Monoamine release during unihemispheric sleep and unihemispheric waking in the fur seal. Sleep (2016) 39:625–36. doi:10.5665/sleep.5540
64. Oleksenko AI, Mukhametov LM, Polyakova IG, Supin AY, Kovalzon VM. Unihemispheric sleep deprivation in bottlenose dolphins. J Sleep Res (1992) 1:40–4. doi:10.1111/j.1365-2869.1992.tb00007.x
65. Li Y, Panossian LA, Zhang J, Zhu Y, Zhan G, Chou YT, et al. Effects of chronic sleep fragmentation on wake-active neurons and the hypercapnic arousal response. Sleep (2014) 37:51–64. doi:10.5665/sleep.3306
66. Nataraj K, Turrigiano G. Regional and temporal specificity of intrinsic plasticity mechanisms in rodent primary visual cortex. J Neurosci (2011) 31:17932–40. doi:10.1523/JNEUROSCI.4455-11.2011
67. Frank MG, Issa NP, Stryker MP. Sleep enhances plasticity in the developing visual cortex. Neuron (2001) 30:275–87. doi:10.1016/S0896-6273(01)00279-3
68. Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, et al. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr Biol (2012) 22:676–82. doi:10.1016/j.cub.2012.02.016
69. Mueller AD, Pollock MS, Lieblich SE, Epp JR, Galea LA, Mistlberger RE. Sleep deprivation can inhibit adult hippocampal neurogenesis independent of adrenal stress hormones. Am J Physiol Regul Integr Comp Physiol (2008) 294:R1693–703. doi:10.1152/ajpregu.00858.2007
70. Mueller AD, Parfyonov M, Pavlovski I, Marchant EG, Mistlberger RE. The inhibitory effect of sleep deprivation on cell proliferation in the hippocampus of adult mice is eliminated by corticosterone clamp combined with interleukin-1 receptor 1 knockout. Brain Behav Immun (2014) 35:182–8. doi:10.1016/j.bbi.2013.10.001
71. Guzman-Marin R, Suntsova N, Methippara M, Greiffenstein R, Szymusiak R, McGinty D. Sleep deprivation suppresses neurogenesis in the adult hippocampus of rats. Eur J Neurosci (2005) 22:2111–6. doi:10.1111/j.1460-9568.2005.04376.x
72. Guzman-Marin R, Bashir T, Suntsova N, Szymusiak R, McGinty D. Hippocampal neurogenesis is reduced by sleep fragmentation in the adult rat. Neuroscience (2007) 148:325–33. doi:10.1016/j.neuroscience.2007.05.030
73. Hairston IS, Little MT, Scanlon MD, Barakat MT, Palmer TD, Sapolsky RM, et al. Sleep restriction suppresses neurogenesis induced by hippocampus-dependent learning. J Neurophysiol (2005) 94:4224–33. doi:10.1152/jn.00218.2005
74. Fujihara H, Sei H, Morita Y, Ueta Y, Morita K. Short-term sleep disturbance enhances brain-derived neurotrophic factor gene expression in rat hippocampus by acting as internal stressor. J Mol Neurosci (2003) 21:223–32. doi:10.1385/JMN:21:3:223
75. Grassi Zucconi G, Cipriani S, Balgkouranidou I, Scattoni R. ‘One night’ sleep deprivation stimulates hippocampal neurogenesis. Brain Res Bull (2006) 69:375–81. doi:10.1016/j.brainresbull.2006.01.009
76. Junek A, Rusak B, Semba K. Short-term sleep deprivation may alter the dynamics of hippocampal cell proliferation in adult rats. Neuroscience (2010) 170:1140–52. doi:10.1016/j.neuroscience.2010.08.018
77. Ingiosi AM, Opp MR, Krueger JM. Sleep and immune function: glial contributions and consequences of aging. Curr Opin Neurobiol (2013) 23:806–11. doi:10.1016/j.conb.2013.02.003
78. Chennaoui M, Gomez-Merino D, Drogou C, Geoffroy H, Dispersyn G, Langrume C, et al. Effects of exercise on brain and peripheral inflammatory biomarkers induced by total sleep deprivation in rats. J Inflamm (Lond) (2015) 12:56. doi:10.1186/s12950-015-0102-3
79. Venancio DP, Suchecki D. Prolonged REM sleep restriction induces metabolic syndrome-related changes: mediation by pro-inflammatory cytokines. Brain Behav Immun (2015) 47:109–17. doi:10.1016/j.bbi.2014.12.002
80. Rosa Neto JC, Lira FS, Venancio DP, Cunha CA, Oyama LM, Pimentel GD, et al. Sleep deprivation affects inflammatory marker expression in adipose tissue. Lipids Health Dis (2010) 9:125. doi:10.1186/1476-511X-9-125
81. Mackiewicz M, Sollars PJ, Ogilvie MD, Pack AI. Modulation of IL-1 beta gene expression in the rat CNS during sleep deprivation. Neuroreport (1996) 7:529–33. doi:10.1097/00001756-199601310-00037
82. Hallett H, Churchill L, Taishi P, De A, Krueger JM. Whisker stimulation increases expression of nerve growth factor- and interleukin-1beta-immunoreactivity in the rat somatosensory cortex. Brain Res (2010) 1333:48–56. doi:10.1016/j.brainres.2010.03.048
83. Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, et al. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience (2008) 156:71–80. doi:10.1016/j.neuroscience.2008.06.066
