 Daniel Agustín Godoy1,2*
Daniel Agustín Godoy1,2* Ali Seifi3David Garza4Santiago Lubillo-Montenegro5Francisco Murillo-Cabezas6
Ali Seifi3David Garza4Santiago Lubillo-Montenegro5Francisco Murillo-Cabezas6
- 1Neurointensive Care Unit, Sanatorio Pasteur, San Fernando del Valle de Catamarca, Argentina
- 2Intensive Care Unit, Hospital San Juan Bautista, Catamarca, Argentina
- 3University of Texas Health Science Center San Antonio, San Antonio, TX, United States
- 4Department of Neurosurgery, University of Texas Health Science Center San Antonio, San Antonio, TX, United States
- 5Intensive Care Unit, Hospital Universitario Na Sa de Candelaria, Santa Cruz de Tenerife, Spain
- 6Intensive Care Unit, Hospital Universitario Virgen del Rocío, Sevilla, Spain
During traumatic brain injury, intracranial hypertension (ICH) can become a life-threatening condition if it is not managed quickly and adequately. Physicians use therapeutic hyperventilation to reduce elevated intracranial pressure (ICP) by manipulating autoregulatory functions connected to cerebrovascular CO2 reactivity. Inducing hypocapnia via hyperventilation reduces the partial pressure of arterial carbon dioxide (PaCO2), which incites vasoconstriction in the cerebral resistance arterioles. This constriction decrease cerebral blood flow, which reduces cerebral blood volume and, ultimately, decreases the patient’s ICP. The effects of therapeutic hyperventilation (HV) are transient, but the risks accompanying these changes in cerebral and systemic physiology must be carefully considered before the treatment can be deemed advisable. The most prominent criticism of this approach is the cited possibility of developing cerebral ischemia and tissue hypoxia. While it is true that certain measures, such as cerebral oxygenation monitoring, are needed to mitigate these dangerous conditions, using available evidence of potential poor outcomes associated with HV as justification to dismiss the implementation of therapeutic HV is debatable and remains a controversial subject among physicians. This review highlights various issues surrounding the use of HV as a means of controlling posttraumatic ICH, including indications for treatment, potential risks, and benefits, and a discussion of what techniques can be implemented to avoid adverse complications.
Introduction
Physicians started to explore hyperventilation (HV) as a way to lower cerebral blood volume (CBV) during the 1920s (1). One of the earliest documented descriptions of this treatment dates back to 1959, when Lundberg reported the use of HV to reduce increased levels of intracranial pressure (ICP) (2). HV induces arteriolar vasoconstriction, which results in decreased cerebral blood flow (CBF) and, consequently, a decrease in CBV (3–5). As time went on, this easily implemented therapy was widely used for the management of intracranial hypertension (ICH) secondary to severe traumatic brain injury (sTBI) (6–8). By the mid-1990s, in neurosurgical centers located within the United States and the United Kingdom, the rate of HV utilization was 83 and 97%, respectively (6, 7). A European database analysis released in 2008 indicated that during the first 24 h after insult, physicians employed prophylactic HV in more than half of their TBI cases (9).
Although HV rapidly and effectively reduces ICP, the effects are transient and have not been associated with the improvement of final patient outcome (10, 11). Because HV can potentially trigger secondary cerebral ischemic lesions and create adverse repercussions that affect other organ systems, and the therapy remains a topic of controversy and vigorous debate (10, 11). Available evidence indicates that intense or prolonged prophylactic HV is detrimental and should be avoided, especially during the acute phase of sTBI; however, the therapy is still recommended as a way to temporarily manage life-threatening elevations of ICP (12). The goal of this review is to provide an update that evaluates the current studies describing HV for ICP control to determine if HV has a role in the management of acute brain injury.
CO2 Physiology: Basic Concepts
Hyperventilation leads to an increase in alveolar ventilation (AV) (13, 14), the volume of air per minute that enters the respiratory zones (bronchioles, alveoli, etc.) that is also available for gas exchange (13). Because a portion of that volume remains in areas where gases do not diffuse into the bloodstream, AV can be determined by the following equation (13):
Alveolar ventilation has an inverse relationship with the alveolar CO2 level; when AV increases, the alveolar CO2 levels decrease (13, 14). However, alveolar CO2 has a direct association with the partial pressure of arterial CO2 (PaCO2), which reflects the balance between the production and elimination of CO2 (13, 14):
A patient’s cellular production of CO2 is dependent on several variables, including diet, exercise, temperature, and hormone activity (thyroid) (13). CO2 production remains relatively stable and constant, except during severe hypermetabolic states. If CO2 transport and cardiac output remain unchanged, PaCO2 levels will be determined inversely by the rate of CO2 elimination through the AV (13).
CO2 is a soluble and diffusible gas that is transported in three different ways: 10–15% of it is dissolved according to the PaCO2 (Henry’s Law); 20–30% of it is bound to plasma proteins and hemoglobin that form carbaminic complexes, and 65–70% of it is converted to bicarbonate/carbonic acid in the red blood cells and plasma (13). This third complex reaction helps maintain equilibrium between bicarbonate and hydrogen (H+) ions (13, 14) (Figure 1).
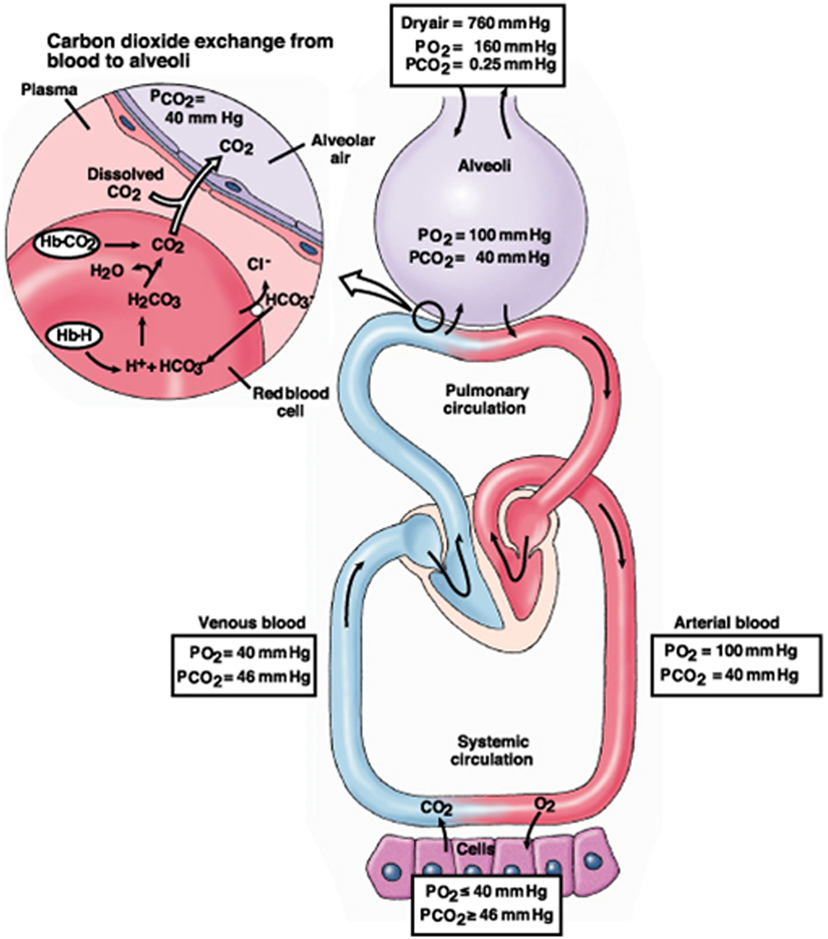
Figure 1. The CO2 physiology from cells to alveoli.
Normal PaCO2 values fluctuate between 35 and 45 mmHg (4.7–6 kPa) at normal body temperature and at a sea level with barometric pressure of 760 mmHg (13, 14).
If body temperature decreases, the solubility of CO2 increases and PaCO2, consequently, decreases (13, 15). PaCO2 decreases 4.5% for each centigrade degree temperature decrease. The opposite occurs when temperature increases (Table 1) (15). At higher altitude, the barometric pressure decreases, stimulating AV, so normal PaCO2 levels are lower (Table 2) (16–20).
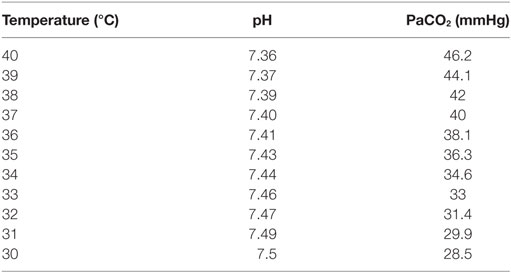
Table 1. Modification of normal PaCO2 values according to changes in central temperature ([ref]15).
