Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by the degeneration of motor neurons. Though many molecular and genetic causes are thought to serve as predisposing or disease propagating factors, the underlying pathogenesis of the disease is not known. Recent discoveries have demonstrated the presence of inflammation propagating substrates in the central nervous system of patients afflicted with ALS. Over the past decade, this hypothesis has incited an effort to better understand the role of the immune system in ALS and has led to the trial of several potential immune-modulating therapies. Here, we briefly review advances in the role of such therapies. The clinical trials discussed here are currently ongoing or have been concluded at the time of writing.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by the dysfunction and loss of motor neurons in the brain and/or spinal cord. The striking clinical heterogeneity seen in ALS implies that a single molecular mechanism is unlikely to be responsible for the onset of the disease. The underlying pathophysiology of ALS is not well understood or characterized to date. The presence of pro-inflammatory markers in those demonstrating symptoms has been recognized but the optimal way to modulate this cause of (and response to) neuronal injury remains to be established.
In ALS, cardinal disruptors of cellular homeostasis were originally thought to be oxidative stress and glutamate-mediated excitotoxicity. More recently, additional pathogenic processes have been identified involving protein (metabolism, misfolding, and aggregation), RNA (altered binding), the endoplasmic reticulum and vesicular transport (1, 2). It has also become evident that inflammation plays a crucial role in mediating neuronal injury and disease progression (3, 4). Thus, targeting the immune system would appear to be a more tractable means of slowing the clinical progression of the disease.
Here, we present a brief overview of inflammation in the central nervous system (CNS) as relevant to ALS, particularly microglial homeostasis (5). We then summarize clinical trials to date of immunomodulatory agents based on these recent insights.
The best-studied animal model of ALS replicates a mutation found in familial ALS (fALS), superoxide dismutase 1 (SOD1). However, only 10–15% of ALS is familial and SOD1 mutations account for just 10–12% of fALS, thus just 1–2% of all ALS in humans (6).
The most common genetic cause of fALS has been identified as a six-nucleotide repeat expansion in the C9orf72 gene (chromosome 9, open-reading frame 72). No single abnormality appears to occur with similar frequency in sALS.
Amyotrophic lateral sclerosis and frontotemporal dementia (FTD) are now often said to be the ends of spectrum, pathologically (7). In the case of ALS-FTD (ALS with FTD), the C9orf72 expansion appears to account for approximately 40% of familial and 5–10% of sporadic cases (8). This appears to have important effects on microglia, discussed below.
Immune System in ALS
For much of the twentieth century the CNS was thought to be ‘immune privileged’ i.e. protected from invasion from inflammatory cells. Activity of the immune system within the CNS is now widely accepted.
A lymphatic system lines the dural sinuses and carries immune cells and fluid to cervical lymph nodes (9). Microglia, closely related to the macrophages found in other organs, reside in the CNS. These cells are capable of screening the entire nervous system for foreign material every few hours (10). While circulating lymphocytes do not pass through an intact blood–brain barrier (BBB), extravasation does occur during periods of inflammation (11). In the discussion below, we refer to proteins with their official NCBI names (followed by more common/historical nomenclature) (12).
Pro- and Anti-inflammatory Immune Phenotypes in ALS
Both microglia (M) and T-cells appear to have central roles in the pathogenesis of ALS. As in other organs, once activated in response to injury or antigen, microglia and helper T-cells (Th) differentiate into a pro-inflammatory (classical, M1 and Th1) phenotype. Once the inciting event has been dealt with, these cells transition to an anti-inflammatory phenotype (‘alternative’, M2 and Th2). This process appears best suited to acute injury; when the pathogenic stimulus cannot be adequately cleared, chronic inflammation develops with persistent M1, Th1 activity that can cause unintended injury to local tissues.
A crucial element in ‘tipping the balance’ from one state to the other, particularly evident in microglia, is the relative activity of NOS2 (inducible nitric oxide synthase) vs. ARG1 (arginase 1); this is shown in Figure 1. This balance is particularly important for microglia, but is also used by other elements of the immune system, including T-cells. The expression of NOS2 is increased by transcription factor nuclear factor kappa B (NF-κB), expression of which is increased by binding of tumor necrosis factor alpha (TNFA) to TNF receptor superfamily member 1A (TNFRSF1A). TNFA can also have antiapoptotic effects and lead to an increase in neurotrophic factors via TNFRSF1B (discussed further below).
Figure 1

Simplified schema of inflammation in amyotrophic lateral sclerosis (ALS). Some of the treatments in this review with relatively ‘specific’ modes of action are also shown.
Microglia
Like macrophages, microglia become activated via the receptor complex CD14 + toll-like receptor 4 (TLR4) in response to antigens such as lipopolysaccharide, proteins released from damaged cells (including SOD1protein aggregates, beta-amyloid and alpha-synuclein), to infiltrative lymphocytes and to signals from the humoral (i.e. antibody-mediated) immune system (Figure 2). Relative to the macrophages of other tissues, microglia are less potent activators of the immune system. This is in part due to their lower expression of protein tyrosine phosphatase, receptor type C (aka CD45), leukocyte common antigen and the major histocompatibility complex (MHC) (13, 14).
Figure 2
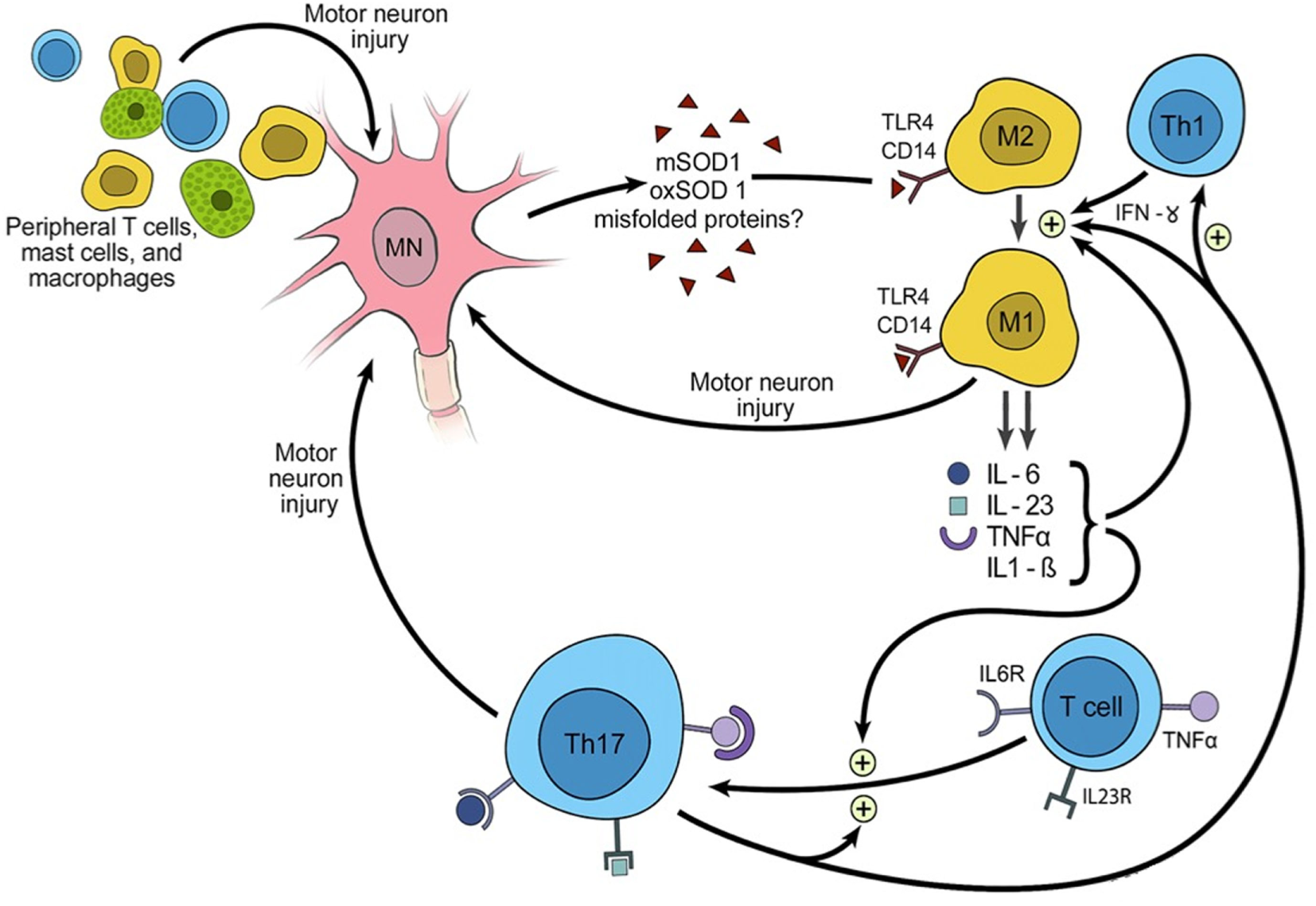
Triggering of inflammation and the role of inflammation in propagating amyotrophic lateral sclerosis.