84. Zhan S, Cai GQ, Zheng A, Wang Y, Jia J, Fang H, et al. Tumor necrosis factor-alpha regulates the hypocretin system via mRNA degradation and ubiquitination. Biochim Biophys Acta (2011) 1812:565–71. doi:10.1016/j.bbadis.2010.11.003
85. Taishi P, Churchill L, Wang M, Kay D, Davis CJ, Guan X, et al. TNFalpha siRNA reduces brain TNF and EEG delta wave activity in rats. Brain Res (2007) 1156:125–32. doi:10.1016/j.brainres.2007.04.072
86. Argente-Arizon P, Guerra-Cantera S, Garcia-Segura LM, Argente J, Chowen JA. Glial cells and energy balance. J Mol Endocrinol (2017) 58:R59–71. doi:10.1530/JME-16-0182
87. Roth AD, Nunez MT. Oligodendrocytes: functioning in a delicate balance between high metabolic requirements and oxidative damage. Adv Exp Med Biol (2016) 949:167–81. doi:10.1007/978-3-319-40764-7_8
88. Nuriya M, Hirase H. Involvement of astrocytes in neurovascular communication. Prog Brain Res (2016) 225:41–62. doi:10.1016/bs.pbr.2016.02.001
89. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol (2017) 79:619–43. doi:10.1146/annurev-physiol-022516-034406
90. Bellesi M, Pfister-Genskow M, Maret S, Keles S, Tononi G, Cirelli C. Effects of sleep and wake on oligodendrocytes and their precursors. J Neurosci (2013) 33:14288–300. doi:10.1523/JNEUROSCI.5102-12.2013
91. Pelluru D, Konadhode RR, Bhat NR, Shiromani PJ. Optogenetic stimulation of astrocytes in the posterior hypothalamus increases sleep at night in C57BL/6J mice. Eur J Neurosci (2016) 43:1298–306. doi:10.1111/ejn.13074
92. Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab (2016) 23:94–102. doi:10.1016/j.cmet.2015.10.010
93. Petit JM, Magistretti PJ. Regulation of neuron-astrocyte metabolic coupling across the sleep-wake cycle. Neuroscience (2016) 323:135–56. doi:10.1016/j.neuroscience.2015.12.007
94. Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, et al. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature (2009) 461:1122–5. doi:10.1038/nature08488
95. Hagewoud R, Whitcomb SN, Heeringa AN, Havekes R, Koolhaas JM, Meerlo P. A time for learning and a time for sleep: the effect of sleep deprivation on contextual fear conditioning at different times of the day. Sleep (2010) 33:1315–22. doi:10.1093/sleep/33.10.1315
96. Graves L, Dalvi A, Lucki I, Blendy JA, Abel T. Behavioral analysis of CREB alphadelta mutation on a B6/129 F1 hybrid background. Hippocampus (2002) 12:18–26. doi:10.1002/hipo.10003.abs
97. Tudor JC, Davis EJ, Peixoto L, Wimmer ME, van Tilborg E, Park AJ, et al. Sleep deprivation impairs memory by attenuating mTORC1-dependent protein synthesis. Sci Signal (2016) 9:ra41. doi:10.1126/scisignal.aad4949
98. Liu C, Kong XZ, Liu X, Zhou R, Wu B. Long-term total sleep deprivation reduces thalamic gray matter volume in healthy men. Neuroreport (2014) 25:320–3. doi:10.1097/WNR.0000000000000091
99. Saletin JM, Goldstein-Piekarski AN, Greer SM, Stark S, Stark CE, Walker MP. Human hippocampal structure: a novel biomarker predicting mnemonic vulnerability to, and recovery from, sleep deprivation. J Neurosci (2016) 36:2355–63. doi:10.1523/JNEUROSCI.3466-15.2016
100. Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife (2016) 5:e13424. doi:10.7554/eLife.13424
101. Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev (2006) 10:49–62. doi:10.1016/j.smrv.2005.05.002
102. Yang G, Lai CS, Cichon J, Ma L, Li W, Gan WB. Sleep promotes branch-specific formation of dendritic spines after learning. Science (2014) 344:1173–8. doi:10.1126/science.1249098
103. Maret S, Faraguna U, Nelson AB, Cirelli C, Tononi G. Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nat Neurosci (2011) 14:1418–20. doi:10.1038/nn.2934
Keywords: locus coeruleus, sleep deprivation, neurodegeneration, vigilance performance, developmental biology
Citation: Zhao Z, Zhao X and Veasey SC (2017) Neural Consequences of Chronic Short Sleep: Reversible or Lasting? Front. Neurol. 8:235. doi: 10.3389/fneur.2017.00235
Received: 18 February 2017; Accepted: 12 May 2017;
Published: 31 May 2017
Edited by:
David Gozal, University of Chicago, United StatesReviewed by:
Priyattam J. Shiromani, Medical University of South Carolina, United StatesRakesh Bhattacharjee, University of California at San Diego, United States
Copyright: © 2017 Zhao, Zhao and Veasey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigrid C. Veasey, dmVhc2V5QG1haWwubWVkLnVwZW5uLmVkdQ==
†These authors are co-first authors and have contributed equally to this work.