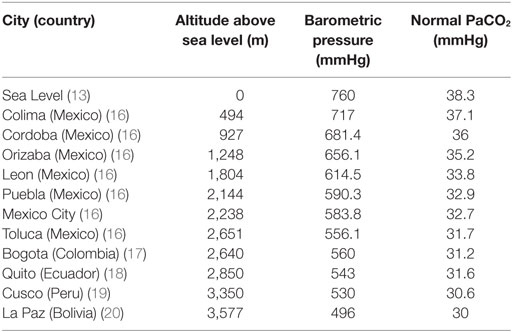
Table 2. The normal PaCO2 according to altitude and barometric pressure.
Changes in Cerebral Physiology During PaCO2 Reduction
The brain is one of the most metabolically active organs in the entire body. Because it lacks reserves of oxygen and glucose, two nutrients that are vital to maintaining vigorous physiochemical activity, it is important for the brain to have some system of continuous delivery through which these substrates can be received (21). This delivery system occurs through the CBF. CBF is so crucial that the brain has developed adaptive mechanisms to maintain adequate and constant flow despite changes in physiological variables or metabolic requirements (21–23). The maintenance of CBF is called “cerebral autoregulation,” and it is primarily achieved by the resistance arterioles (22, 23). By dilating or contracting in response to changes in arterial blood pressure, viscosity, gases, and metabolic demands, the arterioles can regulate CBF (22, 23).
Cerebral Blood Flow
“CO2 reactivity” is the ability cerebral resistance arterioles possess to dilate or contract in response to changes in the partial pressure of arterial CO2. Dilation occurs when the partial pressure of arterial CO2 increases (PaCO2 > 44 mmHg indicates hypercapnia); the vessels contract if the PaCO2 level decreases (PaCO2 < 35 mmHg indicates hypocapnia) (3–5, 10, 11). However, this vascular activity only occurs within the 20–60 mmHg range of PaCO2 (24). The diameter of the vessels will not change if PaCO2 levels rise above or drop below that specific range. Because of these restrictions, when it is plotted on a graph, the autoregulatory curve depicting CBF according to shifting PaCO2 resembles a sigmoid function (Figure 2) (4, 25).
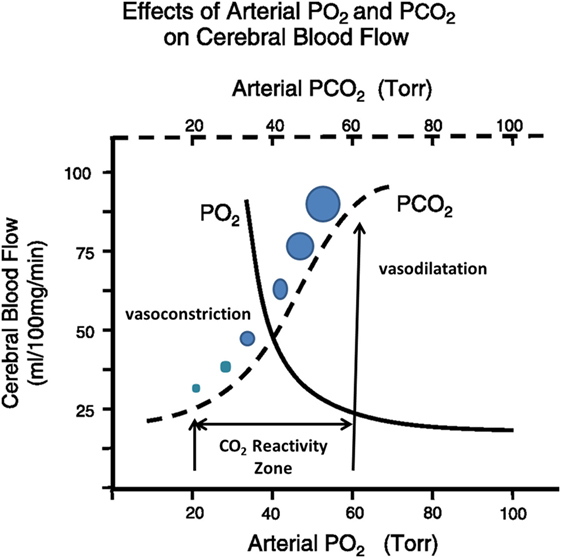
Figure 2. The effects of arterial CO2 on cerebral blood flow.
Changes in vessel diameter elicited by hypocapnia compared to those elicited by hypercapnia are not proportional (5). If the PaCO2 increases to 80 mmHg, vasodilation will increase CBF by 100–200%, causing a release of catecholamines and an increase of metabolic activity. On the other hand, for every millimeter of mercury that the PaCO2 decreases, the CBF will decrease by 3%; thus, PaCO2 levels between 20 and 25 mmHg are associated with a CBF reduction of 40–50% (5, 24).
The pial arterioles responsible for these instances of dilation and contraction are less than 50 µm in diameter. The vascular endothelium reacts to changes in pH that occur within the perivascular space by releasing mediators that regulate the endothelium and vascular smooth muscle (5, 26). These vasoactive factors are thought to include nitric oxide, prostaglandins, cyclic nucleotides, potassium, and calcium (5).
Cerebral blood flow is not normally homogeneous; it varies according to the metabolic rate and activity of each region (27). Cerebrovascular reactivity to CO2 can also vary, depending on location or circumstance (28). During sTBI, especially during the first few hours, CO2 reactivity is exacerbated, especially in areas that are adjacent to contusions or subdural hematomas (29–32). For these reasons, changes in normal levels of CO2 are potentially dangerous secondary insults that can drastically impact brain physiology (11).
Cerebral Blood Volume
In adult humans, normal CBV is 3–4 ml per 100 g of parenchymal tissue (5). Although changes in the diameter of the cerebral blood vessels might alter the total CBV, 70% of the total blood volume contained in the brain corresponds to the venous system (33, 34). Because veins and capillaries do not react to fluctuations in PaCO2, any changes in the CBV following incidents of hypercapnia or hypocapnia can be attributed only to changes in the arterial blood volume (33, 34). It has been estimated that HV reduces CBV by approximately 0.049 ml/100 g per millimeter of mercury CO2 reduction (5). If only 30% of the total CBV is located in the arteries and only pial vessels respond to changes in PaCO2, a hypocapnia-induced CBF decrease of 30% will only result in a CBV decrease of 7% (35). In this manner, a pronounced decrease in CO2 can create a substantial decrease in CBF, but has little effect on the corresponding CBV and ICP (35). It has been suggested that the CBV response to hypocapnia is further diminished during arterial hypotension, specifically when the mean arterial blood pressure (MABP) range is reduced from 154 to 114 Hg (5).
Intracranial Pressure
In accordance with the Monro–Kellie hypothesis, alterations in the CBV will create ICP changes only after spatial compensatory mechanisms are exhausted (10, 11, 14). These compensatory mechanisms include changes in cerebrospinal fluid (CSF) and blood volume principally through increased venous return to the heart and deviation of CSF to the spinal channel. Hypercapnia triggers vasodilation, which leads to an increase in CBV and a subsequent increase in ICP; hypocapnia triggers vasoconstriction, which leads to a decrease in CBV and a subsequent decrease in ICP (10, 11, 14). HV is a therapy that uses the conditions of hypocapnia to trigger vasoconstriction within the resistance arterioles in the cerebral parenchyma in order to reduce ICP. Doing this modifies the absolute value and morphology of the ICP pulse wave by decreasing the P2 (tidal wave) component (36).
Brain Metabolism
Hypocapnia increases cerebral metabolic activity through various mechanisms. It induces the release of excitatory amino acids (N-Methyl-d-aspartate and glutamate) and increases neuronal excitability, glucose consumption, and the metabolic rate of O2 (CMRO2) (11, 14). It also potentiates and prolongs convulsive activity (11, 14).
Cerebral Oxygenation
Hypoxia occurs when the body or a specific region of the body does not receive or is unable to process an adequate amount of oxygen to meet its metabolic demands (37–39). Tissular hypoxia can be local or global, but both variants can be detected using bedside cerebral monitoring that measures either the tissular pressure of O2 (ptiO2) or the venous saturation of O2 in the bulb of the jugular vein (SvjO2) (40). There are four different pathways through which hypocapnia can cause or contribute to tissular hypoxia (37–39):
• Vasoconstriction brought upon by hypocapnia can cause a reduction in CBF, resulting in “ischemic hypoxia” (3, 5, 10, 11, 14).
• A reduction in carbon dioxide levels can impair gas exchange in the lungs, triggering “hypoxemic hypoxia” (10, 11).
• The oxygen–hemoglobin (Hgb) dissociation curve can shift to the left as a result of hypocapnia, which increases the Hgb’s affinity for O2 and hinders the release of O2 into the cells which is also known as “high affinity hypoxia” (10, 11).
• Heightened neuronal excitability and cerebral metabolism brought upon by hypocapnia increases metabolic needs, resulting in hypoxia (11, 14).
The various changes that hypocapnia induces with regard to cerebral physiology are depicted in Figure 3.
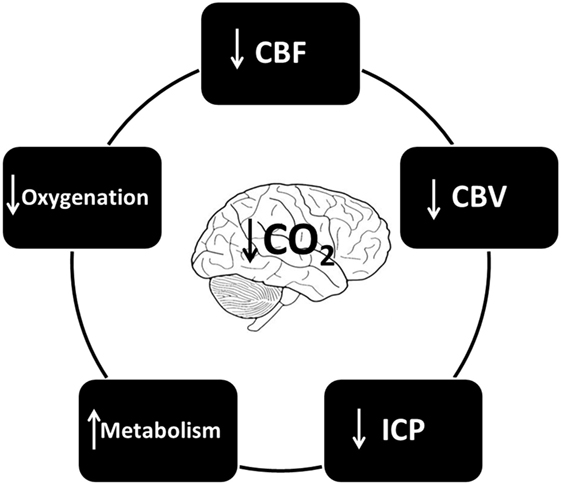
Figure 3. The cerebral effects of hypocapnia.
