 Raimund Helbok*†
Raimund Helbok*† Mario Kofler†
Mario Kofler† Alois Josef Schiefecker
Alois Josef Schiefecker Maxime GaaschVerena RassBettina PfauslerRonny BeerErich Schmutzhard
Maxime GaaschVerena RassBettina PfauslerRonny BeerErich Schmutzhard
- Neurological Intensive Care Unit, Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria
Objective: To review the published literature on the clinical application of cerebral microdialysis (CMD) in aneurysmal subarachnoid hemorrhage (SAH) patients and to summarize the evidence relating cerebral metabolism to pathophysiology, secondary brain injury, and outcome.
Methods: Study selection: Two reviewers identified all manuscripts reporting on the clinical use of CMD in aneurysmal SAH patients from MEDLINE. All identified studies were grouped according to their focus on brain metabolic changes during the early and subacute phase after SAH, their association with mechanisms of secondary brain injury and outcome.
Results: The review demonstrated: (1) limited literature is available in the very early phase before the aneurysm is secured. (2) Brain metabolic changes related to early and delayed secondary injury mechanisms may be used in addition to other neuromonitoring parameters in the critical care management of SAH patients. (3) CMD markers of ischemia may detect delayed cerebral ischemia early (up to 16 h before onset), underlining the importance of trend analysis. (4) Various CMD-derived parameters may be associated with patient outcome at 3–12 months, including CMD-lactate-to-pyruvate-ratio, CMD-glucose, and CMD-glutamate.
Conclusion: The clinical use of CMD is an emerging area in the literature of aneurysmal SAH patients. Larger prospective multi-center studies on interventions based on CMD findings are needed.
Introduction
First reports on monitoring of brain metabolism using cerebral microdialysis (CMD) in patients with subarachnoid hemorrhage (SAH) date back to the year 1992 (1). Despite the long-standing availability of this technique, recommendations for treatment decisions based on CMD monitoring have just been recently published as a consensus statement by clinical experts (2).
Cerebral microdialysis has improved our understanding of pathophysiological mechanisms of early and delayed brain injury in SAH patients by providing metabolic information derived from brain tissue on a cellular level, in addition to intracranial pressure (ICP), cerebral perfusion pressure (CPP), cerebral blood flow (CBF), brain tissue oxygen tension (PbtO2), and electrographic monitoring (electroencephalography and electrocorticography). So far, changes in brain metabolism have been associated with known complications after SAH and may also help to detect impending secondary brain injury early or before they have evolved into irreversibility. Moreover, abnormalities in CMD-derived parameters have been linked to poor brain tissue and functional outcome and may therefore be integrated in the multimodal approach of neuroprognostication. In addition, the effect of commonly applied pharmacological and non-pharmacological treatments on brain metabolism can be studied on an individual level, thereby enhancing the concept of personalized medicine in neurocritical care patients.
Microdialysis Methodology
The principle of CMD is to mimic a capillary blood vessel in the brain for the assessment of local cerebral metabolism (3). A tubular semi-permeable membrane on the tip of the CMD catheter is perfused with an isotonic or colloid-enriched fluid that equilibrates with the extracellular space by simple diffusion. All molecules small enough to pass the membrane follow their electro-chemical gradient into the tube. Established catheters have a membrane length of 1 cm and pore sizes of either 20 or 100 kDa, which do not show differences in the recovery of small molecules (4). A perfusion speed of 0.3 µl/min is recommended in clinical use, which leads to a relative recovery rate (dialyzate concentration divided by true concentration) of about 70% for the most commonly assessed molecules (5).
Criteria for CMD monitoring are not well defined. It can be used as part of the “multimodal neuromonitoring bundle” (Figure 1A) in ventilated (“poor-grade”) patients or in patients with a secondary neurological deterioration. As a primary monitoring device, the probe should be placed in the frontal lobe (anterior/middle cerebral artery watershed), ipsilateral to the ruptured aneurysm, or the maximal blood clot load. When used in patients with secondary deterioration, location can also be guided by local practice to identify tissue at risk (for example, by CT perfusion or transcranial ultrasound) (2, 6). The catheter can be inserted into the brain tissue either by using a cranial access device (bolt) or by tunneling (Figure 1A), penetrating the skull via a craniotomy (Figure 1B) or a twist drill hole. The dialyzate of the first hours should be discarded due to the insertion trauma and dilution effect of the flush sequence filling the system.