The M1 phenotype shows increased expression of pro-inflammatory cytokines such as nitric oxide species, TNFA, and the interleukins (IL) 1B, IL2, and IL6 (4). Receptors and transmembrane proteins involved in antigen presentation are upregulated, including MHC class II, CD86 (B7-2, CTLA-4 counter-receptor B7.2), and the Fc fragment of IgG receptor III (FCGR3; CD16) (15). They also increase production of reactive oxygen species (ROS), which are cytotoxic.
M2 microglia secrete anti-inflammatory cytokines such as IL4, IL10, IL13, TGFB and ARG1 as well as neurotrophic factors such as glial cell line-derived neurotrophic factor, brain-derived neurotrophic factor (BDNF), and insulin-like growth factor-1 (IGF1). IGF1 has the capacity to promote growth and differentiation of ‘neural stem cells’, helping with tissue repair (16, 17). Repair is also mediated via angiogenesis and by remodeling of the extracellular matrix (18).
Thus, neurodegeneration is facilitated by the lack of neurotrophic growth factors and by the continued production of cytotoxic byproducts of a pro-inflammatory response.
One factor favoring the inflammatory phenotype (and contributing to neuronal vulnerability to inflammation) is the C9orf72 expansion. This is in a region which functions as an untranslated promoter in microglia, but is translated by neurons. Under normal circumstances, the highest expression levels in the brain are found in microglia. With the expansion, production of C9orf 72 is impaired in microglia, resulting in impairment of endosome/lysosomal trafficking. When attempting phagocytosis, such cells show an increase in ROS and inflammatory cytokines, thought to be due to impairment of phagosome/lysosome fusion (19). In neurons, the transcribed C9orf72 RNA and the translation of useless dipeptides from the expanded repeat both appear to contribute to susceptibility to inflammation and thus degeneration (8).
Helper T-Cells
Th1 cells secrete pro-inflammatory cytokines such as interferon gamma (IFNG), IL2 and TNFB (beta); these proteins also promote the M1 phenotype of nearby microglia/macrophages. Th2, like M2 cells, secrete the anti-inflammatory cytokines IL4 and IL10; these cytokines also promote the differentiation of microglia/macrophages along M2 lines.
The balance between Th1 and Th2 is influenced heavily by regulatory T-cells (Treg). These are marked by CD4 and IL2RA (IL2 receptor subunit alpha, CD25). Their development and maintenance depend on the transcription factor forkhead box P3 (FOXP3); so-named as mutations in Drosophila cause development of head-like structures at each pole instead of the usual fore/hind-gut.
Regulatory T-cells encourage M2 differentiation and impede the activity of cytotoxic T-lymphocytes (Tc, including natural killer T-cells). Lower circulating levels of Tregs have been shown in those with ALS vs. controls (20). Lower levels of Tregs and of FOXP3 have also been correlated with the rate of clinical progression of ALS and may be a useful prognostic biomarker that could be used to stratify groups in clinical trials (21).
Clinical Trials
Search Methods
We searched the NIH’s ClinicalTrials.gov (NCT), a database of medical studies in human volunteers, with the term “amyotrophic lateral sclerosis” for any trials focused on modifying inflammation. We included trials currently in progress or completed prior to the time of writing. These trials are summarized in Table 1, where they appear in the same order as the treatments discussed below. We classified these treatments based on their use in other pro-inflammatory diseases. They are sorted (broadly) by treatment and by the year the trial began accrual. Some trials are discussed here which were not registered with NCT. Also, some NCT-registered trials do not yet have a corresponding article reporting results and are referenced here with their NCT identifier.
Table 1
| NCT ID | SD | Agent | ra | n | wks | P | RCT | Outcomes | C | R | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary | Secondary | ||||||||||
| Rheumatoid arthritis | |||||||||||
| 01277315 | 2/11 | Anakinra | po | 18 | 4 | 2 | N | S/T | S/T | Y | (26) |
| 02588677 | 4/13 | Masitinib + riluzole | po | 394 | 48 | 2/3 | Y | ALSFRS-R | VC, QoL | Y | NA |
| 02469896 | 11/15 | Tocilizumab | iv | 24 | 16 | 2 | Y | S/T | ALSFRS-R, VC | N | NA |
| Multiple sclerosis | |||||||||||
| 00326625 | 7/06 | Glatiramer acetate | sc | 366 | 52 | 2 | Y | ALSFRS-R | ttDV | Y | (38) |
| 01786174 | 8/13 | Fingolimod | po | 30 | 8 | 2 | Y | ALSFRS-R, VC, FEV1 | T-cell subsets | Y | (47) |
| 02238626 | 8/14 | Ibudilast | po | 120 | 26 | 1b/2a | Y | S/T, ALSFRS-R | VC, MMT, HHD | N | (51) |
| 02525471 | 10/15 | RNS60 (saline) | iv | 18 | 24 | 1 | N | S/T | ALSFRS-R, VC, HHD | N | NA |
| Anti-inflammatory | |||||||||||
| 00355576 | 7/06 | Celecoxib + creatine | po | 86 | 26 | 2 | N | ALSFRS-R | VC, QoL, TGUG | Y | (59) |
| ‘ALS-specific’ | |||||||||||
| 01753076 | 12/12 | Ozanezumab | iv | 304 | 48 | 2 | Y | ALSFRS-R, OS | VC, HHD, QoL | Y | (63) |
| 01091142 | 7/10 | NP001 (chlorite) | iv | 32 | 26 | 1 | N | S/T | Biomarkers | Y | (66) |
| 01281631 | 2/11 | NP001 | iv | 136 | 39 | 2 | Y | ALSFRS-R | VC, TTT, serum IM | Y | (67) |
| 02794857 | 8/16 | NP001 | iv | 120 | 26 | 2 | Y | ALSFRS-R | TTT, serum IM | N | NA |
| Treatment of cancer | |||||||||||
| 00140452 | 2/05 | Thalidomide | po | 18 | 39 | 2 | N | ALSFRS | S/T, QoL, serum IM | Y | (69) |
| 01257581 | 3/11 | Tamoxifen | po | 60 | 42 | 2 | N | ALSFRS-R | VC, TTT, HHD, ATLIS | Y | (73) |
| 00397423 | 12/06 | Granulocyte colony-stimulating factor (G-CSF) | sc | 40 | 52 | 2 | Y | ALSFRS | AARS | Y | (75) |
| 01825551 | 11/12 | G-CSF | sc | 40 | 13 | 2/3 | Y | ALSFRS-R | CMAP, MMT, ALSAQ-40 | Y | (76) |
| 03085706 | 10/10 | Peripheral blood mononuclear cell autotransplantation | sa | 14 | 12 | 1/2 | N | S | Functional independence, balance, dysarthria | Y | NA |
| 02286011 | 11/14 | Bone-marrow mononuclear cells | im | 20 | 104 | 1 | N | S | MUNE, CMAP, MRC | N | NA |
| Transplant rejection | |||||||||||
| 01884571 | 10/13 | Basiliximab, mycophenolate, tacrolimus, methylprednisolone, prednisone | iv, po | 33 | 52 | 2 | N | ALSFRS-R | VC, HHD, serum IM, CSF IM | Y | NA |
Trials of immune-modulating treatments in ALS registered with NCT.