The Systemic Effects of Hypocapnia
Induced hypocapnia affects all organ systems (10, 11, 14). When evaluating hypocapnia for the purposes of potentially administering therapeutic HV, one should also take into account the effects of mechanical ventilation (10, 11, 41, 42). Because patients suffering from sTBI might concomitantly present contused lesions in the lung parenchyma, microaspirations of gastric content, or acute respiratory distress syndrome (ARDS), they may require protective ventilation with low VT and high levels of positive end-expiratory pressure (PEEP) (10, 11, 41, 42). Therapeutic HV is achieved by increasing the RR or VT. Increasing the VT can be detrimental because of the stress it places on the body; it induces alveolar stretching, which causes cytokine release and inflammation both locally and systemically (10, 11, 41–43).
Hypocapnia decreases blood perfusion to renal tissue, gastrointestinal tissue, and skin and skeletal muscle tissue; it also provokes an increase in adhesion and platelet aggregation (10, 11, 14). Low PaCO2 levels incite bronchoconstriction, attenuate hypoxic pulmonary vasoconstriction, inhibit the production of surfactant, and increase the permeability of the alveolo-capillary membrane and upper airways (11, 14). Several variables can compromise gas exchange and increase a patient’s susceptibility to infections. These include: atelectasis, edema, reduced compliance, pulmonary defense mechanism alterations, ventilation–perfusion ratio alterations, and changes to the shunt fraction (11, 14). Respiratory alkalosis (a disturbance in the acid/base balance associated with decreased levels of potassium, calcium, and phosphate in plasma) complicates tissue oxygenation by shifting the O2/Hgb dissociation curve to the left (10, 11).
Hypocapnia-induced vasoconstriction compromises coronary blood flow and increases the risk of coronary spasm. It also increases myocardial metabolic demands, which may increase a patient’s risk for myocardial ischemia. This is especially true if the patient has predisposing factors or a history of heart disease (10, 11, 14). Hypocapnia also promotes reperfusion injury and encourages the development of arrhythmias, specifically, atrial fibrillation (11).
Hypocapnia and Neurotoxicity
Low levels of PaCO2 produce neurotoxic effects by inducing the release of cytotoxic excitatory amino acids, increasing dopamine levels in the basal ganglia, and promoting the incorporation of choline into the phospholipids of cell membranes (44–46).
Hyperventilation and Timing Restrictions
Cerebral arteriolar reactivity to CO2 is dependent on perivascular pH changes (26). HV induces hypocapnic alkalosis, which rapidly triggers buffer mechanisms that attempt to normalize changes made to extracellular space and (CSF) levels (10, 11). During this time, there is a rapid cellular efflux of hydrogen ions (H+) that bind to bicarbonate and generate carbonic acid (H2CO3), which dissociates in water (H2O) and CO2 (10, 11). At the same time, extracellular bicarbonate is exchanged with chloride from the intracellular space (10, 11). These buffer mechanisms are inefficient because they rapidly deplete; if hypocapnia persists, alkalosis will perpetuate.
A slower but more efficient buffer occurs at the proximal renal tubular level, where reabsorption is inhibited at the same time H+ secretion is stimulated (10, 11). These reactions begin minutes after hypocapnic alkalosis originates and are maintained for hours or even days, allowing the CSF and perivascular pH to normalize 6 h after hypocapnia begins; HV naturally becomes less effective after this buffer pathway becomes activated (10, 11). Clinical studies have demonstrated a 40% decrease in CBF when PaCO2 levels are 20 mmHg; but, after 4 h of HV, CBF begins to normalize (47). CO2 levels after HV therapy also requires time to normalize; if PaCO2 rapidly increases, the perivascular pH (normalized by buffer systems) will decrease, causing local acidosis, vasodilatation, and an increase in CBV and ICP (also known as “rebound hyperemia”) (11, 48).
Clinical Evidence of Hyperventilation Therapy in the Management of sTBI
For many years, HV was a key therapy in the control and prevention of posttraumatic ICH (6–8, 49–51). Oertel et al. reported that HV is a potent and more effective tool to lower elevated ICP levels when compared to increase mean arterial pressure (MABP) or decreased brain metabolism with propofol (52). Multiple studies indicate that the mechanism by which HV decreases ICP is vasoconstriction and CBF reduction (3–5, 10, 11). As CBF decreases, the risk of ischemia is a latent danger (8, 10–12). This is a controversial matter that is subject to much debate.
Immediately following trauma, CBF decreases to about 40% and the CMRO2 possibly decreases as well (53–56). After at least 48 h, this period is followed by two consecutive phases of “relative hyperemia” (in which CBF increases above metabolic demands) and vasospasm (57). The posttraumatic brain is extremely susceptible to ischemic damage (58–61). In almost half of all reported sTBI cases, the autoregulatory pressure mechanism is compromised; therefore, CBF becomes “pressure dependent” (58–61). Under different circumstances, the autoregulation pressure curve might shift to the right, which will increase the cerebral perfusion pressure (CPP) limit to help prevent ischemia (58–61). Because the brain needs to achieve a higher CPP during the acute phase of sTBI, it is highly recommended that hypotension should be avoided during this period.
Much like CBF, arteriolar vasoreactivity to CO2 can vary according to region. CO2 reactivity is habitually maintained and exacerbated during the initial phase of trauma, especially in areas adjacent to contusions or subdural hematomas; a close proximity to these areas increases the likelihood of ischemia occurring in those regions (Figure 4) (31, 33, 62). If CO2 reactivity becomes compromised, it is generally by a terminal event associated with poor results (53, 62–64). For these reasons, it is a key point at this stage in treatment to maintain CBF within normal limits to provide adequate CPP (CPP = MABP − ICP) and blood viscosity while avoiding resistance vessel constriction (12, 58–61).
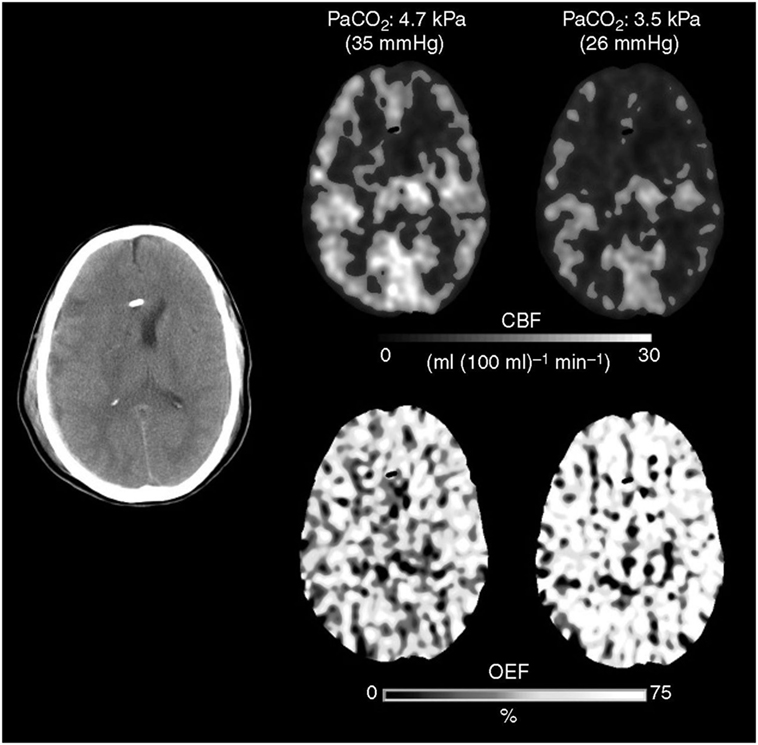
Figure 4. The Xenon CT imaging that showed cerebral blood flow (CBF) decrease and O2 extraction fraction (OEF) increase after hyperventilation.
Hyperventilation and Cerebral Ischemia
Davis was the first to observe the slowing of electroencephalogram waves after HV and attributed this behavior to ischemia (65). Cold evaluated regional CBF (rCBF) using a Xenon (Xe-CT) technique before and after HV between the first day of admittance and 3 weeks post-sTBI (66). Under these conditions, mean ICP was reported to be 19 mmHg and PaCO2 levels changed from 36 to 26 mmHg. The hyperventilated group demonstrated three times as many oligemic regions (defined by a CBF < 20 ml/100 g/min), and their areas of severe CBF reduction (<15 ml/100 g/m) increased from 0.1 to 3%. These observations were more evident in cerebral hemispheres with lower basal CBF. There was also a strong correlation between HV, reductions in rCBF, and poor patient outcome (66).