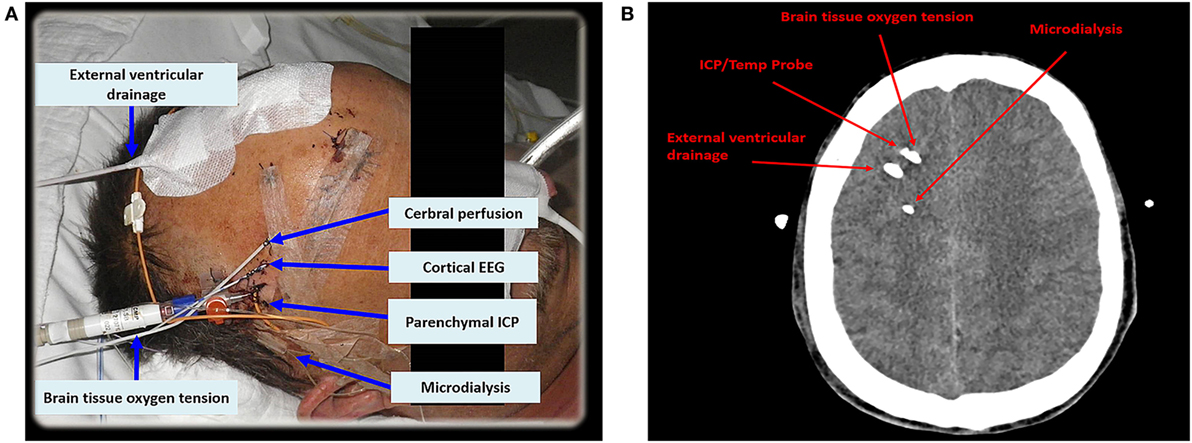
Figure 1. In (A), multimodal neuromonitoring catheters are tunneled in a patient with subarachnoid hemorrhage. (B) Shows neuromonitoring catheters placed in the white matter on an axial computed tomography. EEG, electroencephalography; ICP, intracranial pressure; Temp, temperature.
Interpretation of Data
Concentrations of CMD parameters represent the local metabolic environment surrounding the membrane and cannot be extrapolated to other regions of the brain. Knowledge of catheter location is therefore mandatory for data interpretation. The gold tip of the CMD probe is visible on head CT (Figure 2B), thus its exact location in the brain can be determined and classified by the monitored tissue (gray vs. white matter), lobe, or by the spatial relation to focal pathologies (intralesional/perilesional vs. radiologically normal-appearing brain tissue). The dependency of molecular concentrations on probe location argues for the interpretation of temporal dynamics (trend analysis) in addition to absolute values. Calculating ratios of different CMD parameters (e.g., CMD-lactate-to-CMD-pyruvate-ratio, CMD-LPR) creates variables independent of absolute concentrations and recovery.
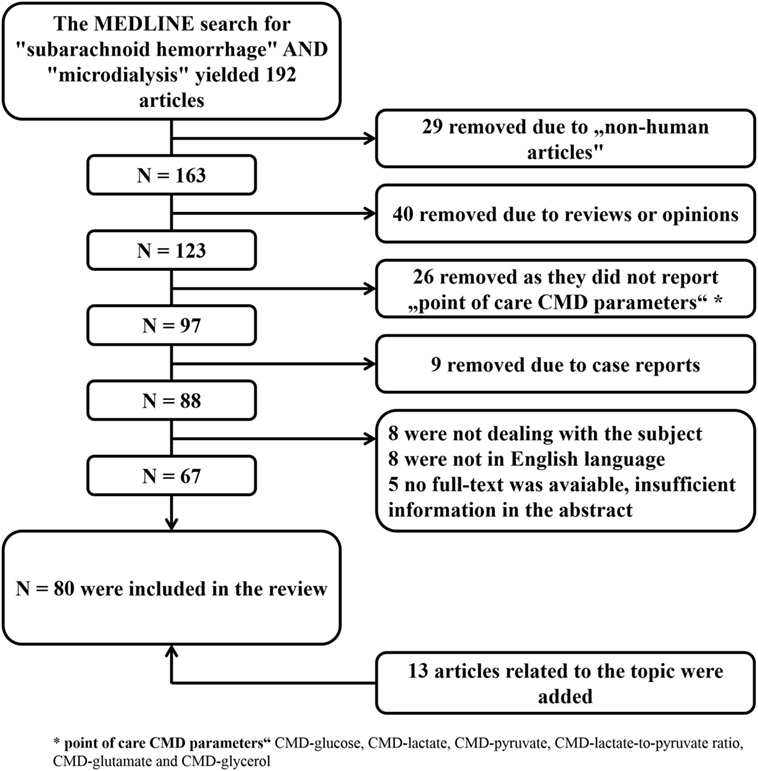
Figure 2. Literature search with selection of articles included in the review. CMD, cerebral microdialysis.