ALS, amyotrophic lateral sclerosis; NCT ID, NIH Clinical Trials identifier; SD, start date (month/year); ra, route of administration; po, oral; iv, intravenous; sc, subcutaneous; sa, subarachnoid space (i.e., into CSF); im, intramuscular; n, number of subjects; wks, duration of trial (weeks); P, Phase (1–3); RCT, randomized, double-blind, placebo-controlled trial? C, completed? (at time of writing); R, reference; NA, not applicable (no results reported yet).
Outcomes—note not all secondary outcomes are listed, for reasons of space.
S, safety; T, tolerability; ALSFRS-R, ALS Functional Rating Scale (revised); VC, vital capacity (any method); FEV1, forced expiratory volume in 1 sec; OS, overall survival; QoL, quality of life; ttDV, time to death or ventillator-dependence; MMT, manual muscle testing; HHD, hand-held dynamometry (muscle strength); TGUG, timed get-up-and-go (sitting to walking); TTT, time to tracheostomy; IM, inflammatory markers; ATLIS, accurate test of limb isometric strength; AARS, Appel ALS Rating Scale; CMAP, compound muscle action potential; ALSAQ-40, ALS Assessment Questionnaire-40; MUNE, motor units estimate (number of motor units); MRC, Medical Research Council (score, muscle strength).
Measures of Outcome
The most commonly used measure of outcome in these trials is the change in the ALS Functional Rating Scale (ALSFRS) over time. In 1999, the scale was revised to include assessments of dyspnea, orthopnea, and the need for ventilatory support. This latter, the ALSFRS (revised) (ALSFRS-R) has been adopted by the majority of clinical investigators since (22). Due to the restrictive effect of ALS on pulmonary function, forced vital capacity (FVC) is also often used as a measure of disease progression.
Drugs Used for Rheumatoid Arthritis
Rheumatoid arthritis (RA) has been noted with a greater frequency than would be expected by chance in the families of those affected by ALS (23). In the case of the individual patient, just seven cases of co-occurring ALS and RA have been reported to date. It is thought that most if not all cases were the result of a chance association; indeed the coincidence of the two conditions would appear to be less than might be expected by chance alone.
Notably, as reported by Padovan et al., in two patients with RA, ALS developed rapidly after the introduction of or increase in dose of infliximab, a TNFA antagonist (24). While these authors acknowledge the limitations of such anecdotal observations, this finding would fit with an upregulation of the antiapoptotic TNFRSF1B in chronic RA, thereby masking a tendency toward ALS in the cases they report. Another case in support of this mechanism is that of a patient with psoriatic arthritis who developed ALS rapidly after starting adalimumab, another antibody targeting TNFA (25).
Anakinra
Interleukin 1 is an endogenous pyrogen and a mediator of autoimmune and infectious diseases (26). Anakinra, a recombinant analog of the endogenous antagonist IL1Ra, has already proven effective in the treatment of refractory RA (27). IL1R antagonists have been shown to extend lifespan in SOD1-G93A mice (28). A pilot study of 17 patients treated with anakinra (at the same dose used for RA, 100 mg daily) for 1 year showed no significant reduction in disease progression, measured with the ALSFRS-R. Though the study did show a decrease in cytokines and fibrinogen during the initial 24 weeks of treatment, there was a ‘rebound’ increase in inflammatory markers during the latter half of the study (29).
Masitinib
Masitinib is a pluripotent tyrosine kinase inhibitor which affects multiple pro-inflammatory mast cell receptors. It inhibits mast cell degranulation and mobility. Studies of masitinib in RA, systemic mastocytosis, mast cell tumors and in Alzheimer’s disease have already shown promising results. In SOD1-G93A rats, masitinib was shown to decrease microglial cell activation via inhibition of colony-stimulating factor 1 receptor (30). Masitinib may also prevent macrophage infiltration into the ventral root by inhibiting KIT (KIT proto-oncogene receptor tyrosine kinase), thereby delaying the ‘dying back’ phenomenon associated with anterior horn cell dysfunction (unpublished data, Emilianos Trias et al.).
A Phase 3 randomized, double-blind, placebo-controlled trial (RDBPCT) is ongoing to determine the efficacy of riluzole plus masitinib vs. placebo in ALS over the course of 48 weeks. The interim results are encouraging, with 50% (191 of 381) of patients having completed the riluzole and masitinib arm. A statistically significant difference was seen in the ALSFRS-R score as well as the secondary end points: change in the Combined Assessment of Function scale and FVC.
Tocilizumab
Interleukin 6 signaling is major inducer of the acute-phase response; its receptor is blocked by the monoclonal antibody tocilizumab. Increased serum IL6 is recognized in autoimmune diseases such as RA, systemic-onset juvenile chronic arthritis and psoriasis as well as patients with sALS. Using peripheral blood mononuclear cells (PBMCs i.e. T and B cells as well as monocytes) from four patients with sALS, tocilizumab was shown to inhibit the pro-inflammatory effects of adding mutant SOD1-G93A (31). In particular, the expression of a number of cytokines was reduced in vitro: IL6, IL1B, TNFA, IFNG and GM-CSF. A Phase 2 RDBPCT investigating the safety and tolerability in adult patients with ALS is ongoing. In addition to safety measures, the study investigates the effect of tocilizumab on novel biomarkers including PBMC gene expression profiles and MRI-PET imaging of activated microglia.
Drugs Used for Multiple Sclerosis (MS)
Like RA and ALS, MS and ALS appear to aggregate within families, while co-occurrence in one individual appears rare. We are aware of just four such case reports to date (32). Microglial activation appears partly responsible for neuronal injury in both ALS and MS (33).
