 Christopher Dardis1*
Christopher Dardis1* Jared Yeo2
Jared Yeo2 Kelly Milton1Lynn S. Ashby1
Kelly Milton1Lynn S. Ashby1 Kris A. Smith3
Kris A. Smith3 Shwetal Mehta4Emad Youssef5
Shwetal Mehta4Emad Youssef5 Jenny Eschbacher6
Jenny Eschbacher6 Kathy Tucker7
Kathy Tucker7 Laughlin Dawes8Neil Lambie9
Laughlin Dawes8Neil Lambie9 Elizabeth Algar10
Elizabeth Algar10 Elizabeth Hovey2,11
Elizabeth Hovey2,11
- 1Department of Neurology, Barrow Neurological Institute, Phoenix, AZ, Unites States
- 2University of New South Wales, Sydney, NSW, Australia
- 3Department of Neurosurgery, Barrow Neurological Institute, Phoenix, AZ, United States
- 4Laboratory of Glial Tumor Biology, Barrow Neurological Institute, Phoenix, AZ, United States
- 5Department of Radiation Oncology, Barrow Neurological Institute, Phoenix, AZ, United States
- 6Department of Pathology, Barrow Neurological Institute, Phoenix, AZ, United States
- 7Hereditary Cancer Clinic, Prince of Wales Hospital, Randwick, NSW, Australia
- 8Department of Diagnostic Radiology, Prince of Wales Hospital, Randwick, NSW, Australia
- 9Department of Anatomical Pathology, Prince of Wales Hospital, Randwick, NSW, Australia
- 10Hudson Institute of Medical Research, Clayton, VIC, Australia
- 11Department of Medical Oncology, Nelune Comprehensive Cancer Center, Prince of Wales Hospital, Randwick, NSW, Australia
We present the first quantitative analysis of atypical teratoid rhabdoid tumors (ATRT) in adults, including two patients from our own institutions. These are of interest as one occurred during pregnancy and one is a long-term survivor. Our review of pathological findings of 50 reported cases of adult ATRT leads us to propose a solely ectodermal origin for the tumor and that epithelial–mesenchymal transition (EMT) is a defining feature. Thus, the term ATRT may be misleading. Our review of clinical findings shows that ATRT tends to originate in mid-line structures adjacent to the CSF, leading to a high rate of leptomeningeal dissemination. Thus, we hypothesize that residual undifferentiated ectoderm in the circumventricular organs, particularly the pituitary and pineal glands, is the most common origin for these tumors. We note that if growth is not arrested soon after diagnosis, or after the first relapse/progression, death is almost universal. While typically rapidly fatal (as in our first case), long-term remission is possible (as in our second). Significant predictors of prognosis were the extent of resection and the use of chemotherapy. Glial differentiation (GFAP staining) was strongly associated with leptomeningeal metastases (chi-squared p = 0.02) and both predicted markedly worse outcomes. Clinical trials including adults are rare. ATRT is primarily a disease of infancy and radiotherapy is generally avoided in those aged less than 3 years old. Treatment options in adults differ from infants in that cranio-spinal irradiation is a viable adjunct to systemic chemotherapy in the adult population. Given the grave prognosis, this combined approach appears reasonable. As effective chemotherapy is likely to cause myelosuppression, we recommend that stem-cell rescue be available locally.
1. Introduction
1.1. History of ATRT
ATRT, a cancer of the CNS, was christened by Rorke et al. in 1996, following a review of 52 pediatric cases (1). We may trace the first appearance of the term “atypical teratoma” to four decades earlier, where it was recognized to occur in the pineal gland (2). The “atypical” refers descriptively to the “teratoid” part of the tumor. This appears to show elements of two germ cell layers (ectoderm, i.e., primitive neuroepithelial, epithelial, and mesoderm, i.e., “rhabdoid”) but is otherwise quite different from classical teratoid tumors. “Rhabdoid” (Greek, “rod-like”) refers to the similarity, on microscopy, to tumors showing skeletal muscle differentiation.
The biology of and treatment strategies for ATRT (focusing on children) have been the subject of a recent, thorough review. Although the prognosis in children has traditionally been dismal, with few long-term survivors, developments over the last 10 years have, at least for children, been encouraging (3). Recently, mutations in the chromatic remodeling complex SMARCB1 (and, as a rare alternative, of SMARCA4) have been identified in the great majority of cases of ATRT (4). Such mutations appear necessary for the development of ATRT; no other consistent genetic abnormalities have been identified.
1.2. ATRT in Adults
While ATRT is the most common malignant CNS tumor in children aged <1, cases in adults (i.e., age >18) are rare—we estimate <1/1,000,000 lifetime risk (5). As such, these patients are presented in detail as case reports or in small case series. There are no molecular markers recognized as unique to adults rather than children; in both cases, loss of INI1 staining via immunohistochemistry (IHC) is considered sufficient for diagnosis in the appropriate context.
1.3. Rhabdoid Tumors
ATRT is often described as analogous to “malignant rhabdoid tumor of the kidney,” a tumor recognized since at least the early 1980s (6). Other CNS tumors with rhabdoid histology include the “epithelioid glioblastoma with rhabdoid component” as well as the “rhabdoid” variants of more common tumors: meningioma, carcinoma, chordoma, and even sarcoma (7).
1.4. EMT
Epithelial–mesenchymal transition (EMT) was initially identified as a phenomenon in developmental biology. The occurrence of EMT in certain cancers has been suggested since 1978, although it only began to receive widespread attention 10 years later (8). The role of EMT in carcinogenesis has been the subject of an extensive review (9). EMT appears to be crucial to the development and maintenance of a pool of slowly dividing, chemoresistant, “cancer stem cells.” We hypothesize that the “rhabdoid” appearance of the tumors above reflects EMT.
2. Case Reports
2.1. A Case in Pregnancy
2.1.1. Presentation
Our first patient was a 29-year-old woman who developed hoarseness during week 20 of her first pregnancy, which deteriorated over the subsequent 5 weeks. Over the course of 1 day, she developed weakness of the right arm and leg. A CT performed elsewhere showed the presence of an extra-axial left cerebello-pontine mass. This was interpreted as a probable meningioma, so further treatment was deferred.
At 35 weeks gestation, she developed status migrainosus and worsening right leg weakness. Her voice had become weaker and she coughed intermittently when swallowing. Paralysis of her left vocal cord was demonstrated via laryngoscopy. Her MRIs at this time are shown in Figures 1 and 2. A cesarean section was performed in anticipation of neurosurgery and a healthy baby boy was delivered.
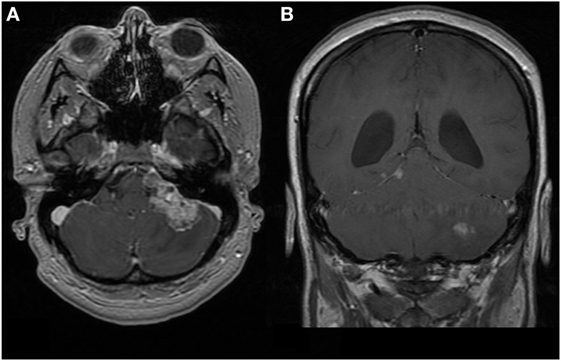
Figure 1. First patient. MRI brain at time of diagnosis, 1.5 T. Images are T1-weighted and Gd-enhanced. (A) Axial: heterogeneous extra-axial enhancement is seen in the left cerebello-pontine angle, with some local compression. (B) Coronal: enhancement is seen along the tentorium cerebelli on the right, which is characteristic of leptomeningeal spread. The left cerebellar lesion appears intraparenchymal.
2.1.2. Initial Treatment
A left retro-sigmoid craniotomy was performed. Pathology, showing loss of immunohistochemical staining for INI1, confirmed ATRT. Her spinal MRI showed leptomeningeal metastases in the cervical and thoracic cord. She rapidly developed hydrocephalus; a shunt and Ommaya reservoir were placed 1 week after surgery.
Multimodal treatment was instituted. Systemic chemotherapy involved two cycles (of 14 days) of ICE (ifosfamide, carboplatin, etoposide). Following the second cycle, she developed pancytopenia and sepsis, and thereafter no further systemic treatment was pursued.
Intrathecal chemotherapy was with liposomal cytarabine 50 mg via Ommaya every 2 weeks; she received 6 doses in total. Intensity-modulated radiotherapy to the cerebellum was 54 Gy in 30 fractions; conformal radiotherapy to the cord at C5–T6 vertebral levels was 30 Gy in 10 fractions. Disease in the brain and upper spinal cord improved overall with treatment, except for one focus in the pre-pontine cistern which grew in a plaque-like fashion.
2.1.3. Treatment at progression
In view of progressive weakness of the legs, she received an additional 30 Gy in 10 fractions to the spinal cord at T12–L4. Within a month of completing this treatment, she developed a flaccid paraplegia with loss of bladder and bowel control and a sensory level at T10. Her MRI confirmed progression (Figure 2). Thereafter, she was cared for by Hospice and died 5 months following her initial surgery.