Multiple clinical studies examining sTBI have confirmed that HV causes significant reductions in CBF (67–69). But observing a reduction in CBF is not enough to accurately diagnose ischemia. To ascertain a valid diagnosis, tissue hypoxia must be associated with any observed decrease in CBF (37–40). Although they are not available in real time at the bedside, it is still important to perform metabolic studies, such as positron emission tomography (PET) (70).
A group from Cambridge conducted a series of studies in which they analyzed the effects of HV in patients with sTBI using PET (71–73). A common denominator among the patients was the absence of ICH (71–73). In the group’s first study, 33 patients were tested within the first week of the trauma, and four were evaluated within the first 24 h of admission (71). The researchers decreased PaCO2 levels from a baseline of 36–29 mmHg. HV elevated CPP and decreased ICP and CBF, which increased the number and volume of areas that reached the hypoperfusion range. However, these changes were not associated with global ischemia since SvjO2 and the arterial–venous difference of O2 (AVDO2) remained within normal limits (71).
In a subsequent study, 13 patients were analyzed during the same posttrauma period to test the hypothesis that diffusion alterations in microcirculation contribute to tissue hypoxia (72). The patients were monitored with PET, O2 extraction fraction (OEF), ptiO2, and pvO2 (72). Again, after inducing HV at a 29 mmHg of PaCO2, CBF decreased. Areas in the range of hypoperfusion and hypoxia (ptiO2 < 10 mmHg) showed less reserve capacity to extract O2, which increased the risk of ischemic damage in these regions (72).
Using a modified PET technique [O2 (15)], Coles et al. evaluated CBF, CMRO2, CBV, and OEF in 30 sTBI patients within 10 days of trauma (73). Hypocapnia (PaCO2 = 29 mmHg) caused a decrease in CBF, an increase in the volume of ischemic areas, and an increase in OEF. CMRO2 increased, but the response was heterogeneous. Twenty-eight percent of hyperventilated individuals showed a marked decrease in CMRO2. CMRO2 correlated with neurophysiological monitoring findings (73).
In another series of sTBI patients with normal ICP values, the effects of HV and mannitol on CBF and metabolic variables were measured using a Doppler ultrasound device and blood samples were taken from a jugular catheter (74). The timing of the study was not specified. The authors reported that, when compared to the 20% (0.5 g/kg) mannitol group, the group hyperventilated at a target PaCO2 of 32 mmHg had a lower CBF and CMRO2; glucose utilization (CMRGlu) and lactate production (CMRL) also increased in this group, which was interpreted by the authors as indicative of anaerobic hyperglycolysis (74).
Using Xe-CT, Stringer analyzed rCBF in 12 individuals with various etiologies, four of which were TBI patients (75). HV was induced heterogeneously with varying expired CO2 values (ETCO2), three of them being lower than 20 mmHg. The study showed a decrease in rCBF. No metabolic parameters were measured (75).
Using thermodiffusion and microdialysis, Marion analyzed rCBF and tissue hypoxia markers in the extracellular fluid of 20 individuals with sTBI before and after HV at a target of 24.6 mmHg (76). Patients maintained normal ICP values. The authors analyzed “apparently healthy areas” adjacent to contusions or subdural hematomas during two-time intervals: 24–36 h and 3–4 days posttrauma (76). After HV, an increase in glutamate, lactate, and the lactate/pyruvate relationship (L/P) was observed to be 10% over basal values. CBF decreased by the same proportion. These changes were seen in both time intervals, but they were more frequently observed during the early stage post-TBI (76).
Using PET, Diringer tested the behavior of CBF, CBV, CMRO2, OEF, and CvO2 after HV under a pre-specified target PaCO2 of 30 mmHg. Patients were analyzed an average of 11 h after sTBI. Of the nine individuals studied, only five had an ICP greater than 20 mmHg (77). Reductions in CBV, CBF, and CvO2 were observed. There was no apparent ischemia or energy dysfunction since CMRO2 remained unchanged at the expense of an increase in OEF (77). Two years later, the same group used the same methodology to compare the effects of HV on patients with, and without ICH on posttrauma days 1 and 5 (78). The results they obtained and the conclusions they reached were no different from those reported in the previous study (78).
Hyperventilation and Cerebral Oxygenation
PaCO2 affects the measurements taken by both global (SvjO2) and regional (ptiO2) oxygenation monitoring methods. HV reduces ICP levels, and clinical studies have demonstrated a simultaneous decrease in SvjO2 values (79–81). When analyzing the impact that ETCO2 levels have on CBF and ptiO2, a direct relationship has been observed between these variables, especially in the range of ETCO2 from 20 to 60 mmHg (82). When HV is more intense and ETCO2 levels are lower, the likelihood of detecting tissue hypoxia using ptiO2 monitoring increases (83). Hypocapnia is one of the secondary insults that is likely to trigger tissue hypoxia (84). The effect HV has on tissue oxygenation becomes more dramatic as time goes on; it has the most impact around 5 days posttrauma (85, 86). This phenomenon is associated with poor results (86–88). Multiple clinical studies have established the deleterious effect HV has on ptiO2 levels. In sTBI, a lower ptiO2 has clearly become an independent predictor of mortality and poor patient outcome (86–89).
Hyperventilation and sTBI Outcome
Only a few studies have established a correlation between patient outcome and HV. Two small studies concluded that mortality and poor functional outcomes were associated with HV when increasing the volume of areas with low CBF into the ischemic range (66) or when ptiO2 levels decreasing along with increasing CO2 reactivity 5 days after trauma (86).
Gordon reported a large retrospective series of patients treated with prolonged hyperventilation (90). 251 patients with sTBI were studied, 51 of whom were hyperventilated (PaCO2 between 25 and 30 mmHg). The time period of HV varied between 6 h and 41 days (mean 10 days). The HV group had a lower mortality (9.8 vs. 32.8%); however, the number of survivors with severe neurological sequela notably increased. Patients who experienced a complete recovery did not differ between groups (90). The authors of the paper gave few details about their methodology; their reported clinical data was also incomplete.
There is only one prospective, controlled, and randomized study that evaluated the final outcome of sTBI patients who were treated with deep and prolonged (5 days) HV (91). Three groups were evaluated in this study: patients who received normoventilation (PaCO2 35 mmHg), patients who received HV at a PaCO2 of 25 mmHg, and patients who received HV and THAM (tromethamine). The THAM acted like a buffer, preventing pH changes within the extracellular cerebral fluid and CSF in order to extend the period during which HV was effective. Prior to randomization, patients were stratified into two groups according to the motor component of the Glasgow Coma Scale (GCS): ≤3 or >3 points. Favorable results at 3 and 6 months of the event were significantly lower in the HV group, especially for patients who had a better clinical status at the time of admission (motor GCS 4–6). After a year had passed from the date of trauma, the differences between the groups were no longer significant. When evaluating CBF (Xe133) and AVDO2, there was no evidence of ischemia in any of the three groups (91).
The study’s conclusions should be interpreted with caution. In the first place, the clinical and tomographic characteristics of the patients were not well balanced between the groups. The number of patients per group was also small, so there could have been statistical errors of type α (false positives). The control group was also hyperventilated with a mean PaCO2 at 31 mmHg. Third, it is apparent that HV was used prophylactically because only 14% of the individuals in the control and HV groups and 5% of the HV and THAM group had high ICP values. Finally, when analyzing the outcome at 12 months posttrauma, the best results correspond to HV + THAM group.
Target PaCO2 in the Management of sTBI
The Cochrane collaboration concludes that there is an insufficient amount of evidence to clearly establish whether hyperventilation therapy in the management of sTBI is beneficial or detrimental (92). In emergent medical situations, Brain Trauma Foundation (BTF) guidelines recommend a brief period of hyperventilation (HV) (15–30 min to target PaCO2 30–35 mmHg) to treat acute neurological deterioration reflecting increased ICP (12). However, in patients with TBI, the targeted PaCO2 of normoventilation is 35–40 mmHg with a pulse oximetry of 95% or greater and/or PaO2 of 80 mmHg or greater (12). If the patient is refractory to all other treatments, including hypertonic saline, sedation, and paralytics, a prolonged period of HV with brain oxygenation monitoring may be required to relieve ICH (12).
Rules to Take into Account before Hyperventilating a Patient with sTBI
Hyperventilation has a place in the management of ICP. For physicians to determine if there is a correct and sufficient indication for treatment with minimal possible risk for the patient, a systematic approach based on current scientific evidence must be undertaken. The authors recommend the following guidelines Figure 5:
• DO NOT hyperventilate prophylactically. HV will not prevent ICP increase, nor will it improve the final outcome.