Glatiramer Acetate (GA)
Glatiramer acetate is comprised of random polymers of glutamic acid (G), lysine (L), alanine (A), and tyrosine (T). It has long been used in the treatment of relapsing–remitting MS. In such patients, treatment with GA leads to an increase in IL10 and IL4 and a decrease in TNFA (34). GA may also increase Treg activity.
The safety of GA in ALS has been demonstrated in a Phase 1 trial of 20 patients and 10 controls, with doses of 20 mg given daily or biweekly to 10 patients each. GA appeared safe and encouraging immunological changes were measured (35).
A Phase 2 RDBPCT with 366 patients used GA 40 mg/day (vs. ×3/week, as is typical in MS). They remained under observation for 52 weeks. No significant difference in the rate of deterioration (ALSFRS-R) or overall survival was seen (36). The authors speculate that differences in BBB permeability between MS and ALS (affecting the pharmacokinetics of GA or the traffic of the immune system into the CNS) may account for these results.
Fingolimod
Fingolimod is an inhibitor of the sphingosine 1-phosphate receptor (S1PR). This receptor is found on immune cells, neural cells, endothelial cells and smooth muscle. It is thought to play a role in angiogenesis, neurogenesis and immune regulation/trafficking. Inhibition of S1PR causes sequestration of lymphocytes within lymph nodes, thereby reducing their numbers in the blood stream. It has already proved to be effective in MS (37).
In SOD1-G93A mice, fingolimod improved survival. Its immunomodulatory effects are mediated through NOS2, IL1B, FOXP3, IL10, ARG1, integrin subunit alpha M (CD11B, part of complement receptor 3) and BDNF (38). An RBDPCT, with 2:1 randomization, in 30 patients with ALS showed the treatment to be safe and tolerable (one patient stopped treatment due to QT prolongation) (39). We are not aware of plans to study this agent further in ALS.
Ibudilast
Ibudilast is a an inhibitor of TLR4 and phosphodiesterase 3 and 4 which has shown immunomodulatory effects in MS by shifting the immune response from Th1 to Th2 (40). In ALS, TLR4 facilitates the transformation of M2 to M1 microglia (41). The safety profile of ibudilast has been established through long use in the treatment of asthma in Japan. It has been shown to reduce the loss of brain volume change in patients with MS (42). A Phase 2 trial of safety and efficacy in patients with ALS has completed accrual and preliminary reports on safety and tolerability are encouraging (43).
RNS60
RNS60 is modified saline; it is mixed with oxygen in a controlled manner to generate stable, electrically charged nanobubbles. This agent is undergoing clinical trials in MS as well as ALS. The electrical charge carried by RNS60 affects membrane ion channels and increases mitochondrial ATP production in cell culture (44, 45). In a mouse model of MS, it has been shown to reduce NO and NOS2 production and thus is anti-inflammatory (46). This effect is thought to be mediated through the inhibition of NF-κB (47). This results in ‘tipping the balance’ toward the alternative (M2, Th2) response.
As the drug’s effects are a result of biological and not chemical activity, preclinical toxicology studies have shown almost no side effects. Safety has been established in three Phase 1 studies, one intravenous and two using inhalation. A Phase 1 trial of intravenous RNS60 has completed patient accrual (NCT02525471). This will investigate its efficacy as measured by the ALSFRS-R and on inflammatory biomarkers.
‘Non-Specific’ Anti-Inflammatory Agents
Intravenous Immunoglobulin (ivIg)
Two small ‘pilot’ studies to assess the effects of a 3-month course of ivIg on the severity of ALS have proved disappointing; this appears to have put an end to investigation of this modality. These studies were performed prior to the advent of the ALSFRS; instead, participants served as their own controls.
The first in nine patients with ALS used ivIg 2 g/kg monthly for 3 months. No change in disease severity was seen using the outcome measure of maximum voluntary isometric contraction (MVIC), as per the Tufts Quantitative Neuromuscular Evaluation system (48). A subsequent study used the same dose and timing in combination with cyclophosphamide 1–2 mg/kg/day in seven patients over 4–13 months. The rate of deterioration was no slower after starting treatment than before, as measured by the means of the Medical Research Council (MRC) score for muscle strength (10 muscles/limb = 40 muscles), a clinical scale for bulbar function (range 1–5) and the modified Rankin disability scale (range 0–5).
Celecoxib
Celecoxib, a cyclooxygenase-2 inhibitor (prostaglandin G/H synthase) is used to treat pain and inflammation; it also decreases the prostaglandin-induced release of glutamate. One RDBPCT enrolled 30 patients with ALS and randomized their treatment (2:1) between celecoxib and placebo for 12 months. No difference was found in disease progression (ALSFRS-R), muscle strength (maximum MVIC), FCV, or in estimates of numbers of motor units (49).
Celecoxib has also been evaluated in a Phase 2 adaptive trial. Here, it was compared with minocycline; both were given with creatine in order to aid mitochondrial function (50). Historical controls were used as a comparison. Planned enrollment was with sequential pools of 60 patients per arm; after one pool per arm, the celecoxib–creatine regimen proved superior to that of minocycline–creatine, although not greatly superior to the historical controls, as assessed by the rate of decline in the ALSFRS-R at 6 months (35).
Agents ‘Specific’ to ALS
Ozanezumab
Activation of the reticulon 4 receptor (RTN4R, aka Nogo receptor) inhibits the growth of axons in mammals following peripheral nervous system injury. RTN4R is also found on macrophages, where, following Wallerian degeneration, it has been shown to mediate the clearance of these cells from the site of injury (51). Additionally, RNT4 (Nogo-A) is overexpressed in the skeletal muscle of patients with ALS (52).
Thus, ozanezumab (an antibody targeting RNT4) may help slow disease progression, either by decreasing the inhibition of axonal growth or by affecting the inflammatory process following neuronal injury. An RDBPCT with 303 patients tested this treatment, given over 48 weeks. No difference in disease progression (ALSFRS-R) or in survival was seen (53). The results are not thought to have been due to inadequate dosing or pharmacokinetic factors.
NP001
Taurine is an amino acid not incorporated into protein, which binds HOCl, as produced by the oxidative burst of activated neutrophils. The product of this reaction, taurine chloramine, is a less toxic oxidizing agent than HOCl; it is also a signaling molecule that contributes to the self-limiting nature of inflammation. Sodium chlorite (NaClO2; in solution with 63 mM chlorite and pH-corrected = NP001, aka WF10) was developed to mimic this effect. In PBMCs stimulated by anti-CD3 (part of the T-cell receptor complex), WF10 reduced IL2 production as well as the nuclear translocation of the transcription factor nuclear factor of activated T-cells 1 (NFATC1).
WF10/NP001 was originally developed for advanced-stage HIV, to suppress the chronic activation of macrophages, which is thought to be partially responsible for neurological injuries in HIV (54, 55). It is also being investigated as a treatment for MS, although work in ALS is farther along than in these other conditions.
A Phase 1 trial in 32 patients showed the treatment to be safe and well tolerated (56). There was a dose-dependent decrease in a marker of monocyte activation, FCGR3. A subsequent RDBPCT with 136 patients showed no effect on ALSFRS-R, although a subset of those with high baseline serum inflammatory markers (including IL6, IFNG and CRP) did show stabilization of disease (57). A follow-up, focused on those with ALS and high baseline CRP, is ongoing.