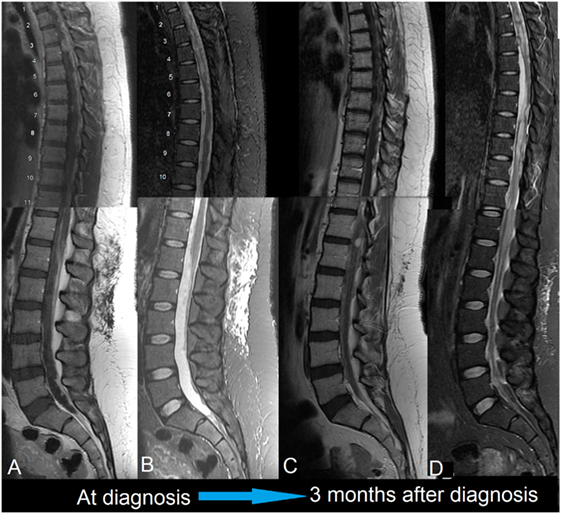
Figure 2. First patient. MRIs of spine, 1.5 T, sagittal. (A) Gd-enhanced: diffuse, nodular enhancement is seen throughout. This is striking dorsally and at the conus medullaris. (B) T2-weighted: the thoracic cord is swollen with intra-medullary edema. This appears to be a reaction to the surrounding leptomeningeal tumor; there may also be an element of cord compression in the thoracic cord. (C) Gd-enhanced: disease in the upper thoracic cord is much improved following radiotherapy. Progression has occurred around the untreated cord caudally. (D) T2-weighted: similarly, intra-medullary edema has improved in the treated areas but worsened elsewhere.
2.2. A Case with a Durable Response to Treatment
2.2.1. Presentation
Our second patient was a 35-year-old man who presented with blurred vision that had been present for 3 months. His MRI at this time, showing a suprasellar mass, is shown in Figure 3.
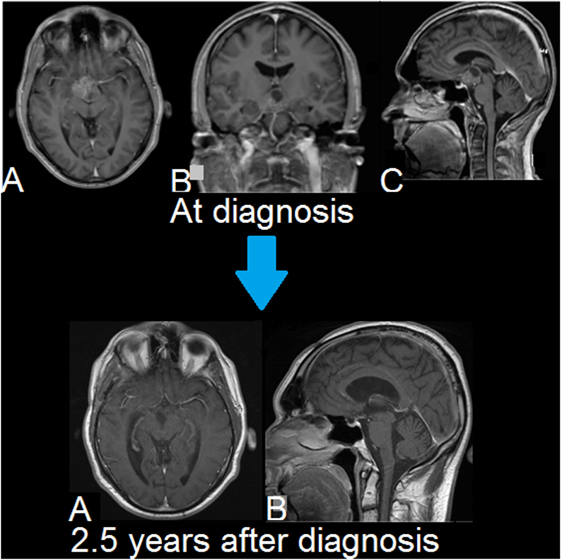
Figure 3. Second patient. MRIs of brain. Upper row (A–C): T1-weighted, Gd-enhanced (magnetization-prepared rapid gradient-echo (MPRAGE)). A suprasellar and interpeduncular lesion is seen with mixed solid and cystic (lobulated) elements. There is heterogeneous enhancement of intermediate intensity. Lower row (A,B): T1-weighted, Gd-enhanced. There is no evidence of tumor recurrence. Smooth linear enhancement is present at the resection site, which is consistent with the long-term changes seen following surgery.
He underwent craniotomy, via a pterional approach, for resection of the tumor. The pathology is shown in Figure 4. This shows how ATRT may be suspected on the basis of hematoxylin and eosin (H&E) staining alone. The diagnosis was confirmed by IHC for INI1.
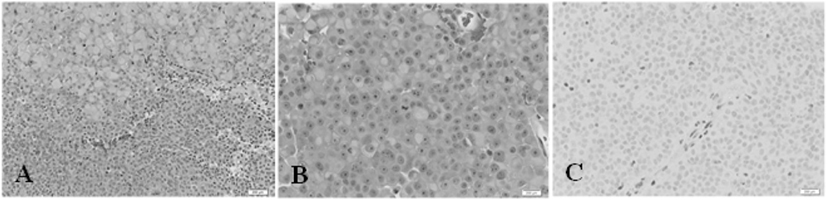
Figure 4. Second patient. Immunohistochemistry. (A) Hematoxylin and eosin (H&E) stain, 200× magnification. The tumor comprises two main elements: sheets of large cells, many with a rhabdoid appearance (top) and small, more primitive-appearing cells (bottom). (B) (H&E) stain, 400×. This shows large cells, many with rhabdoid features and central mitoses. (C) INI1 stain, 400×. There is no nuclear staining in the tumor cells. A positive control is provided by the endothelial cells in the vessel at the center, and in other scattered glial and inflammatory cells.
A homozygous mutation in SMARCB1 was confirmed on Sanger sequencing. This was performed on the fresh-frozen, paraffin-embedded tissue. The sequencing covered the coding region and 5′ and 3′ splice sites from nucleotide 37 to 47,120 in the reference sequence NT_011520.gbk (10). The mutation was described as NM_003073.3: c.1148delC, NP_003064: p.P383fs*96. Occurring in exon 9, this causes a SMARCB1 protein frame-shift and the insertion of additional amino acids at the 3′ end of the protein. This mutation is predicted to lead to inactivation of this protein in the tumor.
2.2.2. Initial Treatment
MRIs performed following surgery demonstrated a nodule in the third ventricle that was believed to be persistent malignancy, as well as a nodule in the upper lumbar spinal cord, most likely a sign of leptomeningeal metastasis. Hence, cranio-spinal radiotherapy was given at a dose of 36 Gy in 20 fractions. Localized boosts were given to the primary site (20 Gy in 12 fractions) and the lumbar spine (8 Gy in 4 fractions). This treatment was completed 3 months following his initial surgery.
He proceeded to receive chemotherapy using the St. Judes ATRT protocol. This was developed for children aged >3 and comprises 4 cycles of cisplatin with high-dose cyclophosphamide and vincristine, with autologous peripheral blood stem-cell transplantation (11). This took 5 months to complete.
Since completing treatment there has been no sign of residual/recurrent disease at 2.5 years follow-up, clinically or on MRI, as seen in Figure 3.
Since his initial resection, he has suffered from panhypopituitarism, a left homonymous hemianopia and short-term memory impairment. He also developed a peripheral neuropathy following chemotherapy, most likely due to vincristine.
3. Methods
3.1. Review of Adult Cases
A literature search was performed using PubMed and Google Scholar to search for all reported cases of ATRT in adults. All 50 cases were tabulated; this is given as Supplementary Material (worksheet d1 in data sheet 1.xlsx). Many of these reports include similar tables; however, we noted a number of inconsistencies in these tables and ours is based on our reading of the original cases. While many prior authors have reported the ‘classical’ features of ATRT via IHC (see below), we are the first to tabulate all pathological variables for each case.
3.1.1. Clinical Variables
In each case, we recorded age, gender, and location as well as presence of leptomeningeal metastases (LM) and details of treatment at initial diagnosis and time of first progression. As above, a meaningful response to treatment was rare if the disease progressed again after “second-line” approaches. ‘Third-line’ treatments of any kind were exceptional and were not recorded.
The diagnosis of LM was based on clinical details and/or imaging features, e.g., in the case report of Wang et al. recurrence was spinal (distal from the original tumor) and appears to have originated from the leptomeninges on MRI, with subsequent intra-medullary spread (12).
A number of nominal (categorical) variables were ‘collapsed’ as this was such a small data set. Location, for example, initially had 15 values; we also analyzed this as a 5- and 3-category variable in order to look for more general patterns. We also assessed whether the location was ‘central’ vs. clearly lateralized and whether the tumor was “next to CSF” based on imaging findings. Similarly, the extent of resection was considered as an ordinal variable (gross total > sub-total resection > biopsy) as well as binary (surgery or not). We analyzed age as binary (i.e., ≥40 years when diagnosed) as well as a continuous numeric variable.
3.1.2. Pathological Variables
We consider here only the pathology at the time of diagnosis of ATRT. We note that only 2/40 (5%) of patients in our series had an autopsy performed (where we could infer this information). A summary table of all pathologic results is given as Supplementary Material (worksheet ihc1 in data sheet 1.xlsx). Most of these variables relate to the presence of certain antigens via IHC.
For IHC, a variety of antibodies and names were used to assess the presence of various elements. We have standardized this by recording the antigen being tested. Keratin staining, in particular, used a range of antibodies and was variously reported by antibody, by weight of the stained keratin, by keratin type or at times as “keratin present.”
3.2. Classification of IHC
3.2.1. Diagnosis of ATRT
Loss of INI1 protein expression (via IHC) is considered a defining feature of ATRT. Partial deletion of chromosome 22 (detected via FISH), where the gene (SMARCB1) is located, is considered as an equivalent test. Both of these tests began to be used widely after the year 2000. While both tests are considered to have a specificity of 100% for ATRT (in the appropriate setting), a negative result does not exclude the diagnosis. Such testing was reported in 23/50 (46%) of the cases we reviewed.
Prior to the advent of INI1 testing, the diagnosis was suspected on the basis of the appearance on standard H&E staining. Staining for the “classical” antigens vimentin, epithelial membrane antigen (EMA), and smooth muscle actin (SMA) was typically used as confirmatory. Both vimentin and SMA are classically associated with mesenchymal cells, although this does not imply a mesodermal origin for such cells. For example, constitutive expression of vimentin is recognized in arguably the most-studied glial cell line, U-251 (13). Again, a negative test result for one of these antigens does not exclude the diagnosis.