The dialyzate is sampled into microvials and analyzed for point of care parameters including CMD-glucose, CMD-lactate, CMD-pyruvate, CMD-glutamate, and CMD-glycerol at the patient’s bedside. A measurement interval of 1 h is commonly used in clinical practice.
Glucose is an important energy substrate for neuronal tissue. Its concentration in the brain depends on systemic supply, diffusivity in the brain tissue, and local consumption. In the process of aerobic glycolysis, it is metabolized to pyruvate and further converted into acetyl-coenzyme A, which is used for mitochondrial energy production. Under conditions of brain tissue hypoxia or mitochondrial dysfunction, pyruvate is fermented into lactate. The LPR reflects the cytoplasmatic redox state and is a marker of anaerobic metabolism and/or mitochondrial dysfunction. The concept of mitochondrial dysfunction arose from observations of impaired cerebral energy metabolism despite normal perfusion and substrate availability. The underlying pathophysiological mechanisms are not sufficiently elucidated. CMD-glutamate has fewer clinical implications, however, elevated concentrations of this excitatory neurotransmitter are considered to be a marker of ischemia and excitotoxicity. Glycerol is a component of neuronal cell membranes, thus CMD-glycerol concentrations are a surrogate marker of cell membrane damage, e.g., under conditions of hypoxia or ischemia.
Materials and Methods
The aim of this review is to summarize the current knowledge of this technique in the critical care management of SAH patients and to discuss its limitations. A MEDLINE search was performed to identify all studies reporting on the clinical use of CMD in aneurysmal SAH patients. The selection process is summarized in Figure 2. All identified studies were grouped according to their focus on the brain metabolic changes during the early and subacute phase after SAH, their association with mechanisms of secondary brain injury and outcome.
Definitions
The early phase was defined as the first 72 h after SAH, commonly referred to as “early brain injury” (EBI) (7). Pathological threshold values of parameters commonly given in the SAH literature are CMD-glucose < 0.7 mmol/l (referred to as neuroglucopenia), CMD-lactate > 4 mmol/l, CMD-pyruvate < 120 μmol/l, CMD-glutamate > 10 μmol/l, CMD-glycerol > 50 μmol/l, and CMD-LPR > 40. A CMD-LPR > 40 is referred to as metabolic distress. Recently, the pattern of mitochondrial dysfunction was defined as CMD-LPR > 30 together with CMD-pyruvate levels > 70 μmol/l. Important metabolic profiles in SAH patients are shown in Table 1.
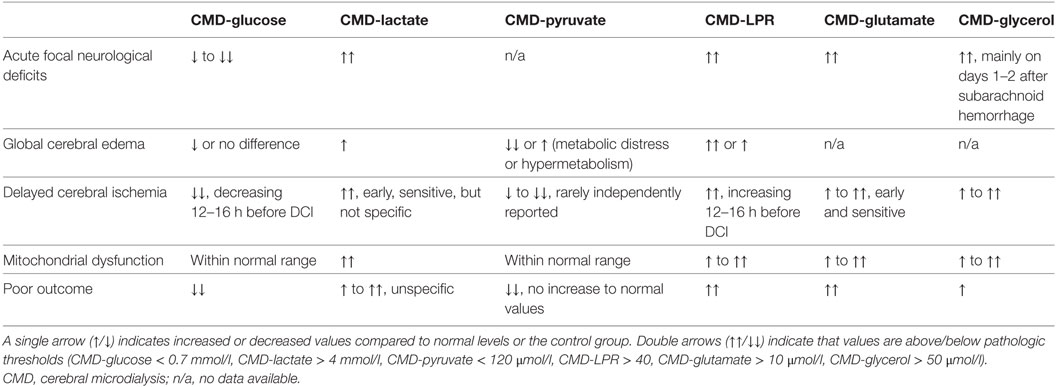
Table 1. Summary of brain metabolic patterns using CMD in SAH patients.
Results
The Clinical Use of CMD in the Acute Phase after SAH
The initial phase after SAH is commonly referred to as “early brain injury” and comprises the first 72 h after the bleeding (7), which is pathophysiologically related to, but temporally separated from the subsequent occurrence of delayed cerebral ischemia (DCI). EBI is the result of the initial hemorrhage leading to a cascade of ischemic injury, global brain swelling and early mitochondrial dysfunction, as well as pressure-related side effects due to parenchymal hematoma or early hydrocephalus.