• DO NOT hyperventilate in the absence of ICH.
• DO NOT hyperventilate for prolonged periods of time. At 4–6 h, buffer systems normalize the pH of the perivascular space, thereby negating the effects of hypocapnia on the cerebral vasculature.
• DO NOT hyperventilate within the first 24 h of sTBI, when the risk of ischemia is greatest.
• DO NOT hyperventilate without oxygenation monitoring. Consider using transcranial Doppler and measuring CO2 levels through the determination of expired ETCO2 levels or arterial gases. Because ischemic hypoxia is a latent and dangerous risk, monitor cerebral oxygenation globally (SvjO2), locally (ptiO2), or both despite the low level of evidence of this recommendation.
• DO NOT suddenly stop HV. Abrupt cessation will increase the risk of ICP elevation rebound.
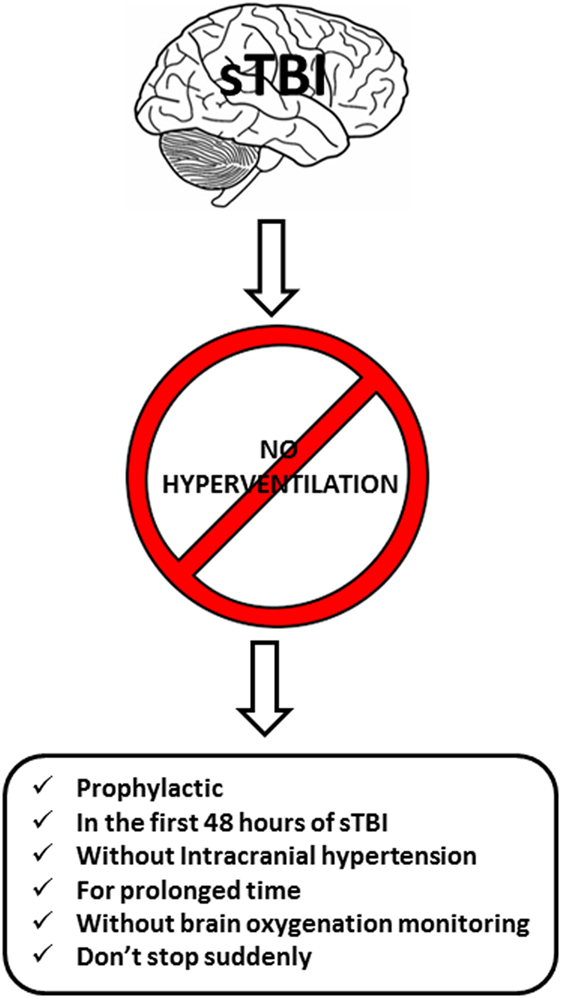
Figure 5. Six DO NOT rules.
Ideal Conditions for Hyperventilation
The cerebral autoregulatory mechanism is compromised in more than half of reported sTBI cases, but HV can help ameliorate those circumstances (93, 94). However, the benefits derived from HV are transient and achievable only through moderate HV. A Doppler study examining 10 patients with sTBI demonstrated that HV at a PaCO2 of 28 mmHg significantly improved cerebral autoregulation, but the benefit was lost when HV was reduced to a PaCO2 of 23 mmHg (95). Another group composed of 30 sTBI individuals with normal ICP values evidenced improvement in autoregulation when PaCO2 levels were temporarily decreased from 38 to 33 mmHg (96).
Hyperventilation is recommended as a temporary measure to reduce high levels of ICP in the following situations:
➢ Herniation syndromes. These are syndromes involving the deterioration of neurological status (mydriasis, abnormal motor postures) secondary to expansive lesions (epidural, subdural hematomas, etc.) as a bridge to surgical resolution (12).
➢ Life-threatening elevations of ICP. For example, type A plateau waves, while investigating triggers and expecting the effect of osmotherapy (12).
➢ Refractory ICH. HV is used in conjunction with second level measures, such as decompressive craniectomy, hypothermia, or high doses of barbiturates (12).
➢ ICH secondary to “hyperemia.”
In approximately 20% of sTBI cases, ICP elevation correlates with high CBF in excess to metabolic demands, mainly in young individuals (53, 97, 98). In these situations, based on the principle of coupling between CBF and CMRO2, the concept of “optimizing HV” emerges in order to lower CBF and, consequently, ICP without modifying CMRO2 (99–102). By implementing this approach alongside cerebral oxygenation monitoring through SvjO2 and associated variables, Cruz et al. calibrate HV to a target PaCO2 that varied from 18 to 30 mmHg. As a result, ICP decreased without modification in CMRO2 while OEF increased (98–102). In another series of sTBI patients, it was reported that HV contributed to the stabilization and improvement of brain glucose uptake (101).
Hyperventilation optimization has its limitations. It is based on global monitoring that does not take into account the regional differences in CBF, metabolism, or CO2 reactivity. ICP compartmentalization and lesion type (focal or diffuse) are similarly disregarded in this manner. There does not exist a consistent definition of “hyperemia,” especially about the monitoring methods available at the bedside. There are also multiple limitations to the method of oxygenation monitoring itself, which can make it difficult to interpret the data obtained from said monitoring.
Hyperventilation Techniques
In Figure 6, we outline a practical algorithm summarizing the concepts one should take into consideration when hyperventilating a patient.
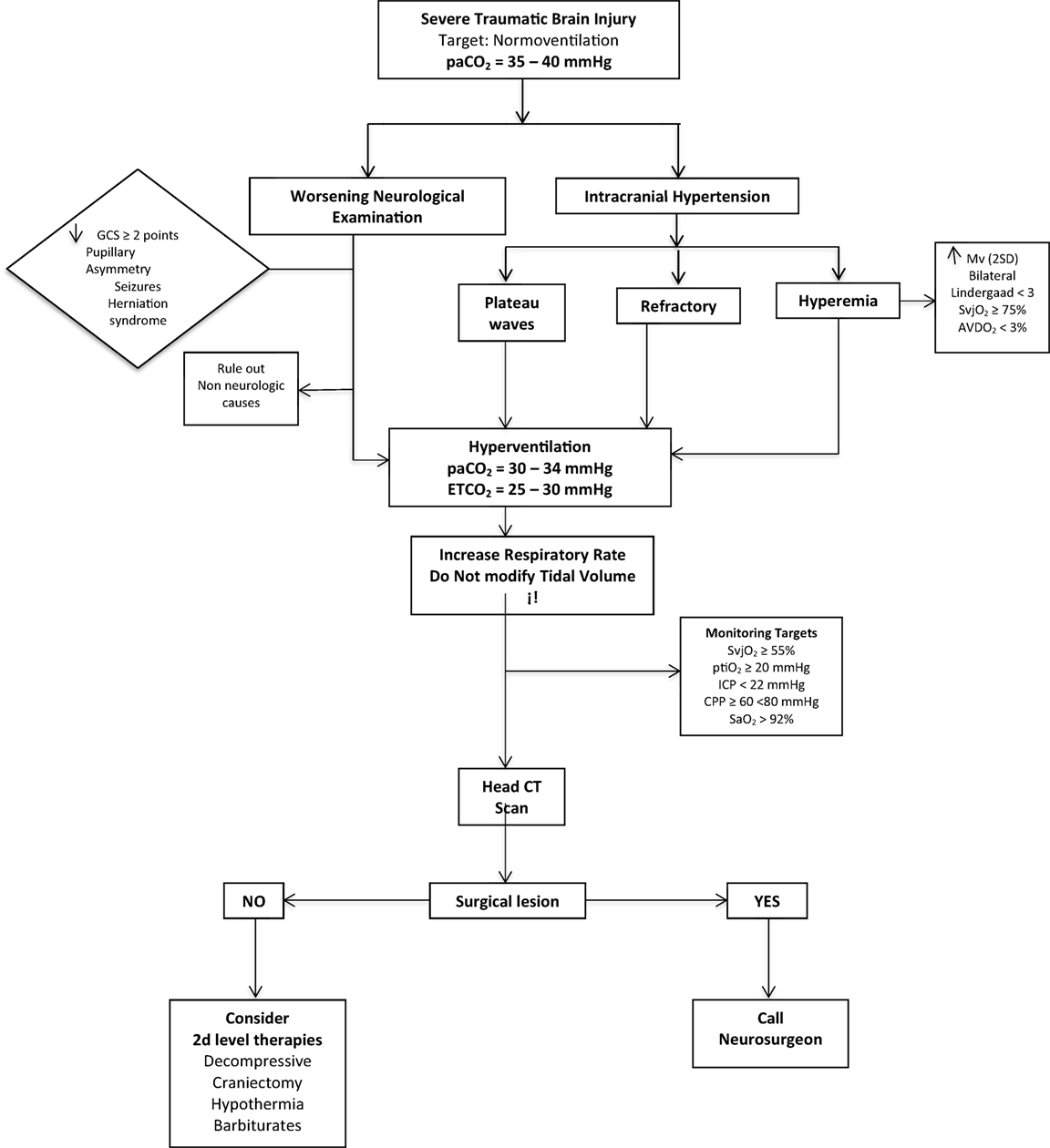
Figure 6. The practical algorithm to perform HV safety. Hyperemia definition: increase in mean velocity (mv) of mean cerebral artery in TCD (TRANSCRANIAL DOPPLER) of more than 2 standard deviations.