3.2.2. IHC and Differential Diagnosis
While the diagnosis of ATRT is often quickly suspected in an infant, the initial differential diagnosis in adults is typically far wider. Thus, the range of IHC stains employed in adult cases is far greater and more variable. In total, 81 tests (using IHC) were performed in our case series. In 19 (23%) cases, the antigen of interest was tested for in just one specimen.
IHC often included tests for metastasases, germ cell tumors, neuro-endocrine tumors, and other primary tumors of the nervous system (glial, medulloblastoma, neuroblastoma). Testing for a wider range of antigens (in adults vs. children) gives us a greater insight into the pathophysiology of ATRT.
Other possible differential diagnoses in adults may include primitive neuroectodermal tumor (PNET), choroid plexus carcinoma, rhabdoid meningioma, and germinoma. However, these entities generally have more characteristic immunohistochemical staining patterns with specific markers than the poly-phenotypic patterns seen in ATRT; also INI1 staining is retained.
3.2.3. Differential: Rhabdoid Glioblastoma
Tumors resembling ATRT, staining with glial fibrillary acidic protein (GFAP) as well as vimentin, SMA and EMA have been suggested to represent rhabdoid glioblastoma (GB) rather than ATRT (7). EMT in the setting of GB appears plausible as an entity distinct from ATRT. GB with EMT may have been misdiagnosed as adult ATRT (prior to the advent of testing for INI1), although this appears unlikely given the clinical and histological findings in our cases.
3.2.4. Germ Cell Layers
We used IHC to determine the putative germ cell layer of origin of each specimen. In so doing, we were guided by the tissues that are known to stain for an antigen under “normal” conditions. The website accompanying the textbook “Pathology Outlines” was particularly helpful in this regard (14).
Some stains are not restricted to one germ cell layer. An example is keratin 8, which predominantly occurs in ectodermal tissue but is also recognized in endodermal tissue (15).
3.2.5. Classification
We thus classified pathological findings into the following groups:
1. ‘ATRT specific’
• ‘Defining’ features (loss of INI1 or del(22))
• ‘Classical’ features (vimentin, EMA, SMA)
2. Ectoderm
• Neural crest (neuronal, glial, neuro-endocrine, melanoma markers)
• Other ectodermal
3. Endoderm
4. Keratins
5. Mesoderm
• Mesenchymal
• Leukemia/lymphoma markers
6. Germ cell
7. Others (found in a variety of tissues).
3.3. Quantitative Analysis
Data analysis was performed using R (RRID:SCR_001905) (16–18). The complete analysis is available as Supplementary Material (data sheet 2.pdf). Nominal variables are given as fractions (percentage) and continuous variables as median (range).
We did not attempt to correct for multiple hypothesis testing, as the present work is exploratory (19). As the sample size is small, a priori we considered p values of <0.1 to be potentially worth reporting.
3.3.1. Associations
We began by looking at the significance of all two-variable associations, using standard measures of strength and significance of association as follows:

3.3.2. Survival analysis
Time-to-event data were analyzed used the proportional-hazards model (PHM). We looked at the following outcomes:
1. time to progression
2. overall survival
3. time from progression to death (to assess the effects of second-line treatments).
Given the small number of observations and deaths, over-fitting was a problem with multivariate PHMs.
Survival times are reported as a median with 95% confidence intervals (CI). Effect size is given as hazard ratio (HR, relative to control) and the p value is from the score test (or Wald test for multivariate models).
3.3.3. Missing Data
Given such a large quantity of missing data, we tried to compensate in a number of ways:
• Recursive partitioning ‘looks ahead’ at survival to find the best split for the given data. It is particularly well suited to data sets where there is near-perfect prediction of outcome by certain variables. It also allows for a clear ordering of variables in terms of predictive importance (20).
• “Multivariate imputation by chained equations” uses repeated regression on the existing variables to estimate missing values. The technique does have a tendency to strengthen existing associations rather than developing new insights (21).
• The “nested cohort” design allows for more ‘accurate’ estimates from a PHM where a covariate is observed for only some subjects (the cohort). Sampling is stratified by a variable that is available on all cohort members.
This allows for frequency matching on confounders, or oversampling on the extremes of surrogate [markers] for exposures, to improve efficiency (22).
That is, this technique allows one to control for major confounders in order to arrive at a more meaningful estimate of effect size and probability. In our case, we sought to better determine the effects of various treatments using a sampling scheme based on:
1. the presence of LM or GFAP staining—as these were the major confounders of treatment
2. the occurrence of death when the last recorded follow-up was performed—to try to control for censoring, i.e., the fact that certain patients were not followed up until death had occurred.
4. Results
4.1. Demographics
The 50 patients reviewed (including the present cases) had a median age of 32 (18–65); 27 (54%) were females. Of those of childbearing age, 3/20 (15%) were pregnant when the diagnosis was made.
4.2. IHC
Some of the more significant findings on IHC are given in Table 1. As above, the complete table, with explanations, is given in (data sheet 1.xlsx).
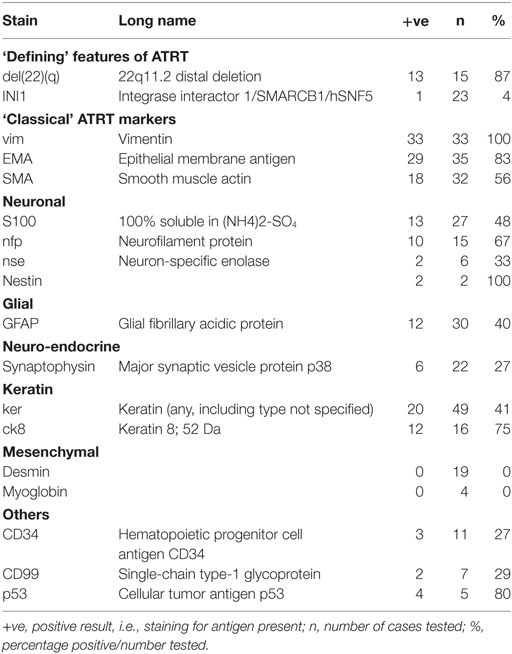
Table 1. Immunohistochemistry (IHC) of adult ATRT.
Defining features of ATRT were found in almost all cases where these were assessed: loss of INI1 in 23/24 (96%), del22q in 13/15 (87%). It is tempting to consider those cases where these were absent as “false negatives”; we will return to this in the discussion.
Of the “classical” features, only vimentin was universally positive (33/33) in our series. Germ cell markers, where assessed, were uniformly negative. Keratins were variably expressed; keratin 8 was found in the highest proportion of samples assessed, 12/16 (75%). As above, we cannot use this help attribute a germ cell layer of origin to a specimen (15).
4.2.1. Nestin
Markers of neuronal, glial, and neuro-endocrine differentiation were relatively common (c. 25–75%). Of particular interest is the presence of nestin in 2/2 cases studied. This is considered a marker of ‘neuroepithelial stem cells,’ is active in embryogenesis, and is downregulated in maturity, when expression of neurofilament protein and GFAP occur.
Interestingly, nestin is also expressed in developing muscle (presomitic mesoderm), to be replaced by desmin in maturity. Yet, desmin was universally absent in our sample (0/19). Other markers of mature muscle differentiation were also lacking, as well as the myogenic regulatory factor MYOD1 (0/2 cases), which is involved in myogenesis.
4.2.2. CD34
The hematopoietic system is thought to be derived from mesoderm; as such the presence of markers more typically used in the diagnosis of leukemia and lymphoma in our sample may at first appear anomalous. However, CD34 is recognized to occur in tumors of neuroepithelial origin, particularly those of childhood (23). It is also expressed on endothelial cells.
4.2.3. CD99
CD99 has been less well studied in CNS tumors. It is recognized to occur in “primary neuroepithelial tumors of the kidney,” an entity we believe to be closely related to ATRT (24). It is also present in ependymoma, peripheral primitive neuroectodermal tumor, and in the rare chordoid glioma, all of which are of neuroepithelial origin (25–27).
4.3. Associations
A number of striking associations are given in Table 2. Additional relationships of significance statistically but of debatable importance biologically are given as in the Supplementary Material as data sheet 2.pdf.
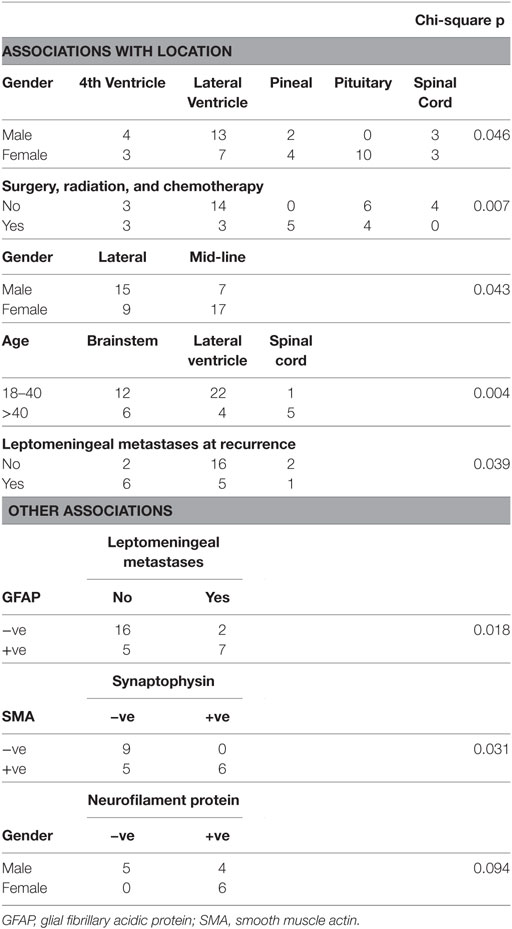
Table 2. Significant (p < 0.1) two-variable associations.