Procedural Monitoring
One of the most feared complications early after SAH is aneurysm rebleeding, with the highest risk of occurrence within the first 24 h (8). Importantly, in most studies, metabolic monitoring using CMD started after the aneurysm had been secured (time to monitoring is given in Tables 3–8). While no CMD data are available during coiling, several studies have investigated changes in cerebral metabolism during aneurysm surgery (Table 2) (9–13). Commonly used CMD sampling intervals between 10 and 60 min seem to be too imprecise to depict periprocedural changes in brain metabolism (9–11), although increases in CMD-LPR and CMD-glutamate levels following prolonged artery clipping and ischemic complications were reported (9, 10). The largest study, including 38 aneurysmal SAH patients, found no significant differences in metabolic markers of brain tissue ischemia during transient artery occlusion (median occlusion time was 14 min) (11). In two patients with a prolonged occlusion time of more than 30 min, a pronounced increase of CMD-glutamate was observed (11). The feasibility of rapid-sampling microdialysis (obtaining values up to every 30 s) was investigated during temporal lobe retraction and transient artery occlusion (13). While metabolic changes reached a maximum after 3–10 min during temporal lobe retraction, increasing CMD-lactate and decreasing CMD-glucose levels were observed until clip removal during artery occlusion (13). A higher sampling frequency in CMD monitoring may help to define a threshold for ischemia to prevent irreversible tissue damage during aneurysm obliteration.
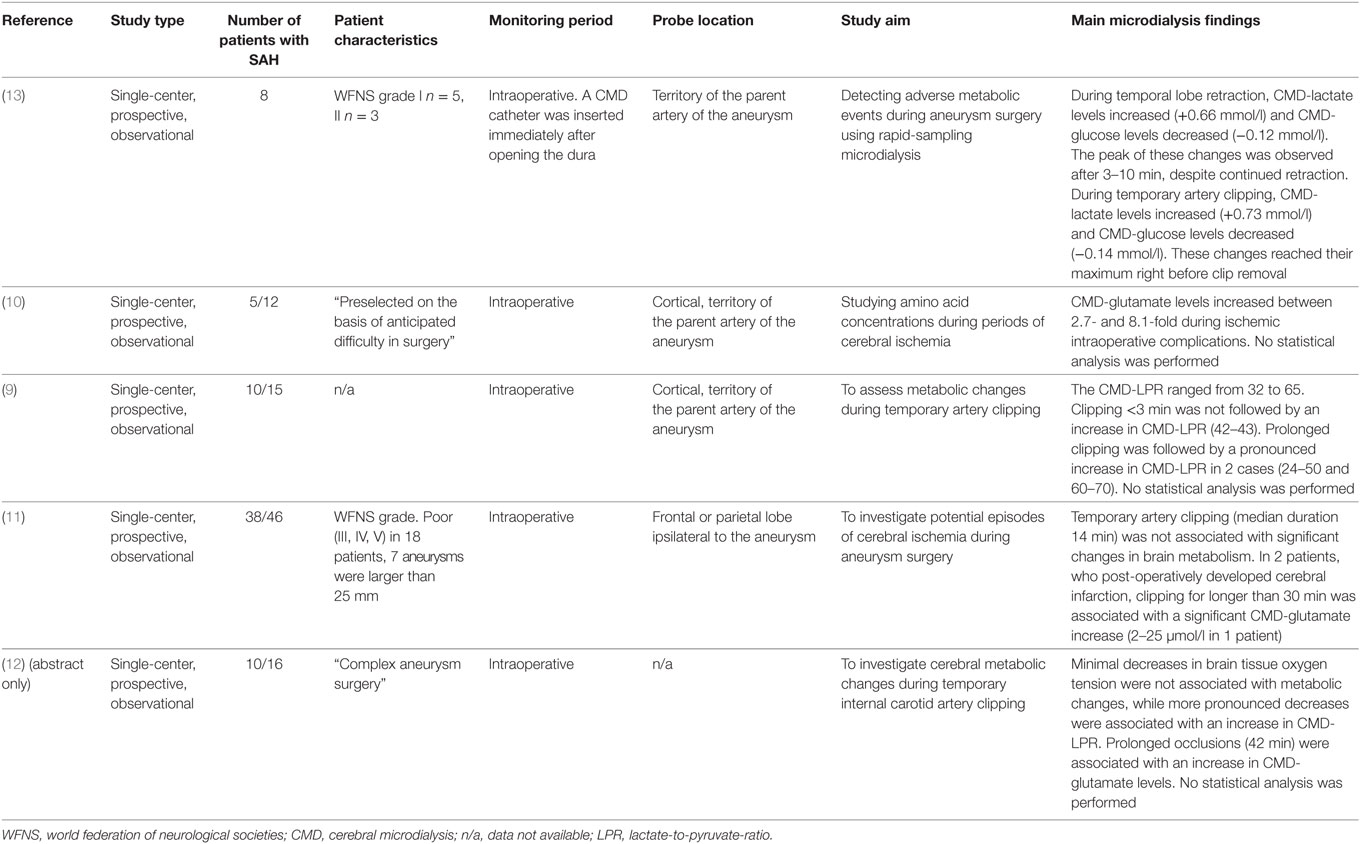
Table 2. Brain metabolism during aneurysm surgery.