Conclusion
Patients with an elevated ICP require emergent intervention to prevent deleterious consequences. Under certain conditions, when cerebrovascular CO2 reactivity is intact, HV can be used temporarily to induce hypocapnia to elicit arteriolar vasoconstriction with the aim of decreasing CBF and, ultimately, ICP. The cerebral effects of hypocapnia are transient. Because profound and prolonged HV carries the risk of ischemia, it is important that the therapy be closely monitored to prevent any adverse cerebral effects.
Hyperventilation has serious systemic consequences. It should not be implemented during the first 24 h of trauma when CBF is markedly reduced. Prophylactic HV or HV without an indication of elevated ICP will not yield any benefits. Current recommendations suggest that a brief period of HV (15–30 min) with a PaCO2 target of 30–35 mmHg and CPP target of 60–70 mmHg coupled with close oxygenation neuromonitoring is an effective method of controlling ICH during the acute-phase of sTBI.
Author Contributions
All authors contribute to the preparation of the manuscript in the same manner.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, BA, and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
References
1. Diringer M. Hyperventilation in head injury: what have we learned in 43 years? Crit Care Med (2002) 30:2142–3. doi:10.1097/00003246-200209000-00032
2. Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl (1960) 36:1–193.
3. Raichle ME, Plum F. Hyperventilation and cerebral blood flow. Stroke (1972) 3:566–75. doi:10.1161/01.STR.3.5.566
4. Heffner JE, Sahn SA. Controlled hyperventilation in patients with intracranial hypertension. Application and management. Arch Intern Med (1983) 143:765–9. doi:10.1001/archinte.1983.00350040155022
5. Brian JE. Carbon dioxide and the cerebral circulation. Anesthesiology (1998) 88:365–86. doi:10.1097/00000542-199805000-00029
6. Ghajar J, Hariri R, Narayan R. Survey of critical care management of comatose, head injured patients in the United States. Crit Care Med (1995) 23:560–7. doi:10.1097/00003246-199503000-00023
7. Matta B, Menon D. Severe head injury in the United Kingdom and Ireland: a survey of practice and implications for management. Crit Care Med (1996) 24:1743–8. doi:10.1097/00003246-199610000-00023
8. Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury. J Neurotrauma (2007) 24(Suppl 1):S1–106. doi:10.1089/neu.2007.9999
9. Neumann JO, Chambers IR, Citerio G, Enblad P, Gregson BA, Howells T, et al. The use of hyperventilation therapy after traumatic brain injury in Europe: an analysis of the brain IT database. Intensive Care Med (2008) 34:1676–82. doi:10.1007/s00134-008-1123-7
10. Stochetti N, Maas AI, Chieragato A, van der Plas AA. Hyperventilation in head injury. Chest (2005) 127:1812–27. doi:10.1378/chest.127.5.1812
11. Curley G, Kavanagh BP, Laffey JG. Hypocapnia and the injured brain: more harm than benefit. Crit Care Med (2010) 38:1348–59. doi:10.1097/CCM.0b013e3181d8cf2b
12. Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery (2017) 80:6–15. doi:10.1227/NEU.0000000000001432
13. West JB, Luks AM. Respiratory Physiology: The Essentials. 10th ed. Philadelphia, USA: Wolters Kluwer (2016).
15. Stoelting RK, Hillier SC. Acid-base balance. In: Stoelting RK, Hillier SC, editors. Pharmacology and Physiology in Anesthetic Practice. Philadelphia: Lippincott Williams and Wilkins (2006). p. 794–802.
16. Vazquez Garcia JC, Perez Padilla R. Valores gasométricos estimados para las principales poblaciones y sitios a mayor altitud. Rev Inst Nal Enf Resp Mex (2000) 13:6–13.
17. Restrepo J, Reyes P, Vasquez P, Ardila M, Diaz Granados B. Gasometria arterial y alveolar en adultos sanos a nivel de Bogota. Acta Med Colomb (1982) 7:461–6.
18. Llanos M, Villanagua B, Garelli Z, Freund P, Finol D, Gonzalez A, et al. Investigacion en Medicina Critica. Interpretacion de los gases sanguíneos arteriales a nivel de Quito, Ecuador. Biomed J (2016) 1:1–9.
19. Pereira Victorio CJ, Huamanquispe Quintana J, Castelo Tamayo LE. Gasometria arterial en adultos clínicamente sanos a 3350 metros de altutud. Rev Peru Med Exp Salud Publica (2014) 31:473–9.
20. Hinojosa Campero WE. Gasometria arterial y adaptación a la altura. Rev Med Cient (2011) 2:39–45.
21. Godoy DA, Canitrot Ugarte M. Physiological basis for the correct interpretation of different situations in acute cerebral injury. In: Godoy DA, editor. Intensive Care in Neurology and Neurosurgery (Vol. 1, Chap. 3), Torino, Italy: Seed (2013). p. 53–68.
22. Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke (1989) 20:45–52. doi:10.1161/01.STR.20.1.45
23. Czosnyka M, Brady K, Reinhard M, Smielewski P, Steiner LA. Monitoring of cerebrovascular autoregulation: facts, myths, and missing links. Neurocrit Care (2009) 10:373–86. doi:10.1007/s12028-008-9175-7
24. Cold GE. Cerebral blood flow in acute head injury. The regulation of cerebral blood flow and metabolism during the acute phase of head injury, and its significance for therapy. Acta Neurochir Suppl (Wien) (1990) 49:1–64.
25. Harper AM, Glass HI. Effect of alterations in the arterial carbondioxide tension on the blood flow through the cerebral cortex at normal and low arterial blood pressures. J Neurol Neurosurg Psychiatry (1965) 28:449–52. doi:10.1136/jnnp.28.5.449
26. Kontos HA, Raper AJ, Patterson JL. Analysis of vasoactivity of local pH, PCO2 and bicarbonate on pial vessels. Stroke (1977) 8:358–60. doi:10.1161/01.STR.8.3.358
27. Ingvar DH, Cronqvist S, Ekberg R, Risberg J, Hoedt-Rasmussen K. Normal values of regional cerebral blood flow in man, including flow and weight estimated of gray and white matter. Acta Neurol Scand Suppl (1965) 14:72.
28. Ito H, Yokoyama I, Lida H, Kinoshita T, Hatazawa J, Shimosegawa E, et al. Regional differences in cerebral vascular response to PaCO2 changes in humans measured by positron emission tomography. J Cereb Blood Flow Metab (2000) 20:1264–70. doi:10.1097/00004647-200008000-00011
29. Bouma GJ, Muizelaar JP. Cerebral blood flow, cerebral blood volume, and cerebrovascular reactivity after severe head injury. J Neurotrauma (1992) 9(Suppl 1):S333–48.
30. Bouma GJ, Muizelaar JP, Stringer WA, Choi SC, Fatouros P, Young HF. Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J Neurosurg (1992) 77:360–8. doi:10.3171/jns.1992.77.3.0360
31. McLaughlin MR, Marion DW. Cerebral blood flow and vasoresponsivity within and around cerebral contusions. J Neurosurg (1996) 85:871–6. doi:10.3171/jns.1996.85.5.0871
32. Marion DW, Firlik A, McLaughlin MR. Hyperventilation therapy for severe traumatic brain injury. New Horiz (1995) 3:439–47.