4.3.1. Demographics and Location
Where the tumor was in or next to the pituitary, 10/11 cases were in females (91%). The predilection of adult ATRT for this location has been recognized, although the sex-specificity seen here is novel (28).
Tumors of the spinal cord were associated with increasing age (Pearson’s r = 0.43, F test p = 0.002).
4.3.2. IHC
Tumors with LM when diagnosed were much more likely to show GFAP staining (n = 30, chi-squared p = 0.02), implying that glial differentiation is a major risk factor for this complication.
All tumors that showed synaptophysin staining also stained for SMA (n = 20, chi-squared, p = 0.03). Synaptophysin was the only neuroendocrine marker commonly assessed in the sample; this finding suggests that differentiation along such lines is associated with a greater tendency to manifest a mesenchymal phenotype.
Findings regarding neural differentiation were difficult to interpret. Neurofilament protein (NFP) staining was present in all female patients (6/6) vs. 4/9 males (chi-square p < 0.1). Strangely, staining for NFP and neurospecific enolase (NSE) appeared mutually exclusive, i.e., where one was positive the other was negative, although there were only 4 subjects where both were checked. No cases where NFP staining was absent stained for GFAP (n = 15).
4.4. Treatment
4.4.1. Surgery
Surgery (SX) at diagnosis was a cornerstone of treatment; in only one case was this not undertaken, where imaging initially suggested vestibular schwannoma and radiotherapy was used instead (12). Gross total resection (GTR) was reported in 10/43 (23%) of cases. This was most common for those tumors located next to a lateral ventricle. Subtotal resection was most common for tumors of the pituitary and pineal gland.
4.4.2. Adjuvant Treatment
Radiotherapy (RT) was undertaken in 33/42 (79%) and systemic chemotherapy (CT) in 17/42 (40%). Cranio-spinal irradiation (CSI) was performed in 5/34 (15%). All three approaches (SX, RT, and CT) were used in 15/42 cases (36%). This multimodality approach was most commonly used for those tumors next to a lateral ventricle (Table 2).
Intrathecal CT was used in just two cases other than our first patient. In contrast to our use of liposomal cytarabine, these two patients were treated with small quantities of methotrexate: three and one dose(s), respectively (29, 30).
4.4.3. Treatment at recurrence
When disease re-occurred/progressed, lower proportions of patients received treatment. Additional SX was attempted in 12/26 (46%), RT in 13/26 (50%) and CT in 11/26 (42%).
4.5. Survival
The median time to progression (TTP) was 5 months (95% CI 3–18). Overall survival (OS) was 23 months (14–56). Time from progression until last observation or death (time from progression to death, TPD), in the 27 patients where this was applicable and available, was 8 months (5–28). The fractions of patients without progression, and surviving, at various points in time from diagnosis are shown in Table 3.

Table 3. Percentages of patients without progression, and surviving, at timepoints following diagnosis.
4.5.1. Long-term Survivors
Apart from our second patient, two others are reported to have survived for more than 3 years (31, 32). Unlike our patient, both re-occurred on multiple occasions following their initial treatment. The first was a frontal lesion that required complete excision 6 times (and once in the contralateral frontal lobe) as well as receiving radiotherapy, Gamma Knife radiosurgery and systemic chemotherapy; after 7 years of relapsing disease, she remained disease free for another 10 years. The other was a parietal lesion which re-occurred 3 times over the course of 9 years and which appeared stable 1 year after most recent treatment. Interestingly, this patients’ tumor was diagnosed and treated as glioma, at presentation and at first re-occurrence. The diagnosis of ATRT was subsequently made in retrospect due to the absence of INI1 by IHC in all specimens.
4.5.2. Clinical Variables and IHC
Significant predictors of survival in PHMs are shown in Table 4. Of note, LM and GFAP staining appear as significant predictors in almost all of these models. While LM and GFAP are correlated, LM appears to be the more important predictor, particularly in multivariable PHMs. The effect of LM on OS is shown in Figure 5.
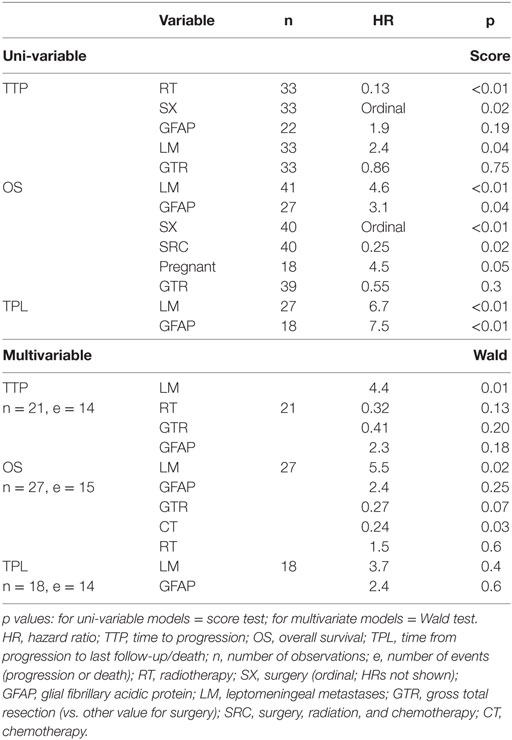
Table 4. Leptomeningeal metastases and GFAP staining are significant predictors in most proportional-hazards models.
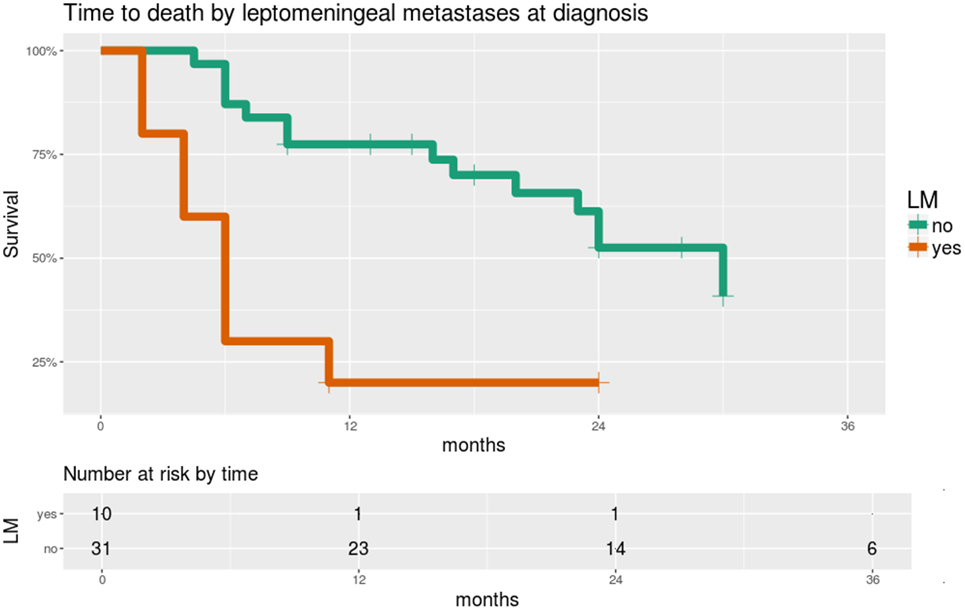
Figure 5. Overall survival is significantly worse in patients with leptomeningeal metastases (LM) at the time of diagnosis. Time scale is shown in months. Hazard ratio 4.4, score test p = 0.0005.
4.5.3. Treatments
“Over-fitting” was a problem when using surgery as an ordinal-scale variable in multivariate models. Instead, we give this as “gross total resection?” (GTR, yes/no). Regarding OS, only SX and multimodality treatment (SX, RT, and CT, HR = 0.3) were significant predictors. Both GTR and CT were significant predictors in the multivariable PMH for OS.
In assessing the effects of treatment, we acknowledge that we are not “comparing like with like,” i.e., patients for whom a certain treatment is not possible will be expected to have a worse outcome. Regarding surgery, GTR is generally the goal, where possible; those for whom this is not feasible will tend to have a worse prognosis, no matter the approach to surgery. Similarly, those for whom CT is impossible will tend to have poorer survival outcomes. However, significant comorbidities or disabilities due to disease (thus limiting the use of CT) were exceptional at the time when CT was commenced.
4.6. Recursive Partitioning
The most important predictor of OS was clearly LM; this is shown in Figure 6. When LM was excluded from the analysis, multimodality therapy (SX, RT, and CT) became the most significant variable (Supplementary Material, data sheet 2.pdf).