Treatments Used Primarily in Patients with Cancer
Thalidomide
Thalidomide is an immunomodulating agent whose effects on TNFA have been shown to be beneficial in SOD1-G93A mice (58). A trial using 400 mg/day, with outcomes available in 18 patients, showed no improvement in ALSFRS or FCV vs. historical controls. (This dose is similar to that used for refractory chronic graft-versus-host disease and to that used in the multi-agent treatment of multiple myeloma.) Treatment with thalidomide was associated with a number of adverse effects, including deep vein thrombosis and bradycardia (59). Bradycardia appears common, with another study reporting a rate of 50% in 18 patients, when used in combination with riluzole (60).
Tamoxifen
A competitive inhibitor of estrogen receptors, tamoxifen continues to be used as adjuvant treatment for breast cancer as well as endometrial carcinoma. Slowing of ALS progression was reported in a woman who started tamoxifen treatment for breast cancer at the 2004 International ALS/MND Symposium. Its anti-inflammatory effect may result from inhibition of protein kinase C (3). A Phase 2 trial in 60 patients reported an increase in survival in those using more than 20 mg/day vs. lower doses (61).
Another Phase 2 trial randomized patients to daily creatine 30 mg or tamoxifen 40 vs. 80 mg. In those taking the higher dose of tamoxifen, disease progression was reduced (ALSFRS-R), an effect which remained significant after controlling for gender, site of disease onset and VC. These results were presented by Atassi et al. at the Northeast ALS Consortium Webinar 2013.1
Granulocyte Colony-Stimulating Factor (G-CSF, Filgrastim)
This compound stimulates the proliferation and differentiation of granulocytes; it has been shown to play a part in neuro- and angio-genesis as well as modulation of the immune system. It is commonly used in myelotoxic chemotherapy to prevent or treat neutropenia. In SOD1-G93A mice, use of pegylated G-CSF increased survival and reduced microgliosis (62).
A pilot study, with 10 patients available for complete assessment, used a dose of 2 g/kg for 5 days. While the first 3 months of showed a significant reduction in the rate of decline (ALSFRS), there was a ‘rebound’ worsening in the subsequent 3 months (63). The decline in compound muscle action potential (CMAP) was also reduced during the first 3 months and remained unchanged thereafter.
An RDBPCT in 10 patients used a dose of 10 μg/kg on days 1–10 and 20–25 (64). No difference in disease progression at 100 days (ALSFRS) was noted. There was a smaller decline in fractional anisotropy of the brain in the treated group. This MRI finding, which measures the ‘directionality’ of water flow, is considered a marker of axonal myelination (65).
Based on these encouraging results, a larger RDBPCT was performed with 40 patients, using G-CSF at 5 μg/kg/q12h for 5 days; they were observed for 3 months. There was no change in disease progression, as measured with the ALSFRS-R, the ALS Assessment Questionnaire-40, manual muscle testing, and CMAPs (66). Although the duration of the trial was relatively short, the authors note “previous studies suggest that a longer duration of follow-up is unlikely to confer any important clinical benefit with currently administered doses of G-CSF.” The small sample size was based on a large expected difference in the change in ALSFRS-R, which may in retrospect have been a little optimistic. Another trial of G-CSF in 40 patients appears to have been completed, although no results are available (NCT00397423).
White Blood Cell Support
Isolation of mononuclear cells from blood and bone marrow has long been used to provide support for patients undergoing myelotoxic chemotherapy or marrow transplantation. Thus, such facilities are likely to be available wherever such treatments are used.
A single-center, Phase 1 RDBPCT to assess the rate of adverse events related to the intramuscular infusion of autologous mononuclear cells of bone marrow is currently underway and scheduled to complete at the end of 2017 (NCT02286011). A similar study was completed to access the deliverance of PBMC transplantation into the subarachnoid space, but has not yet reported results (NCT03085706).
Another Phase 1 trial with three patients used leukapheresis followed by isolation and expansion of Tregs. They received four infusions of these Tregs every 2 weeks, with simultaneous subcutaneous IL2 (to expand T-cells). This modality appears safe and clinical benefits were noted for up to 1 month after the infusions were stopped (67).
Treatments Used as Part of Organ Transplantation
Cyclosporine
Cyclosporine, which reduces production of IL2, is an oral agent which does not appear to cross the intact BBB. When infused into the lateral cerebral ventricle of SOD1-G93A mice, cyclosporine leads to improved survival (68). An RDBPCT of 74 patients used variable dosing to achieve a target serum level of 400–600 ng/mL. This level is slightly higher than that typically used after solid organ transplant (100–400 ng/mL). A slower rate of progression was seen in men who started to receive treatment within 18 months of symptom onset. However, those who entered the study later in the disease course than 18 months showed no improvement (69).
Total Lymphoid Irradiation
Irradiating the thymus, spleen and lymph nodes (while shielding non-lymphoid organs) is a treatment used typically in preparation for organ transplant and occasionally for severe cases of autoimmune diseases such as RA. One RDBPCT with 61 patients showed no difference, after 2 years observation, in muscle strength (via MRC or dynamometer), swallowing (time to swallow 4 oz water), walking (time to walk 15 and 25 feet) or survival (70).
Prevention of Transplant Rejection
A variety of agents, typically in combination, are used to prevent acute graft-versus-host disease in transplant recipients. An observation in ALS patients undergoing neural stem cell injections into the spinal cord lead to the hypothesis that the immune-suppression required for the injections was, at least in part, responsible for the therapeutic effects. A Phase 2 trial with 33 patients to assess this has been completed (NCT01884571). This used basiliximab (anti-IL2) with mycophenolate (an inhibitor of nucleotide synthesis, which T- and B-cells rely upon) and tacrolimus (inhibits translocation of NFATC1 and thus transcription of IL2, TNFA, IFNG and NOS2 among other effects) as well as steroids.
Discussion
The majority of the agents we reviewed appear safe and well tolerated; many are already in use for other conditions and so good information is available regarding side effects and monitoring. The most promising approaches identified in our review included NP001, masitinib–riluzole, celecoxib–creatine and tamoxifen–creatine [(58, 66, 71), NCT02588677]. All of these treatments await confirmation in larger RCTs.
As ALS progresses, so does the risk of infection, particularly aspiration pneumonia. Also, the part played by ‘tractable/modifiable inflammation’ relative to ‘degeneration’, as a cause of clinical disability, is likely to decrease, although this requires further investigation. In the case of secondary-progressive MS, where this transition has been better studied, CNS inflammation does not appear to cease, but becomes more difficult to address as much of the inflammation it is ‘hidden’ behind the BBB (72).
Thus, the risk/benefit ratio to immunosuppression in ALS is likely to increase with disease progression. A number of the treatments above are strongly immune-suppressing and the risk of infection associated with such medicines is well recognized in treating other autoimmune disorders, particularly in rheumatology (71). The risk of cancer with long-term immunosuppression in the setting of solid organ transplant has also been acknowledged. With a follow-up of 15 years after transplant, the sex- and age-standardized incidence ratio (i.e., number of cancers observed/number of cancers expected) has been estimated at 2.2 (73).