33. Ito H, Ibaraki M, Kanno I, Fukuda H, Miura S. Changes in the arterial fraction of human cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab (2005) 25:852–7. doi:10.1038/sj.jcbfm.9600076
34. Ito H, Kanno I, Iida H, Hatazawa J, Shimosegawa E, Tamura H, et al. Arterial fraction of cerebral blood volume in humans measured by positron emission tomography. Ann Nucl Med (2001) 15:111–6. doi:10.1007/BF02988600
35. Fortune JB, Feustel PJ, deLuna C, Graca L, Hasselbarth J, Kupinski AM. Cerebral blood flow and blood volume in response to O2 and CO2 changes in normal humans. J Trauma (1995) 39:463–71. doi:10.1097/00005373-199509000-00012
36. Cardoso ER, Rowan JO, Galbraith S. Analysis of the cerebrospinal fluid pulse wave in intracranial pressure. J Neurosurg (1983) 59:817–21. doi:10.3171/jns.1983.59.5.0817
37. Siggaard-Andersen O, Ulrich A, Gothgen IH. Classes of tissue hypoxia. Acta Anaesthesiol Scand Suppl (1995) 107:137–42. doi:10.1111/j.1399-6576.1995.tb04348.x
38. Zauner A, Daugherty WP, Bullock MR, Warner DS. Brain oxygenation and energy metabolism: part I-biological function and pathophysiology. Neurosurgery (2002) 51:289–301. doi:10.1097/00006123-200208000-00003
39. Marín-Caballos AJ, Murillo-Cabezas F, Domínguez-Roldan JM, Leal-Noval SR, Rincón-Ferrari MD, Muñoz-Sánchez MÁ. Monitorización de la presión tisular de oxígeno (PtiO2) en la hipoxia cerebral: aproximación diagnóstica y terapéutica. Med Intensiva (2008) 32:81–90. doi:10.1016/S0210-5691(08)70912-4
40. Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on Multimodality Monitoring in Neurocritical Care: a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Neurocrit Care (2014) 21(Suppl 2):S1–26. doi:10.1007/s12028-014-0041-5
41. Marhong J, Fan E. Carbon dioxide in the critically ill: too much or too little of a good thing. Respir Care (2014) 59:1597–605. doi:10.4187/respcare.03405
42. Go SL, Singh JM. Pro/con debate: should PaCO2 be tightly controlled in all patients with acute brain injuries? Crit Care (2013) 17:202. doi:10.1186/cc11389
43. Mazzeo AT, Fanelli V, Mascia L. Brain-lung crosstalk in critical care: how protective mechanical ventilation can affect the brain homeostasis. Minerva Anestesiol (2013) 79:299–309.
44. Graham EM, Apostolou M, Mishra OP, Delivoria-Papadopoulos M. Modification of the N-methyl-d-aspartate (NMDA) receptor in the brain of newborn piglets following hyperventilation induced ischemia. Neurosci Lett (1996) 218:29–32. doi:10.1016/0304-3940(96)13114-1
45. Pastuszko P, Wilson DF. Activation of tyrosine hydroxylase in striatum of newborn piglets in response to hypocapnic ischemia and recovery. Adv Exp Med Biol (1997) 411:65–73. doi:10.1007/978-1-4615-5865-1_8
46. Mykita S, Golly F, Dreyfus H, Freysz L, Massarelli R. Effect of CDP-choline on hypocapnic neurons in culture. J Neurochem (1986) 47:223–31. doi:10.1111/j.1471-4159.1986.tb02853.x
47. Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol (1970) 23:394–403. doi:10.1001/archneur.1970.00480290014002
48. Muizelaar JP, van der Poel HG, Li ZC, Kontos HA, Levasseur JE. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg (1988) 69:923–7. doi:10.3171/jns.1988.69.6.0923
49. Bruce DA, Alavi A, Bilaniuk L, Dolinskas C, Obrist W, Uzzell B. Diffuse cerebral swelling following head injuries in children: the síndrome of malignant brain edema. J Neurosurg (1981) 54:170–8. doi:10.3171/jns.1981.54.2.0170
50. Becker DP, Gardner S. Intensive management of head injury. In: Wilkins RH, Rengachary SS, editors. Neurosurgery (Vol. 2), New York: McGraw-Hill (1985). p. 1593–9.
51. Marshall LF, Marshall S. Medical management of intracranial pressure. 2nd ed. In: Cooper PR, editor. Head Injury. Baltimore: Williams & Wilkins (1987). p. 177–96.
52. Oertel M, Kelly DF, Lee JH, Mc Arthur DL, Glenn TC, Vespa P, et al. Efficacy of hyperventilation, blood pressure elevation, and metabolic suppression therapy in controlling intracranial pressure after head injury. J Neurosurg (2002) 97:1045–53. doi:10.3171/jns.2002.97.5.1045
53. Obrist WD, Langfitt TW, Jaggi JL, Cruz J, Gennarelli TA. Cerebral blood flow and metabolism in comatose patients with acute head injury. J Neurosurg (1984) 61:241–53. doi:10.3171/jns.1984.61.2.0241
54. Bouma GJ, Muizelaar JP, Choi SC, Newlon PG, Young HF. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg (1991) 75:685–93. doi:10.3171/jns.1991.75.5.0685
55. Jaggi JL, Obrist WD, Gennarelli TA, Langfitt TW. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg (1990) 72:176–82. doi:10.3171/jns.1990.72.2.0176
56. Marion DW, Darby J, Yonas H. Acute regional cerebral blood flow changes caused by severe head injuries. J Neurosurg (1991) 74:407–14. doi:10.3171/jns.1991.74.3.0407
57. Martin NA, Patwardhan RV, Alexander MJ, Africk CZ, Lee JH, Shalmon E, et al. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. J Neurosurg (1997) 87:9–19. doi:10.3171/jns.1997.87.1.0009
58. Chesnut RM. Care of central nervous system injuries. Surg Clin North Am (2007) 87:119–56. doi:10.1016/j.suc.2006.09.018
59. Stochetti N, Maas AI. Traumatic intracranial hypertension. N Engl J Med (2014) 370:2121–30. doi:10.1056/NEJMra1208708
60. Perez-Barcena J, Lompart-Pou JA, O’Phelan KH. Intracranial pressure monitoring and management of intracranial hypertension. Crit Care Clin (2014) 30:735–50. doi:10.1016/j.ccc.2014.06.005
61. Kirkman MA, Smith M. Intracranial pressure monitoring, cerebral perfusion pressure estimation, and ICP/CPP-guided therapy: a standard of care or optional extra after brain injury? Br J Anaesth (2014) 112(1):35–46. doi:10.1093/bja/aet418
62. Paolini A, Rodriguez G, Betetto M, Simini G. Cerebral hemodynamic response to CO2 after severe head injury: clinical and prognostic implications. J Trauma (1998) 44:495–500. doi:10.1097/00005373-199803000-00012
63. Marion DW, Bouma GJ. The use of stable xenon-enhanced computed tomography studies of cerebral blood flow to define changes in cerebral carbon dioxide vasoresponsivity caused by a severe head injury. Neurosurgery (1991) 29:869–73. doi:10.1097/00006123-199112000-00011
64. Tenjin H, Yamaki T, Nakagawa Y, Kuboyama T, Ebisu T, Kobori N, et al. Impairment of CO2 reactivity in severe head injury patients: an investigation using thermal diffusion method. Acta Neurochir (Wien) (1990) 104:121–5. doi:10.1007/BF01842829
65. Davis H, Wallace WM. Factors affecting changes produced in electroencephalogram by standardized hyperventilation. Arch Neurol Psychiat (1942) 47:606–25. doi:10.1001/archneurpsyc.1942.02290040096005
66. Cold GE. Does acute hyperventilation provoke cerebral oligaemia in comatose patients after acute head injury? Acta Neurochir (Wien) (1989) 96:100–6. doi:10.1007/BF01456166
67. Skippen P, Seear M, Poskitt K, Kestle J, Cochrane D, Annich G, et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med (1997) 25:1402–9. doi:10.1097/00003246-199708000-00031
68. Dahl B, Bergholt B, Cold GE, Astrup J, Mosdal B, Jensen K, et al. CO(2) and indomethacin vasoreactivity in patients with head injury. Acta Neurochir (Wien) (1996) 138:265–73. doi:10.1007/BF01411736
69. Sioutos PJ, Orozco JA, Carter LP, Weinand ME, Hamilton AJ, Williams FC. Continuous regional cerebral cortical blood flow monitoring in head-injured patients. Neurosurgery (1995) 36:943–9. doi:10.1227/00006123-199505000-00009
70. Coles JP, Fryer TD, Smielewski P, Rice K, Clark JC, Pickard JD, et al. Defining ischemic burden after traumatic brain injury using 15O PET imaging of cerebral physiology. J Cereb Blood Flow Metab (2004) 24:191–201. doi:10.1097/01.WCB.0000100045.07481.DE
71. Coles JP, Minhas PS, Fryer TD, Smielewski P, Aigbirihio F, Donovan T, et al. Effect of hyperventilation on cerebral blood flow in traumatic head injury: clinical relevance and monitoring correlates. Crit Care Med (2002) 30:1950–9. doi:10.1097/00003246-200209000-00002
72. Menon DK, Coles JP, Gupta AK, Fryer TD, Smielewski P, Chatfield DA, et al. Diffusion limited oxygen delivery following head injury. Crit Care Med (2004) 32:1384–90. doi:10.1097/01.CCM.0000127777.16609.08
73. Coles JP, Fryer TD, Coleman MR, Smielewski P, Gupta AK, Minhas PS, et al. Hyperventilation following head injury: effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med (2007) 35:568–78. doi:10.1097/01.CCM.0000254066.37187.88
74. Soustiel JF, Mahamid E, Chistyakov A, Shik V, Benenson R, Zaaroor M. Comparison of moderate hyperventilation and mannitol for control of intracranial pressure control in patients with severe traumatic brain injury – a study of cerebral blood flow and metabolism. Acta Neurochir (Wien) (2006) 148:845–51. doi:10.1007/s00701-006-0792-7
75. Stringer WA, Hasso AN, Thompson JR, Hinshaw DB, Jordan KG. Hyperventilation induced cerebral ischemia in patients with acute brain lesions: demonstration by xenon CT. AJNR Am J Neuroradiol (1993) 14:475–84.