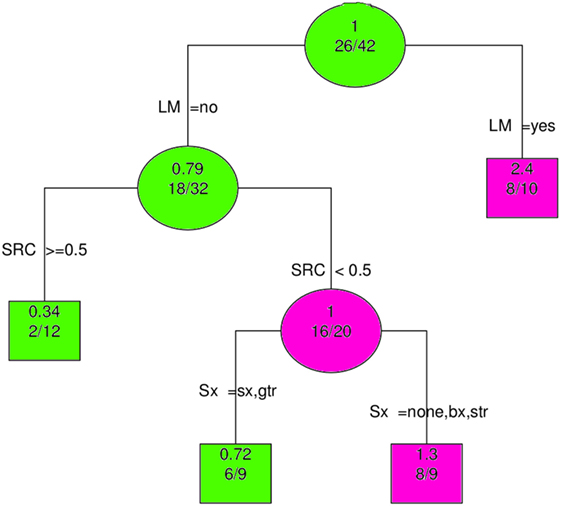
Figure 6. Recursive partitioning analysis shows LM to be the most important predictor of overall survival. Abbreviations: LM, leptomeningeal metastases; SRC, surgery, radiation and chemotherapy (≥0.5 means all modalities employed); Sx, Surgery; gtr, gross total resection; sx, surgery (extent unspecified); str, subtotal resection; bx, biopsy; none, no surgery. Key: circles, branching nodes; squares, terminal nodes; green, better outcomes; pink, worse outcomes; upper, no. predicted event rate; lower, no. no. events/no. at risk.
4.7. Multiple Imputation and “Nested” PHM
Applying these techniques to the potentially important predictors already identified tended to confirm the results above (data sheet 2.pdf).
The effects of the most common initial treatments on OS, when controlling for LM and GFAP (and death, in both cases), are shown in Table 5.
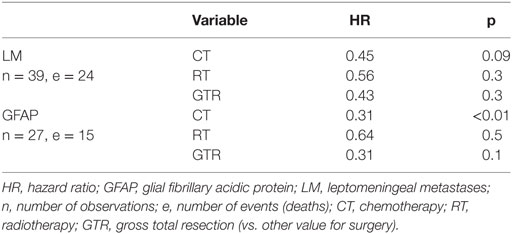
Table 5. Chemotherapy is the most important predictor of overall survival, when controlling for leptomeningeal metastases and GFAP staining (using ‘nested cohort’ proportional hazards models).
5. Discussion
5.1. Pathogenesis
5.1.1. Location at Diagnosis and Implications for Anatomical Origin
ATRT in adults has a predilection for mid-line structures, particularly the pineal and pituitary glands. These are circumventricular organs, which are now recognized as a source of neural stem cells in adults (33).
ATRT may be said to be the prototypical tumor of infancy, in having the earliest onset and highest perinatal incidence of such tumors. Tumors of infancy in general are thought to arise from cells that have not completed the process of terminal differentiation and continue to undergo hyperplasia from the latter half of pregnancy to age 3 or so (34).
Our case series suggests that there remains a pool of slowly dividing ‘ectodermal stem cells’ in at least some of these circumventricular organs that remains capable of acquiring carcinogenic mutations throughout most of adulthood (35). The proximity of these organs to CSF may explain the close association of these tumors with such locations and their tendency to develop LM. Of note, when ATRT occurs in a cerebral lobe, it typically appears in communication with a lateral ventricle.
We suspect that in many cases the tumor has already spread via CSF prior to diagnosis. This would help to explain the proposal of such entities as “primary diffuse cerebral leptomeningeal ATRT” and “ATRT arising from the acoustic nerve”; these structures appear unlikely to be points of origin for ATRT (12, 36).
The strong association between pituitary involvement and female gender is consistent with the greater mitotic activity of the pituitary in females throughout the lifespan (37). We acknowledge there remains much work to be done in this area.
5.1.2. IHC and Implications for Cell Type of Origin
As summarized in Table 1, findings on IHC suggest a tumor of ectoderm origin with EMT. For example, the combination of EMA (epithelial, 83%), vimentin (mesenchymal, 100%), and complete absence of markers of mature mesenchyme (desmin, myoglobin, 0%) are characteristic of such a phenotype.
In this regard, the name “ATRT” is somewhat misleading in that it is clearly not a teratoma and does not contain elements originating from mesoderm or showing skeletal muscle differentiation.
Classification of IHC by cell type and germ layer in adult ATRT has also been undertaken by Raisanen et al. (38). Their table is similar to our own, albeit with a smaller sample size. The authors do not attempt to infer any implications for pathogenesis from their table.
5.1.3. Germline Mutations and del22q
Loss of INI1 is not sufficient to cause ATRT. This is shown by the lack of complete penetrance of ATRT in germline mutations (distal 22q11 microdeletion syndromes) (39). None of the cases of ATRT in this series, including our own, had germline analysis performed to look for a mutations or deletion of chromosome 22q. The only case we reviewed where this seemed likely was our case with a prolonged response to treatment. The patent had macrocephaly and a history of spinal schwannoma as a teenager. None of the other reported a personal or family history of dysmorphism or of tumors seen in the rhabdoid-predisposition syndrome.
Nonetheless, it has been recognized that germline mutations in SMARCB1 germline mutations are not always inherited and unaffected adult carriers are recognized (40). In this series of 100 patients, those with germline mutations tended to present at a much younger age, at a median of 5 months (range birth—5 years) vs. 18 months (birth—17 years) for those with acquired mutations. However, in 4 of 7 cases of inherited mutations, the parents appeared completely unaffected. The authors also recognize germline/gonadal mosaicism in their sample, i.e., the mutation must have been present within parental gametes but not somatic cells.
Such germline mutations result in a phenotype known as “rhabdoid predisposition syndrome.” In addition to ATRT, carriers appear predisposed to rhabdoid tumors (renal and extra-renal), PNET, medulloblastoma, choroid plexus carcinoma and schwannoma (41). These authors also suggest the mutation as a cause for cases of Li–Fraumeni-like syndrome (patients with a childhood cancer or “sarcoma” aged <45 and a family history of cancer at a young age, in those lacking germline TP53 mutations).
Thus, we recommend somatic analysis in addition to analysis of the tumor, even in adult patients (42). Analysis of parental somatic and germline DNA also appears desirable.
5.1.4. INI1, del22q, and Epigenetic Subgroups
The only consistent genetic change in these tumors appears to be loss of INI1 due to SMARCB1 mutations (which are variable) (4, 43). Mutation of SMARCA4 is recognized as an alternative inciting event in rare cases, perhaps accounting for the single patient in our series with preserved INI1 expression. The lack of other consistent candidate oncogenic mutations has led to the recognition that epigenetic changes are crucial to pathogenesis. Three such epigenetic subgroups are now recognized (44). There were no adults in this recent work characterizing subgroups (age range: birth to 9.5 years). The subgroups are named based on the pathways most commonly upregulated, here shown with some of the genes whose expression is characteristically increased:

Phenotypically, the ATRT-TYR subgroup is strongly associated with a supratentional location vs. ATRT-MYC; ATRT-SHH appears in both locations.
Further characterization of phenotype by subgroup may be facilitated by preserving the methylation status of DNA in the tumor specimen. We suggest that fresh-frozen material be obtained for this purpose where practical. Technical advances continue to make methylation profiling more practical in formalin fixed, paraffin-embedded samples.
5.1.5. Chromatin Remodeling and EMT
EMT is known to result in a more ‘open’ chromatic structure (9). This is necessary to facilitate de-differentiation, i.e., the transcription of mRNA, which is typically unavailable to the differentiated epithelial cell. This includes a shift in energy (ATP) production from oxidative phosphorylation to glycolysis. This is necessary for the supply of energy of biosynthetic precursors, of a balanced redox status and appears necessary to maintain the undifferentiated state.
Chromatin remodeling complexes such as INI1 have been implicated in various cancers (45). INI1 is now recognized as a tumor suppressor, due to its mutation in tumors including choroid plexus carcinoma, medulloblastoma, primitive neuroectodermal tumor, and chronic myeloid leukemia. INI1 is a member of the SWI2/SNF2 remodeling complex, which is ATP dependent.
Loss of function of these chromatin remodeling complexes enables the development of the ‘open’ structure required for EMT. It remains unclear why loss of INI1 leads to diverging epigenetic phenotypes.
5.2. Clinical Trials
Ongoing and recently completed trials for this condition are tabulated in the recent review by Fruhwald et al. (3). The dearth of clinical trials enrolling adults >21 years old, just seven at the time of writing, is shown in Table 6.