In other inflammatory conditions with progressive disability, patients appear willing to accept an early risk of complications in return for preventing later disability. In the case of a survey of MS patients, in return for approximately 5 years free of relapse and disability, patients were willing to accept a risk of approximately 1% for each of progressive multifocal leukoencephalopathy, leukemia and liver failure (74). Similar patient risk/benefit preferences have not been studied for ALS. Such work will likely need to wait for robust estimates for a treatment with proven efficacy.
The immune-suppressing treatment with the greatest potential short-term risk and arguably the greatest prospect of long-term control remains autologous bone marrow transplantation (ABMT). Preliminary experience of safety and efficacy in MS has proved encouraging, with transplant-related mortality rates of 0.3% (349 patients transplanted after 2005) and 0% (119 patients treated with a low-intensity regimen) (75). ABMT is not under investigation in ALS, to our knowledge.
Many of the drugs discussed above have also proved effective in psoriasis, a common inflammatory condition that also injures ectoderm-derived cells and is characterized by a chronic Th1 response, but that has received almost no attention in relation to ALS. While psoriasis tends to develop at a far younger age, any association (or lack thereof) in an individual or family affected by ALS could improve our understanding of both conditions. Of course, affecting inflammation within the relatively ‘immune-privileged’ CNS (vs. skin) has proved to be more challenging, in part due to accessibility and differences in pharmacokinetics.
The problem in translating work in mice into trials in humans in ALS is well recognized and has been reviewed by Mitsumoto et al. (5). SOD1-G93A remains the predominant mouse model. Given the rarity of SOD1-G93A as a cause of fLAS, trials conducted in those affected have proved difficult to perform and arguably would be limited relevance to sALS. Importantly, this mouse model has a different cause than sALS; the latter appears to have a variety of predisposing and precipitating factors, all of which converge on motor neuron death. This phenomenon, whereby a variety of pathological processes converge on a common phenotype, is also seen in Parkinson’s and (arguably) in Alzheimer’s disease. In both conditions, a subtype characterized by inflammation (vs. other metabolic abnormalities) has been proposed (76, 77). Splitting or stratifying such ‘hot’ vs. ‘cold’ disease subtypes appears to be a rational strategy, particularly while investigating anti-inflammatory and immune-suppressing treatments.
Focusing on the mediators of disease progression (vs. inciting mechanisms), particularly the immune system, thus offers the promise of treatments which are applicable to a wide range of patients with ALS.
Despite these challenges and the number of negative results, work by trialists has done much to improve our understanding of ALS in untreated patients. The ALS Patient Care Database should also prove valuable in this regard (78).
Whether ALS may be said to have a ‘natural history’ is debatable, as continuous improvements in supportive care have led to better outcomes for those affected. This is particularly true in the case of preventing infections, pressure ulcers, malnutrition (with feeding via gastrostomy if necessary) and respiratory failure (with non-invasive ventilation). Bearing this in mind, we hope that a ‘pooled’ cohort of patients receiving placebo as part of a clinical trial will become available. Such patients could serve as historical controls for new treatments in early stages of development. However, the quality of supportive care would need to be accounted for in such a pooled group; as a surrogate, the date and location of those receiving placebo on trial could be used.
Conclusion
A number of basic science and clinical research studies have demonstrated a correlation between the immune system and ALS pathology. Despite these findings, the optimal targets have yet to be elucidated. It remains to be seen if inflammation is of similar importance in all forms of ALS. Improving our knowledge of inflammatory biomarkers that correlate with ALS progression may speed the development of such immune-modulating treatments.
Statements
Author contributions
SK, LA and RK all contributed to drafting the manuscript, reviewing references, and generating table and figures. SL provided helpful suggestions for improvement. CD prepared the final version of the manuscript, table and figures. All the authors have read and approved the final version.
Funding
SK and LA were funded by the Howard Hughes Medical Institute.
Acknowledgments
The authors wish to thank Neuroscience Publications at the Barrow Neurological Institute for their assistance in preparing Figure 2.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
1
HetzCMollereauB. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci (2014) 15:233–49.10.1038/nrn3689
2
ParakhSSpencerDMHalloranMASooKYAtkinJD. Redox regulation in amyotrophic lateral sclerosis. Oxid Med Cell Longev (2013) 2013:408681.10.1155/2013/408681
3
OrsiniMOliveiraABNascimentoOJMReisCHMLeiteMAAde SouzaJAet alAmyotrophic lateral sclerosis: new perspectives and update. Neurol Int (2015) 7:5885.10.4081/ni.2015.5885
4
LewisC-AManningJRossiFKriegerC. The neuroinflammatory response in ALS: the roles of microglia and T cells. Neurol Res Int (2012) 2012:803701.10.1155/2012/803701
5
MitsumotoHBrooksBRSilaniV. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved?Lancet Neurol (2014) 13:1127–38.10.1016/S1474-4422(14)70129-2
6
TaylorJPBrownRHJrClevelandDW. Decoding ALS: from genes to mechanism. Nature (2016) 539:197–206.10.1038/nature20413
7
RadfordRAMorschMRaynerSLColeNJPountneyDLChungRS. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front Cell Neurosci (2015) 9:414.10.3389/fncel.2015.00414
8
LallDBalohRH. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest (2017) 127:3250–8.10.1172/JCI90607
9
LouveauASmirnovIKeyesTJEcclesJDRouhaniSJPeskeJDet alStructural and functional features of central nervous system lymphatic vessels. Nature (2016) 523:337–41.10.1038/nature14432
10
NimmerjahnAKirchhoffFHelmchenF. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308:1314–8.10.1126/science.1110647
11
NeumannHMedanaIMBauerJLassmannH. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci (2002) 25:313–9.10.1016/S0166-2236(02)02154-9
12
Gene. Bethesda, MD: National Library of Medicine (US), National Center for Biotechnology Information (2004). Available from: http://www.ncbi.nlm.nih.gov/gene/
13
LullMEBlockML. Microglial activation and chronic neurodegeneration. Neurotherapeutics (2010) 7:354–65.10.1016/j.nurt.2010.05.014
14
AloisiF. Immune function of microglia. Glia (2001) 36:165–79.10.1002/glia.1106
15
CherryJDOlschowkaJAO’BanionMK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation (2014) 11:98.10.1186/1742-2094-11-98
16
ZieglerANLevisonSWWoodTL. Insulin and IGF receptor signalling in neural-stem-cell homeostasis. Nat Rev Endocrinol (2015) 11:161–70.10.1038/nrendo.2014.208
17
Joseph D’ErcoleAYeP. Expanding the mind: insulin-like growth factor I and brain development. Endocrinology (2008) 149:5958–62.10.1210/en.2008-0920
18
VarinAGordonS. Alternative activation of macrophages: immune function and cellular biology. Immunobiology (2014) 7:630–41.10.1016/j.imbio.2008.11.009
19
O’RourkeJGBogdanikLYáñezALallDWolfAJMuhammadAKet alC9orf72 is required for proper macrophage and microglial function in mice. Science (2016) 351:1324–9.10.1126/science.aaf1064
20
RentzosMEvangelopoulosESeretiEZouvelouVMarmaraSAlexakisTet alAlterations of T cell subsets in ALS: a systemic immune activation?Acta Neurol Scand (2012) 125:260–4.10.1111/j.1600-0404.2011.01528.x
21
HenkelJSBeersDRWenSRiveraALToennisKMAppelJEet alRegulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med (2014) 5(1):64–79.10.1002/emmm.201201544
22
CedarbaumJMStamblerNMaltaEFullerCHiltDThurmondBet alThe ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci (1999) 31:13–21.10.1016/S0022-510X(99)00210-5
23
AppelSHStockton-AppelVStewartSSKermanRH. Amyotrophic lateral sclerosis. Associated clinical disorders and immunological evaluations. Arch Neurol (1986) 43:234–8.10.1001/archneur.1986.00520030026007
24
PadovanMCaniattiLMTrottaFGovoniM. Concomitant rheumatoid arthritis and amyotrophic lateral sclerosis: report of two new cases and review of literature. Rheumatol Int (2011) 31:715–9.10.1007/s00296-010-1760-3
25
BougeaAAnagnostouEStamboulisEKararizouE. Amyotrophic lateral sclerosis developing during adalimumab therapy for psoriatic arthritis. Rev Neurol (2014) 170:228–9.10.1016/j.neurol.2013.11.004
26
GarlandaCDinarelloCAMantovaniA. The interleukin-1 family: back to the future. Immunity (2013) 39:1003–18.10.1016/j.immuni.2013.11.010
27
MertensMSinghJA. Anakinra for rheumatoid arthritis. Cochrane Database Syst Rev (2009) (1):CD005121.10.1002/14651858.CD005121.pub3
28
MeissnerFMolawiKZychlinskyA. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc Natl Acad Sci U S A (2010) 29:13046–50.10.1073/pnas.1002396107
29
MaierADeigendeschNMullerKWeishauptJHKrannichARohleRet alInterleukin-1 antagonist Anakinra in amyotrophic lateral sclerosis – a pilot study. PLoS One (2015) 10:e0139684.10.1371/journal.pone.0139684
30
TriasEIbarburuSBarreto-NunezRBabdorJMacielTTGuilloMet alPost-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation (2016) 13:177.10.1186/s12974-016-0620-9
31
MizwickiMTFialaMMagpantayLAzizNSayreJLiuGet alTocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am J Neurodegener Dis (2012) 1:305–15.