76. Marion DW, Puccio A, Wisniewski SR, Kochanek P, Dixon CE, Bullian L. Effect of hyperventilation on extracellular concentrations of glutamate, lactate, pyruvate and local cerebral blood flow in patients with severe traumatic brain injury. Crit Care Med (2002) 30:2619–25. doi:10.1097/00003246-200212000-00001
77. Diringer MN, Yundt K, Videen TO, Adams RE, Zazulia AR, Deibert E, et al. No reduction in cerebral metabolism as a result of early moderate hyperventilation following severe traumatic brain injury. J Neurosurg (2000) 92:7–13. doi:10.3171/jns.2000.92.1.0007
78. Diringer MN, Videen TO, Yundt K, Zazulia AR, Aiyagari V, Dacey RG, et al. Regional cerebrovascular and metabolic effects of hyperventilation after severe traumatic brain injury. J Neurosurg (2002) 96:103–8. doi:10.3171/jns.2002.96.1.0103
79. Unterberg AW, Kiening KL, Härtl R, Bardt T, Sarrafzadeh AS, Lanksch WR. Multimodal monitoring in patients with head injury: evaluation of the effects of treatment on cerebral oxygenation. J Trauma (1997) 42:S32–7. doi:10.1097/00005373-199705001-00006
80. Thiagarajan A, Goverdhan PD, Chari P, Somasunderam K. The effect of hyperventilation and hyperoxia on cerebral venous oxygen saturation in patients with traumatic brain injury. Anesth Analg (1998) 87:850–3. doi:10.1213/00000539-199810000-00019
81. Rangel-Castilla L, Lara LR, Gopinath S, Swank PR, Valadka A, Robertson C. Cerebral hemodynamic effects of acute hyperoxia and hyperventilation after severe traumatic brain injury. J Neurotrauma (2010) 27:1853–63. doi:10.1089/neu.2010.1339
82. Hemphill JC III, Knudson MM, Derugin N, Morabito D, Manley GT. Carbon dioxide reactivity and pressure autoregulation of brain tissue oxygen. Neurosurgery (2001) 48:377–83. doi:10.1227/00006123-200102000-00028
83. Carrera E, Schmidt JM, Fernandez L, Kurtz P, Merkow M, Stuart M, et al. Spontaneous hyperventilation and brain tissue hypoxia in patients with severe brain injury. J Neurol Neurosurg Psychiatry (2010) 81:793–7. doi:10.1136/jnnp.2009.174425
84. Sarrafzadeh AS, Kiening KL, Callsen TA, Unterberg AW. Metabolic changes during impending and manifest cerebral hypoxia in traumatic brain injury. Br J Neurosurg (2003) 17:340–6. doi:10.1080/02688690310001601234
85. Gopinath SP, Valadka AB, Uzura M, Robertson CS. Comparison of jugular venous oxygen saturation and brain tissue Po2 as monitors of cerebral ischemia after head injury. Crit Care Med (1999) 27:2337–45. doi:10.1097/00003246-199911000-00003
86. Carmona Suazo JA, Maas AI, van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ. CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med (2000) 28:3268–74. doi:10.1097/00003246-200009000-00024
87. Fandino J, Stocker R, Prokop S, Imhof HG. Correlation between jugular bulb oxygen saturation and partial pressure of brain tissue oxygen during CO2 and O2 reactivity tests in severely head-injured patients. Acta Neurochir (Wien) (1999) 141:825–34. doi:10.1007/s007010050383
88. Dings J, Meixensberger J, Amschler J, Hamelbeck B, Roosen K. Brain tissue Po2 in relation to cerebral perfusion pressure, TCD findings and TCD-CO2-reactivity after severe head injury. Acta Neurochir (Wien) (1996) 138:425–34. doi:10.1007/BF01420305
89. van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ, Suazo JA, Hogesteeger C, et al. Brain oxygen tension in severe head injury. Neurosurgery (2000) 46:868–76. doi:10.1227/00006123-200004000-00018
90. Gordon E. Controlled respiration in the management of patients with traumatic brain injuries. Acta Anaesth Scand (1971) 15:193–208. doi:10.1111/j.1399-6576.1971.tb05461.x
91. Muizelaar JP, Marmarou A, Ward JD, Kontos HA, Choi SC, Becker DP, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg (1991) 75:731–9. doi:10.3171/jns.1991.75.5.0731
92. Roberts I, Schierhout G. Hyperventilation therapy for acute traumatic brain injury. Cochrane Database Syst Rev (1997) 4(4):CD000566. doi:10.1002/14651858.CD000566 Updated 2009
93. Bouma GJ, Muizelaar JP, Bandoh K, Marmarou A. Blood pressure and intracranial pressure volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg (1992) 77:15–9. doi:10.3171/jns.1992.77.1.0015
94. Hlatky R, Valadka AB, Robertson CS. Intracranial pressure response to induced hypertension: role of dynamic pressure autoregulation. Neurosurgery (2005) 57:917–23. doi:10.1227/01.NEU.0000180025.43747.fc
95. Newell DW, Weber JP, Watson R, Aaslid R, Winn HR. Effect of transient moderate hyperventilation on dynamic cerebral autoregulation after severe head injury. Neurosurgery (1996) 39:35–43. doi:10.1227/00006123-199609000-00056
96. Steiner LA, Balestreri M, Johnston AJ, Coles JP, Chatfield DA, Pickard JD, et al. Effects of moderate hyperventilation on cerebrovascular pressure-reactivity after head injury. Acta Neurochir Suppl (2005) 95:17–20. doi:10.1007/3-211-32318-X_4
97. Kelly DF, Kordestani RK, Martin NA, Nguyen T, Hovda DA, Bergsneider M, et al. Hyperemia following traumatic brain injury: relationship to intracranial hypertension and outcome. J Neurosurg (1996) 85:762–71. doi:10.3171/jns.1996.85.5.0762
98. Muizelaar JP, Marmarou A, DeSalles AA, Ward JD, Zimmerman RS, Li Z, et al. Cerebral blood flow and metabolism in severely head-injured children. Part 1: relationship with GCS score, outcome, ICP, and PVI. J Neurosurg (1989) 71:63–71. doi:10.3171/jns.1989.71.1.0063
99. Cruz J. Combined continuous monitoring of systemic and cerebral oxygenation in acute brain injury: preliminary observations. Crit Care Med (1993) 21:1225–32. doi:10.1097/00003246-199308000-00025
100. Cruz J, Jaggi JL, Hoffstad OJ. Cerebral blood flow, vascular resistance, and oxygen metabolism in acute brain trauma: redefining the role of cerebral perfusion pressure? Crit Care Med (1995) 23:1412–7. doi:10.1097/00003246-199508000-00016
101. Cruz J. An additional therapeutic effect of adequate hyperventilation in severe acute brain trauma: normalization of cerebral glucose uptake. J Neurosurg (1995) 82:379–85. doi:10.3171/jns.1995.82.3.0379
Keywords: hyperventilation, intracranial hypertension, intracranial pressure, hypocapnia, cerebral ischemia, cerebral hypoxia, severe traumatic brain injury
Citation: Godoy DA, Seifi A, Garza D, Lubillo-Montenegro S and Murillo-Cabezas F (2017) Hyperventilation Therapy for Control of Posttraumatic Intracranial Hypertension. Front. Neurol. 8:250. doi: 10.3389/fneur.2017.00250
Received: 06 March 2017; Accepted: 19 May 2017;
Published: 17 July 2017
Edited by:
Fernando Testai, University of Illinois at Chicago, United StatesReviewed by:
Benjamin Aaron Emanuel, Keck School of Medicine of USC, United StatesMinjee Kim, Northwestern University Feinberg School of Medicine, United States
Baback Arshi, University of Illinois at Chicago, United States
Copyright: © 2017 Godoy, Seifi, Garza, Lubillo-Montenegro and Murillo-Cabezas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Agustín Godoy, ZGFnb2RveXRvcnJlc0B5YWhvby5jb20uYXI=