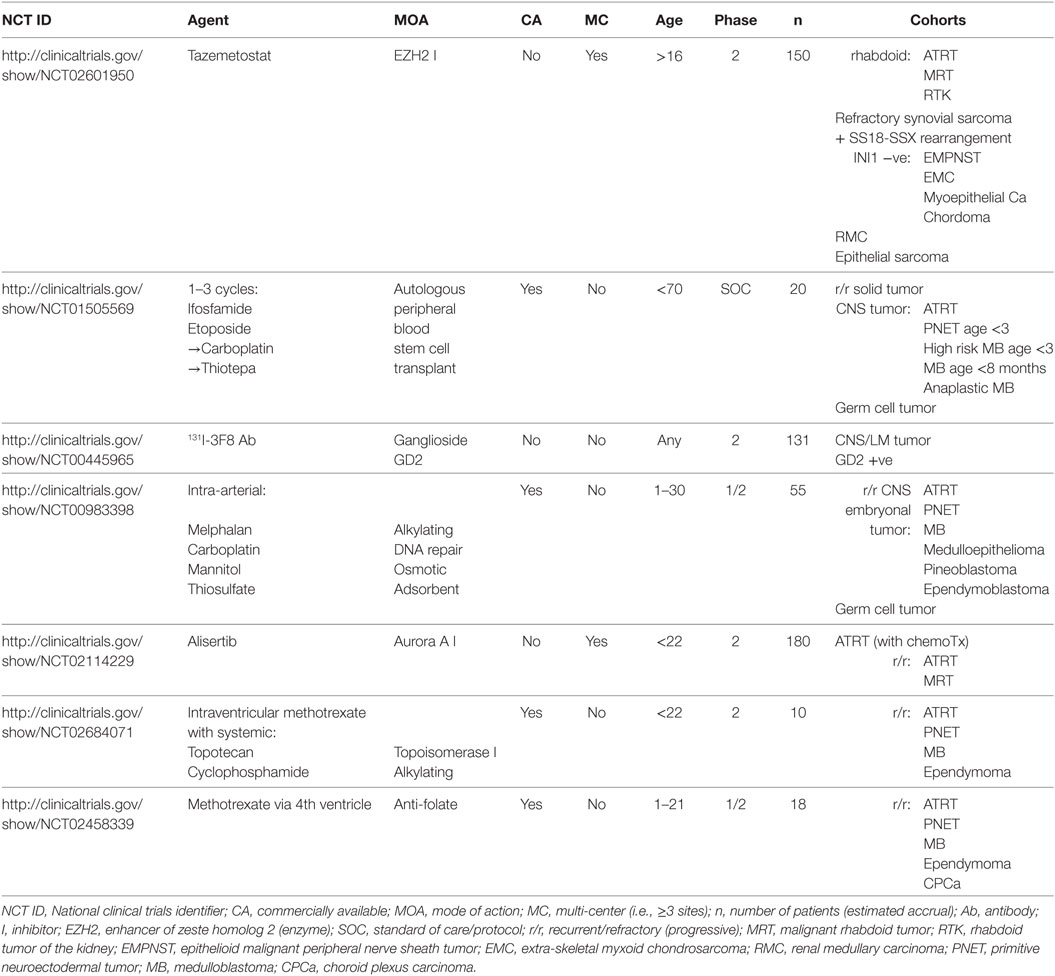
Table 6. Ongoing clinical trials enrolling patients aged >21.
As a rare condition, even in children, there is some justification for ‘lumping’ ATRT with similar tumors. This is particularly rational when the intervention is relatively non-specific, e.g., radiotherapy, traditional ‘cytotoxic’ chemotherapy (and soon, perhaps, immunotherapy).
In the case of more ‘targeted’ treatments, the pool of analogous diseases will be much smaller. Two of the seven trials may be said to be target aspects of the biology of ATRT. It is striking that the trial of alisertib aims to inhibit aurora kinase A, an enzyme that has been implicated in EMT. This trial includes malignant rhabdoid tumors (MRT), which show EMT but do not lose INI1 protein (46). MRTs, as well as rhabdoid tumors of the kidney (also showing EMT), are included in the trial of tamezostat (an EZH2 inhibitor). While beyond the scope of the current work, we suggest that the term “rhabdoid” is synonymous with EMT. Interestingly, the trial of tamezostat considers other tumors lacking INI1 to be sufficiently similar to be worth including.
The other five trials that include ATRT are relatively ‘non-specific’ and compensate for its low incidence by combining it with other ‘serious’ tumors, typically also of childhood. For example, four of five include PNET and medulloblastoma.
5.3. Treatment
As ATRT is primarily a disease of infancy, co-ordination of treatment between pediatric and adult oncology services appears appropriate.
5.3.1. Chemotherapy
ATRT usually occurs before CSI can be given safely, i.e., in those aged <3. Thus, CT has traditionally been a cornerstone of treatment. We suggest that a primary guiding factor be penetration into CSF (given the high prevalence of LM) as well as into the intraparenchymal tumor. As these tumors tend to disrupt the blood–brain barrier (BBB) and are often next to parts of the brain with no BBB, penetration of CT into normal brain parenchyma does not appear to be of prime concern.
In the rare cases when ATRT is localized and can be completely excised, local RT appears appropriate; arguably, a protocol similar to that used for glioblastoma can be adopted in such cases.
The most common approach to CT typically involves cytotoxic and relatively non-specific agents. Ifosfamide, carboplatin, and etoposide (ICE) was the most common regimen in our series, a protocol typically used for sarcoma and lymphoma. These agents have reasonable brain and CSF penetration (47–49). However, given the grave prognosis of this condition, a more aggressive approach appears warranted in adults, as in our second case.
5.3.2. Radiotherapy
Several reports demonstrated the beneficial role of adjuvant RT after surgical resection in patients with ATRT (11, 50, 51). However, RT is a significant challenge in those younger than 3 years old (52).
The benefit of combining RT with SX in ATRT has been shown by Lau et al. (53). In a sample of 171 pediatric and 3 adult cases, they report an increase in median survival from 1.9 ± 0.4 to 5.9 ± 0.7 years in those who had RT in addition to SX. Buscariollo et al. confirmed the value of RT in another retrospective series, this with 144 patients, where median survival improved from 6 to 34 months (p < 0.001) (54).
Once a decision on the use of RT has been made for a patient with ATRT, factors such as timing, dosing, and technique need to be considered.
Chen et al. reported improved disease-free survival in patients who had a total dose more than 50 Gy (55). Significant improvement in median survival was reported in patients who had a shorter interval between SX and RT. RT delivered early vs. latein the course of treatment has also been associated with improved survival (11).
Proton therapy may be especially attractive in younger patients, in order to minimize the risk of late complications. Encouraging results with local conformal proton therapy have been reported by Bernstein et al. (56). Nine of ten patients with ATRT (with a median age of 1.8 years) had no evidence of disease, with a median follow-up of 27.3 months.
As distal relapse is associated with higher mortality and as the majority of distant relapses occur within the CNS, CSI is recommended is those older than 3 years old: 23.4 Gy to the neuraxis (with 54 Gy to the tumor bed) is recommended inpatients older than 3 years old. Younger patients have been treated using either no adjuvant RT or local-only RT, with a trend toward improved survival with the addition of RT.
5.3.3. A Combined Approach
Promising results have been already been shown in children >3 with PNET undergoing CSI followed by high-dose CT and in the setting of recurrent CNS tumors (57, 58). Stem-cell rescue was required with these more aggressive regiments, either from peripheral blood or bone marrow. Given its known role as a radiosensitizer and excellent brain and CSF penetration, the use of temozolomide during radiotherapy also appears reasonable (59). Early use of intrathecal chemotherapy also appears rational. As it is well tolerated in long-term use, it may also be an acceptable approach in those patients who would not tolerate more aggressive forms of CT.
6. Conclusion
ATRT in adults carries a grave prognosis, particularly in those cases with leptomeningeal spread. We postulate that ATRT is a tumor of neuroectodermal origin that demonstrates mesenchymal transition.
Our cases indicate the possibility of improved outcome with more aggressive therapy. CSI should always be considered in adults. The use of stem-cell rescue allows for the use of more aggressive chemotherapeutic regimens than has hitherto been the case.
Consent
Written, informed consent was obtained from both patients specifically for the publication of these case reports.
Author Contributions
CD drafted the manuscript and performed the statistical analysis. CD and KM wrote the first case report and performed the literature review. JY and EH wrote the second case report. EA performed the Sanger sequencing. All the authors contributed to data interpretation and to the final draft of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fneur.2017.00247/full#supplementary-material.
Additional information is provided in supplemental files:
• data sheet 1.xlsx A spreadsheet with the following worksheets:
• d1 Data set used for analysis.
• k1 Key to data set.
• ihc Immunohistochemistry.
• data sheet 2.pdf Statistical analysis.
Case reports and case series reviewed, but not specifically cited above (60–89).