32
TrojsiFSagnelliACirilloGPiccirilloGFemianoCIzzoFet alAmyotrophic lateral sclerosis and multiple sclerosis overlap: a case report. Case Rep Med (2012) 2012:324685.10.1155/2012/324685
33
YiangouYFacerPDurrenbergerPChessellIPNaylorABountraCet alCOX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol (2006) 6:12.10.1186/1471-2377-6-12
34
BosterABartoszekMPO’ConnellCPittDRackeM. Efficacy, safety, and cost-effectiveness of glatiramer acetate in the treatment of relapsing-remitting multiple sclerosis. Ther Adv Neurol Disord (2011) 4:319–32.10.1177/1756285611422108
35
GordonPHCheungY-KLevinBAndrewsHDoorishCMacarthurRBet alA novel, efficient, randomized selection trial comparing combinations of drug therapy for ALS. Amyotroph Lateral Scler (2008) 9:212–22.10.1080/17482960802195632
36
MeiningerVDroryVELeighPNLudolphARobberechtWSilaniV. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: a double- blind, randomized, multicentre, placebo-controlled trial. Amyotroph Lateral Scler (2009) 10:378–83.10.3109/17482960902803432
37
BrinkmannVBillichABaumrukerTHeiningPSchmouderRFrancisGet alFingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov (2010) 9:883–97.10.1038/nrd3248
38
PotenzaRLDe SimoneRArmidaMMazziottiVPezzolaAPopoliPet alFingolimod: a disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics (2016) 13:918–27.10.1007/s13311-016-0462-2
39
BerryJPaganoniSAtassiNGoyalNRivnerMSimpsonEet alA phase IIa double-blind, placebo-controlled study to evaluate the safety of oral fingolimod in patients with amyotrophic lateral sclerosis (ALS) (P7.067). Neurology (2015) 84(14 Suppl). Available from: http://www.neurology.org/content/84/14_Supplement/P7.067.abstract
40
FengJMisuTFujiharaKSakodaSNakatsujiYFukauraHet alIbudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Mult Scler (2004) 10:494–8.10.1191/1352458504ms1070oa
41
AppelSHZhaoWBeersDRHenkelJS. The microglial-motoneuron dialogue in ALS. Acta Myol (2011) 30:4–8.
42
BarkhofFHulstHEDrulovicJUitdehaagBMMatsudaKLandinRet alIbudilast in relapsing-remitting multiple sclerosis: a neuroprotectant?Neurology (2010) 74:1033–40.10.1212/WNL.0b013e3181d7d651
43
BrooksBRBravverESanjakMLangfordVMooreLSmithMet alAdaptive design single center phosphodiesterase type 4 (PDE4) inhibitor – ibudilast (MN-166-ALS-1201) phase 1b/2a clinical trial double-blind (DB) with open label extension (OLE) [NCT02238626] for amyotrophic lateral sclerosis (ALS) patients [1] not requiring non-invasive ventilation (no NIV) up to 5 years (Early Cohort – EC) and [2] requiring non-invasive ventilation (NIV) up to 10 years (advanced NIV cohort – ANC) from disease onset – report of clinical trial DB, OLE and post-treatment cessation epochs – per-protocol (PP) treatment completion associated with improved survival and post treatment cessation loss of muscle strength (P3.127). Neurology (2017) 88(16 Suppl). Available from: http://www.neurology.org/content/88/16_Supplement/P3.127.abstract
44
ChoiJCMegaTLGermanSWoodABWatsonRL. Electrokinetically altered normal saline modulates ion channel activity. Biophys J (2012) 102:683a.10.1016/j.bpj.2011.11.3712
45
ChoiSYuEKimDSSugimoriMLlinásRR. RNS60, a charge-stabilized nanostructure saline alters Xenopus laevis oocyte biophysical membrane properties by enhancing mitochondrial ATP production. Physiol Rep (2015) 3:e12261.10.14814/phy2.12261
46
MondalSMartinsonJAGhoshSWatsonRPahanK. Protection of Tregs, suppression of Th1 and Th17 cells, and amelioration of experimental allergic encephalomyelitis by a physically-modified saline. PLoS One (2012) 7:e51869.10.1371/journal.pone.0051869
47
KhasnavisSJanaAFRoyAFMazumderMFBhushanBFWoodTFet alSuppression of nuclear factor-kappaB activation and inflammation in microglia by physically modified saline. J Biol Chem (2012) 287:29529–42.10.1074/jbc.M111.338012
48
DalakasMCSteinDPOteroCSekulECuplerEJMcCroskyS. Effect of high-dose intravenous immunoglobulin on amyotrophic lateral sclerosis and multifocal motor neuropathy. Arch Neurol (1994) 51:861–4.10.1001/archneur.1994.00540210031010
49
CudkowiczMEShefnerJMSchoenfeldDAZhangHAndreassonKIRothsteinJDet alTrial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol (2006) 60:22–31.10.1002/ana.20903
50
KlivenyiPFerranteRJMatthewsRTBogdanovMBKleinAMAndreassenOAet alNeuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med (1999) 5:347–50.10.1038/6568
51
DavidSFryEJLópez-ValesR. Novel roles for Nogo receptor in inflammation and disease. Trends Neurosci (2008) 31:221–6.10.1016/j.tins.2008.02.002
52
BergesABullmanJBatesSKrullDWilliamsNChenC. Ozanezumab dose selection for amyotrophic lateral sclerosis by pharmacokinetic-pharmacodynamic modelling of immunohistochemistry data from patient muscle biopsies. PLoS One (2015) 10:e0117355.10.1371/journal.pone.0117355
53
MeiningerVGengeAvan den BergLHRobberechtWLudolphAChioAet alSafety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol (2017) 16:208–16.10.1016/S1474-4422(16)30399-4
54
McGrathMSKahnJOHerndierBG. Development of WF10, a novel macrophage-regulating agent. Curr Opin Investig Drugs (2002) 3:365–73.