References
1. Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg (1996) 85(1):56–65. doi: 10.3171/jns.1996.85.1.0056
2. Russell DS. “Ectopic pinealoma”: its kinship to atypical teratoma of the pineal gland; report of a case. J Pathol Bacteriol (1954) 68(1):125–9. doi:10.1002/path.1700680115
3. Fruhwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol (2016) 18(6):764–78. doi:10.1093/neuonc/nov264
4. Hasselblatt M, Isken S, Linge A, Eikmeier K, Jeibmann A, Oyen F, et al. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer (2013) 52(2):185–90. doi:10.1002/gcc.22018
5. Woehrer A, Slavc I, Waldhoer T, Heinzl H, Zielonke N, Czech T, et al. Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer (2010) 116(24):5725–32. doi:10.1002/cncr.25540
6. Schmidt D, Harms D, Zieger G. Malignant rhabdoid tumor of the kidney. Histopathology, ultrastructure and comments on differential diagnosis. Virchows Arch A Pathol Anat Histopathol (1982) 398(1):101–8. doi:10.1007/BF00585617
7. Mutou J, Hirose Y, Ikeda E, Yoshida K, Nakazato Y, Kawase T. Malignant brain tumor with rhabdoid features in an adult. Neurol Med Chir (Tokyo) (2011) 51(6):449–54. doi:10.2176/nmc.51.449
8. Kahn LB, Uys CJ, Dale J, Rutherfoord S. Carcinoma of the breast with metaplasia to chondrosarcoma: a light and electron microscopic study. Histopathology (1978) 2(2):93–106. doi:10.1111/j.1365-2559.1978.tb01698.x
9. Li L, Li W. Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Ther (2015) 150:33–46. doi:10.1016/j.pharmthera.2015.01.004
10. Nucleotide Database. NCBI Reference Sequence: NT_011520.13. Technical report, National Center for Biotechnology Information. [NCBI:NT_011520] (2016). Available from: http://www.ncbi.nlm.nih.gov/nuccore/NT_011520
11. Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol (2005) 23(7):1491–9. doi:10.1200/JCO.2005.05.187
12. Wang X, Liu X, Lin Z, Chen Y, Wang P, Zhang S. Atypical teratoid/rhabdoid tumor (AT/RT) arising from the acoustic nerve in a young adult: a case report and a review of literature. Medicine (Baltimore) (2015) 94(4):e439. doi:10.1097/MD.0000000000000439
13. Fuchs E. Threads between useful and useless. Curr Biol (1991) 1(5):284–6. doi:10.1016/0960-9822(91)90085-B
15. Hosoya A, Kwak S, Kim EJ, Lunny DP, Lane EB, Cho SW, et al. Immunohistochemical localization of cytokeratins in the junctional region of ectoderm and endoderm. Anat Rec (Hoboken) (2010) 293(11):1864–72. doi:10.1002/ar.21233
16. Core Team R. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing (2015).
17. Xie Y. knitr: A General-Purpose Package for Dynamic Report Generation in R. R package version 1.12-3 (2016).
18. Dardis C, Woolf EC, Scheck AC. Towards reproducible research: from data analysis (in R) to a typeset laboratory notebook (as. pdf) using the text editor emacs with the ‘mp’ package. F1000Research (2015) 4:483. doi:10.12688/f1000research.6800.2
19. Perneger TV. What’s wrong with Bonferroni adjustments. BMJ (1998) 316(7139):1236–8. doi:10.1136/bmj.316.7139.1236
20. Therneau T, Atkinson B, Ripley B. rpart: Recursive Partitioning. R package version 4.1-2 (2013).
21. van Buuren S, Groothuis-Oudshoorn K. Mice: multivariate imputation by chained equations in R. J Stat Softw (2011) 45(3):1–67. doi:10.18637/jss.v045.i03
22. Katki H, Mark S. Survival analysis for cohorts with missing covariate information. R J (2008) 8(1):14–9.
23. Chappe C, Padovani L, Scavarda D, Forest F, Nanni-Metellus I, Loundou A, et al. Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol (2013) 23(5):574–83. doi:10.1111/bpa.12048
24. Parham DM, Roloson GJ, Feely M, Green DM, Bridge JA, Beckwith JB. Primary malignant neuroepithelial tumors of the kidney: a clinicopathologic analysis of 146 adult and pediatric cases from the National Wilms’ Tumor Study Group Pathology Center. Am J Surg Pathol (2001) 25(2):133–46. doi:10.1097/00000478-200102000-00001
25. Mahfouz S, Aziz AA, Gabal SM, el Sheikh S. Immunohistochemical study of CD99 and EMA expression in ependymomas. Medscape J Med (2008) 10(2):41.
26. Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer (1991) 67(7):1886–93. doi:10.1002/1097-0142(19910401)67:7<1886::AID-CNCR2820670712>3.0.CO;2-U
27. Romero-Rojas AE, Diaz-Perez JA, Ariza-Serrano LM. CD99 is expressed in chordoid glioma and suggests ependymal origin. Virchows Arch (2012) 460(1):119–22. doi:10.1007/s00428-011-1170-2
28. Regan J, Tehrani M, Rodriguez F, Watson J. Adult At/Rt: predilection for the pituitary? J Neurol Neurosurg (2015) 2(1):110. doi:10.19104/jnn.2015.110
29. Kawaguchi T, Kumabe T, Watanabe M, Tominaga T. Atypical teratoid/rhabdoid tumour with leptomeningeal dissemination in an adult. Acta Neurochir (Wien) (2004) 146(9):1033–8. doi:10.1007/s00701-004-0313-5
30. Liebigt S, Florschutz A, Arndt N, Stock K, Renner C. Atypical teratoid/rhabdoid tumor of the pineal region in a young adult male patient: case report and review of the literature. J Neurol Surg A Cent Eur Neurosurg (2017) 78(1):92–8. doi:10.1055/s-0036-1583180
31. Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M. Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol (2008) 88(3):321–30. doi:10.1007/s11060-008-9571-z
32. Takahashi K, Nishihara H, Katoh M, Yoshinaga T, Mahabir R, Kanno H, et al. Case of atypical teratoid/rhabdoid tumor in an adult, with long survival. Brain Tumor Pathol (2011) 28(1):71–6. doi:10.1007/s10014-010-0008-y
33. Bennett L, Yang M, Enikolopov G, Iacovitti L. Circumventricular organs: a novel site of neural stem cells in the adult brain. Mol Cell Neurosci (2009) 41(3):337–47. doi:10.1016/j.mcn.2009.04.007
34. Mora J. What is a pediatric tumor? Clin Oncol Adolesc Young Adults (2012) 2:7–15. doi:10.2147/COAYA.S29791
35. Calvo JL, Boya J, Carbonell AL, Garcia-Maurino JE. Cell proliferation in the developing rat pineal gland. A bromodeoxyuridine immunohistochemical study. Histol Histopathol (2000) 15(4):1005–10.
36. El-Nabbout B, Shbarou R, Glasier CM, Saad AG. Primary diffuse cerebral leptomeningeal atypical teratoid rhabdoid tumor: report of the first case. J Neurooncol (2010) 98(3):431–4. doi:10.1007/s11060-009-0094-z
37. Levy A. Physiological implications of pituitary trophic activity. J Endocrinol (2002) 174(2):147–55. doi:10.1677/joe.0.1740147
38. Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol (2005) 15(1):23–8. doi:10.1111/j.1750-3639.2005.tb00096.x
39. Lafay-Cousin L, Payne E, Strother D, Chernos J, Chan M, Bernier FP. Goldenhar phenotype in a child with distal 22q11.2 deletion and intracranial atypical teratoid rhabdoid tumor. Am J Med Genet A (2009) 149A(12):2855–9. doi:10.1002/ajmg.a.33119
40. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer (2011) 56(1):7–15. doi:10.1002/pbc.22831
41. Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet (1999) 65(5):1342–8. doi:10.1086/302639
42. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res (1999) 59(1):74–9.
43. Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest (2012) 122(8):2983–8. doi:10.1172/JCI64400
44. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell (2016) 29(3):379–93. doi:10.1016/j.ccell.2016.02.001
45. Davis PK, Brackmann RK. Chromatin remodeling and cancer. Cancer Biol Ther (2003) 2(1):22–9. doi:10.4161/cbt.189
46. Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu L, et al. Inhibition of aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis (2008) 29(10):1930–7. doi:10.1093/carcin/bgn176
47. Jacobs S, McCully CL, Murphy RF, Bacher J, Balis FM, Fox E. Extracellular fluid concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemother Pharmacol (2010) 65(5):817–24. doi:10.1007/s00280-009-1085-7
48. Kaijser GP, De Kraker J, Bult A, Underberg WJ, Beijnen JH. Pharmacokinetics of ifosfamide and some metabolites in children. Anticancer Res (1998) 18(3B):1941–9.