55
DardisC. Acute motor axonal neuropathy in a patient with prolonged CD4 depletion due to HIV: a local variant of macrophage activation syndrome?Oxf Med Case Rep (2015) 2:200–2.10.1093/omcr/omv009
56
MillerRGZhangRBlockGKatzJBarohnRKasarskisEet alNP001 regulation of macrophage activation markers in ALS: a phase I clinical and biomarker study. Amyotroph Lateral Scler Frontotemporal Degener (2014) 15:601–9.10.3109/21678421.2014.951940
57
MillerRGBlockGKatzJSBarohnRJGopalakrishnanVCudkowiczMet alRandomized phase 2 trial of NP001-a novel immune regulator: safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm (2015) 2:e100.10.1212/NXI.0000000000000100
58
KiaeiMPetriSKipianiKGardianGChoiD-KChenJet alThalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci (2006) 26:2467–73.10.1523/JNEUROSCI.5253-05.2006
59
StommelEWCohenJAFadulCECogbillCHGraberDJKingmanLet alEfficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: a phase II open label clinical trial. Amyotroph Lateral Scler (2009) 10(5–6):393–404.10.3109/17482960802709416
60
MeyerTMaierABorisowNDullingerJSSplettstosserGOhlraunSet alThalidomide causes sinus bradycardia in ALS. J Neurol (2008) 255:587–91.10.1007/s00415-008-0756-3
61
RentzosMMichalopoulouMNikolaouCRombosADimitrakopoulosA. Serum levels of soluble intercellular adhesion molecule-1 (s-ICAM-1) and soluble endothelial leukocyte adhesion molecule-1(s-ELAM-1) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord (2005) 6(2):118–21.10.1080/14660820410021311a
62
PollariESavchenkoEJaronenMKanninenKMalmTWojciechowskiSet alGranulocyte colony stimulating factor attenuates inflammation in a mouse model of amyotrophic lateral sclerosis. J Neuroinflammation (2011) 8:74.10.1186/1742-2094-8-74
63
ZhangYWangLFuYSongHZhaoHDengMet alPreliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sclerosis. Amyotroph Lateral Scler (2009) 10(5–6):430–1.10.3109/17482960802588059
64
DuningTSchiffbauerHWarneckeTMohammadiSFloelAKolpatzikKet alG-CSF prevents the progression of structural disintegration of white matter tracts in amyotrophic lateral sclerosis: a pilot trial. PLoS One (2011) 6(3):e17770.10.1371/journal.pone.0017770
65
AlexanderALLeeJELazarMFieldAS. Diffusion tensor imaging of the brain. Neurotherapeutics (2007) 4:316–29.10.1016/j.nurt.2007.05.011
66
AmirzagarNNafissiSTafakhoriAModabberniaAAmirzargarAGhaffarpourMet alGranulocyte colony-stimulating factor for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled study of Iranian patients. J Clin Neurol (2015) 11:164–71.10.3988/jcn.2015.11.2.164
67
ThonhoffJBeersDZhaoWLayLBrownDBerryJet alA phase 1 study assessing the infusion of expanded autologous regulatory T lymphocytes in subjects with amyotrophic lateral sclerosis (P3.137). Neurology (2017) 88(16 Suppl). Available from: http://www.neurology.org/content/88/16_Supplement/P3.137.short
68
KeepMElmerEFongKSCsiszarK. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res (2001) 894:327–31.10.1016/S0006-8993(01)02012-1
69
AppelSHStewartSSAppelVHaratiYMietlowskiWWeissWet alA double-blind study of the effectiveness of cyclosporine in amyotrophic lateral sclerosis. Arch Neurol (1988) 45:381–6.10.1001/archneur.1988.00520280027011
70
DrachmanDBChaudhryVCornblathDKunclRWPestronkAClawsonLet alTrial of immunosuppression in amyotrophic lateral sclerosis using total lymphoid irradiation. Ann Neurol (1994) 35:142–50.10.1002/ana.410350205
71
GilesJTBathonJM. Serious infections associated with anticytokine therapies in the rheumatic diseases. J Intensive Care Med (2004) 19:320–34.10.1177/0885066604267854
72
StadelmannCWegnerCBrückW. Inflammation, demyelination, and degeneration – recent insights from MS pathology. Biochim Biophys Acta (2011) 1812:275–82.10.1016/j.bbadis.2010.07.007
73
SerrainoDPiselliPBusnachGBurraPCitterioFArbustiniEet alRisk of cancer following immunosuppression in organ transplant recipients and in HIV-positive individuals in southern Europe. Eur J Cancer (2007) 43:2117–23.10.1016/j.ejca.2007.07.015
74
JohnsonFRVan HoutvenGOzdemirSHassSWhiteJFrancisGet alMultiple sclerosis patients’ benefit-risk preferences: serious adverse event risks versus treatment efficacy. J Neurol (2009) 256(4):554–62.10.1007/s00415-009-0084-2
75
BurmanJFoxRJ. Autologous hematopoietic stem cell transplantation for MS: safer than previously thought. Neurology (2017) 88:2072–3.10.1212/WNL.0000000000003995
76
BrockmannKSchulteCSchneiderhan-MarraNApelAPont-SunyerCVilasDet alInflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur J Neurol (2017) 24:427–e6.10.1111/ene.13223
77
BredesenDE. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging (2015) 7:595–600.10.18632/aging.100801
78
The ALS C.A.R.E. Program. Worcester, MA: Center for Outcomes Research, University of Massachusetts Medical School (1995). Available from: http://www.outcomes-umassmed.org/ALS/
Summary
Keywords
amyotrophic lateral sclerosis, immunotherapy, microglial activation, neuroinflammation, SOD1
Citation
Khalid SI, Ampie L, Kelly R, Ladha SS and Dardis C (2017) Immune Modulation in the Treatment of Amyotrophic Lateral Sclerosis: A Review of Clinical Trials. Front. Neurol. 8:486. doi: 10.3389/fneur.2017.00486
Received
08 June 2017
Accepted
31 August 2017
Published
25 September 2017
Volume
8 - 2017
Edited by
Marina Bentivoglio, University of Verona, Italy
Reviewed by
Eduardo Nobile-Orazio, Università degli Studi di Milano, Italy; Gennaro Pagano, King’s College London, United Kingdom
Updates
Copyright
© 2017 Khalid, Ampie, Kelly, Ladha and Dardis.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher Dardis, christopherdardis@gmail.com
Specialty section: This article was submitted to Movement Disorders, a section of the journal Frontiers in Neurology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.