49. Muldoon LL, Soussain C, Jahnke K, Johanson C, Siegal T, Smith QR, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol (2007) 25(16):2295–305. doi:10.1200/JCO.2006.09.9861
50. Balaton AJ, Vaury P, Videgrain M. Paravertebral malignant rhabdoid tumor in an adult. A case report with immunocytochemical study. Pathol Res Pract (1987) 182(5):713–8. doi:10.1016/S0344-0338(89)80130-X
51. Arita K, Sugiyama K, Sano T, Oka H. Atypical teratoid/rhabdoid tumour in sella turcica in an adult. Acta Neurochir (Wien) (2008) 150(5):491–5. doi:10.1007/s00701-008-1500-y
52. Squire SE, Chan MD, Marcus KJ. Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol (2007) 81(1):97–111. doi:10.1007/s11060-006-9196-z
53. Lau CS, Mahendraraj K, Chamberlain RS. Atypical teratoid rhabdoid tumors: a population-based clinical outcomes study involving 174 patients from the surveillance, epidemiology, and end results database (1973–2010). Cancer Manag Res (2015) 7:301. doi:10.2147/CMAR.S88561
54. Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a surveillance, epidemiology, and end results analysis. Cancer (2012) 118(17):4212–9. doi:10.1002/cncr.27373
55. Chen YW, Wong TT, Ho DM, Huang PI, Chang KP, Shiau CY, et al. Impact of radiotherapy for pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience). Int J Radiat Oncol Biol Phys (2006) 64(4):1038–43. doi:10.1016/j.ijrobp.2005.10.001
56. Bernstein KDA, Sethi R, Trofimov A, Zeng C, Fullerton B, Yeap BY, et al. Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys (2013) 86(1):114–20. doi:10.1016/j.ijrobp.2012.12.004
57. Strother D, Ashley D, Kellie SJ, Patel A, Jones-Wallace D, Thompson S, et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem-cell rescue for patients with newly diagnosed medulloblastoma or supratentorial primitive neuroectodermal tumor after craniospinal radiotherapy: results of a collaborative study. J Clin Oncol (2001) 19(10):2696–704. doi:10.1200/JCO.2001.19.10.2696
58. Finlay JL, Goldman S, Wong MC, Cairo M, Garvin J, August C, et al. Pilot study of high-dose thiotepa and etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS tumors. The Children’s Cancer Group. J Clin Oncol (1996) 14(9):2495–503. doi:10.1200/JCO.1996.14.9.2495
59. Zhu W, Zhou L, Qian JQ, Qiu TZ, Shu YQ, Liu P. Temozolomide for treatment of brain metastases: a review of 21 clinical trials. World J Clin Oncol (2014) 5(1):19–27. doi:10.5306/wjco.v5.i1.19
60. Arrazola J, Pedrosa I, Mendez R, Saldana C, Scheithauer BW, Martinez A. Primary malignant rhabdoid tumour of the brain in an adult. Neuroradiology (2000) 42(5):363–7. doi:10.1016/j.jocn.2009.02.011
61. Ashraf R, Bentley RC, Awan AN, McLendon RE, Ragozzino MW. Implantation metastasis of primary malignant rhabdoid tumor of the brain in an adult (one case report). Med Pediatr Oncol (1997) 28(3):223–7. doi:10.1002/(SICI)1096-911X(199703)28:3<223:AID-MPO14>3.0.CO;2-F
62. Bruch LA, Hill DA, Cai DX, Levy BK, Dehner LP, Perry A. A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors. Hum Pathol (2001) 32(2):156–62. doi:10.1053/hupa.2001.21572
63. Byram D. Regarding Weiss et al., IJROBP 41:103-109; 1998. Int J Radiat Oncol Biol Phys (1999) 45(1):247. doi:10.1017/CBO9781107415324.004
64. Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol (2007) 84(2):217–22. doi:10.1007/s11060-007-9361-z
65. Cossu A, Massarelli G, Manetto V, Viale G, Tanda F, Bosincu L, et al. Rhabdoid tumours of the central nervous system. Report of three cases with immunocytochemical and ultrastructural findings. Virchows Arch A Pathol Anat Histopathol (1993) 422(1):81–5. doi:10.1007/BF01605137
66. Dreyfuss Z, Simpson T. A pediatric tumor causing a neurosurgical emergency in an adult. Am J Respir Crit Care Med (2016) 193:A1965.
67. Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, et al. The DNA sequence of human chromosome 22. Nature (1999) 402(6761):489–95. doi:10.1038/990031
68. Erickson ML, Johnson R, Bannykh SI, de Lotbiniere A, Kim JH. Malignant rhabdoid tumor in a pregnant adult female: literature review of central nervous system rhabdoid tumors. J Neurooncol (2005) 74(3):311–9. doi:10.1007/s11060-004-7560-4
69. Fisher BJ, Siddiqui J, Macdonald D, Cairney AE, Ramsey D, Munoz D, et al. Malignant rhabdoid tumor of brain: an aggressive clinical entity. Can J Neurol Sci (1996) 23(4):257–63. doi:10.1017/S0317167100038191
70. Gorayski P, Boros S, Ong B, Olson S, Foote M. Radiation-induced primary cerebral atypical teratoid/rhabdoid tumour in an adult. J Clin Neurosci (2013) 20(10):1466–8. doi:10.1016/j.jocn.2013.03.041
71. Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E. Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol (1992) 83(4):445–8. doi:10.1007/BF00713540
72. Ingold B, Moschopulos M, Hutter G, Seeger H, Rothlisberger B, Landolt H, et al. Abdominal seeding of an atypical teratoid/rhabdoid tumor of the pineal gland along a ventriculoperitoneal shunt catheter. Acta Neuropathol (2006) 111(1):56–9. doi:10.1007/s00401-005-1112-7
73. Kachhara R, Retnam TM, Kumar S, Nair S, Bhattacharya RN, Krishnamoorthy T, et al. Rhabdoid tumor of the thalamus. Neurol India (2003) 51(2):273–4.
74. Kanoto M, Toyoguchi Y, Hosoya T, Kuchiki M, Sugai Y. Radiological image features of the atypical teratoid/rhabdoid tumor in adults: a systematic review. Clin Neuroradiol (2015) 25(1):55–60. doi:10.1007/s00062-013-0282-2
75. Kuge A, Kayama T, Tsuchiya D, Kawakami K, Saito S, Nakazato Y, et al. [Suprasellar primary malignant rhabdoid tumor in an adult: a case report]. No Shinkei Geka (2000) 28(4):351–8.
76. Lutterbach J, Liegibel J, Koch D, Madlinger A, Frommhold H, Pagenstecher A. Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol (2001) 52(1):49–56. doi:10.1023/A:1010683416555
77. Moretti C, Lupoi D, Spasaro F, Chioma L, Di Giacinto P, Colicchia M, et al. Sella turcica atypical teratoid/rhabdoid tumor complicated with lung metastasis in an adult female. Clin Med Insights Case Rep (2013) 6:177–82. doi:10.4137/CCRep.S12834
78. Park HG, Yoon JH, Kim SH, Cho KH, Park HJ, Kim SH, et al. Adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor treated with a multimodal approach: a case report. Brain Tumor Res Treat (2014) 2(2):108–13. doi:10.14791/btrt.2014.2.2.108
79. Pimentel J, Silva R, Pimentel T. Primary malignant rhabdoid tumors of the central nervous system: considerations about two cases of adulthood presentation. J Neurooncol (2003) 61(2):121–6. doi:10.1023/A:1022135518846
80. Rezanko T, Tunakan M, Kahraman A, Sucu HK, Gelal F, Akkol I. Primary rhabdoid tumor of the brain in an adult. Neuropathology (2006) 26(1):57–61. doi:10.1111/j.1440-1789.2006.00624.x
81. Roy S, Mallik C, Maiti S, Chaudhuri T. Temporal lobe atypical teratoid/rhabdoid tumor in a 24-year old adult female. South Asian J Cancer (2013) 2(4):210. doi:10.4103/2278-330X.119909
82. Samaras V, Stamatelli A, Samaras E, Stergiou I, Konstantopoulou P, Varsos V, et al. Atypical teratoid/rhabdoid tumor of the central nervous system in an 18-year-old patient. Clin Neuropathol (2009) 28(1):1–10. doi:10.5414/NPP28001
83. Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: a case report and review of the literature. Surg Neurol Int (2014) 5:75. doi:10.4103/2152-7806.133105
84. Shonka NA, Armstrong TS, Prabhu SS, Childress A, Choi S, Langford LA, et al. Atypical teratoid/rhabdoid tumors in adults: a case report and treatment-focused review. J Clin Med Res (2011) 3(2):85–92. doi:10.4021/jocmr535w
85. Sinha P, Ahmad M, Varghese A, Parekh T, Ismail A, Chakrabarty A, et al. Atypical teratoid rhabdoid tumour of the spine: report of a case and literature review. Eur Spine J (2015) 24(Suppl 4):S472–84. doi:10.1007/s00586-014-3445-1
86. Souki C, Abdel-Rhaman M, Qasem A, Al-Hussaini M. Atypical teratoid rhabdoid tumor in adulthood. Clin Neuropathol (2014) 33(3):245–50. doi:10.5414/NP300683
87. Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M. Pineal malignant rhabdoid tumor with chondroid formation in an adult. Pathol Int (1999) 49(12):1114–8. doi:10.1046/j.1440-1827.1999.00988.x
88. Takei H, Adesina AM, Mehta V, Powell SZ, Langford LA. Atypical teratoid/rhabdoid tumor of the pineal region in an adult. J Neurosurg (2010) 113(2):374–9. doi:10.3171/2009.10.JNS09964
Keywords: teratoma, rhabdoid tumor, adult, pregnancy, statistical data analysis, statistical data interpretation, etiology
Citation: Dardis C, Yeo J, Milton K, Ashby LS, Smith KA, Mehta S, Youssef E, Eschbacher J, Tucker K, Dawes L, Lambie N, Algar E and Hovey E (2017) Atypical Teratoid Rhabdoid Tumor: Two Case Reports and an Analysis of Adult Cases with Implications for Pathophysiology and Treatment. Front. Neurol. 8:247. doi: 10.3389/fneur.2017.00247
Received: 28 February 2017; Accepted: 18 May 2017;
Published: 20 June 2017
Edited by:
Gordon Li, Stanford University, United StatesReviewed by:
Michael C. Fruehwald, Children’s Hospital Augsburg, GermanyOfelia Cruz, Hospital Sant Joan de Déu Barcelona, Spain
Copyright: © 2017 Dardis, Yeo, Milton, Ashby, Smith, Mehta, Youssef, Eschbacher, Tucker, Dawes, Lambie, Algar and Hovey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher Dardis, Y2hyaXN0b3BoZXJkYXJkaXNAZ21haWwuY29t