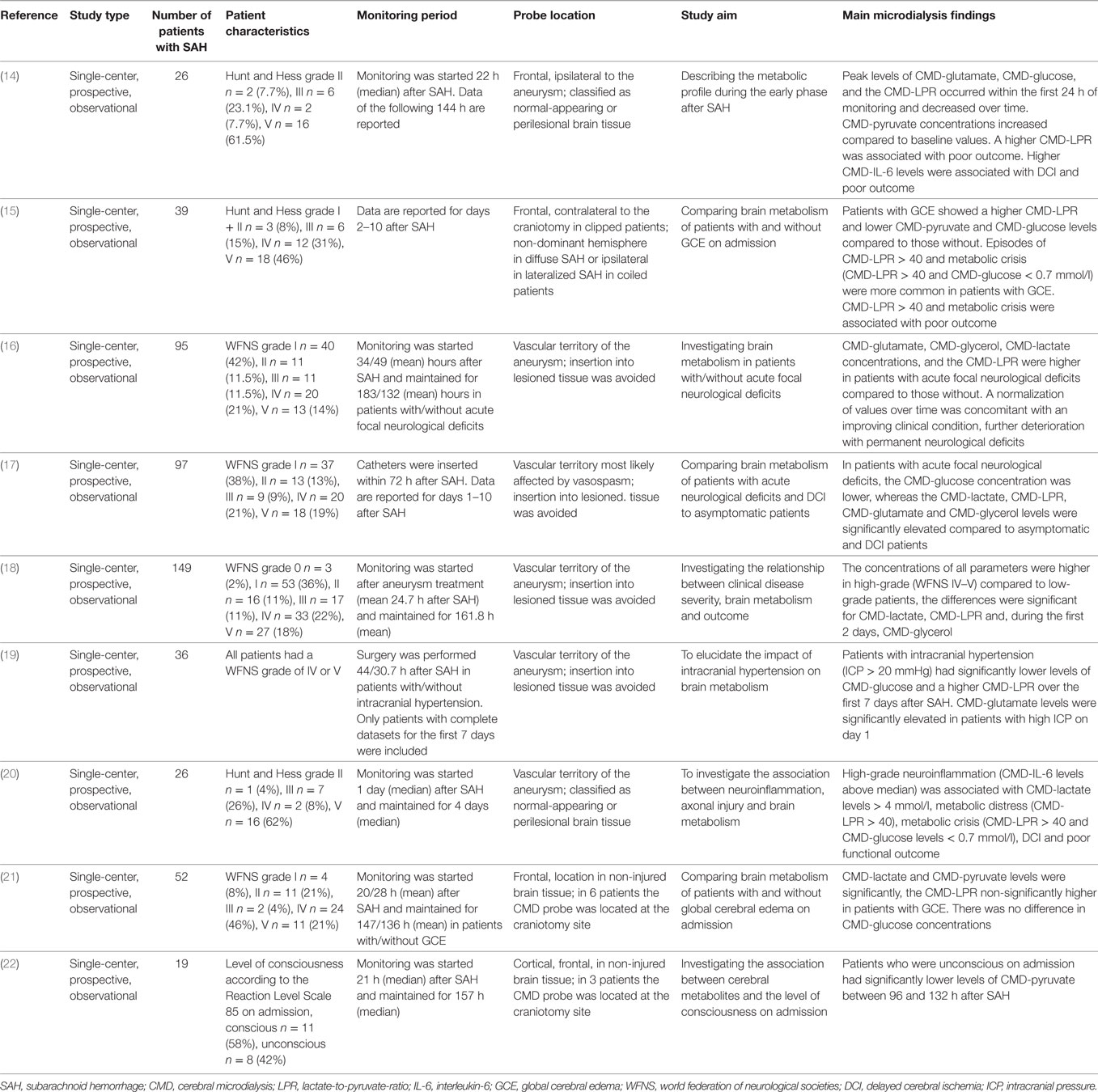
Table 3. The clinical use of CMD in the acute phase after SAH.
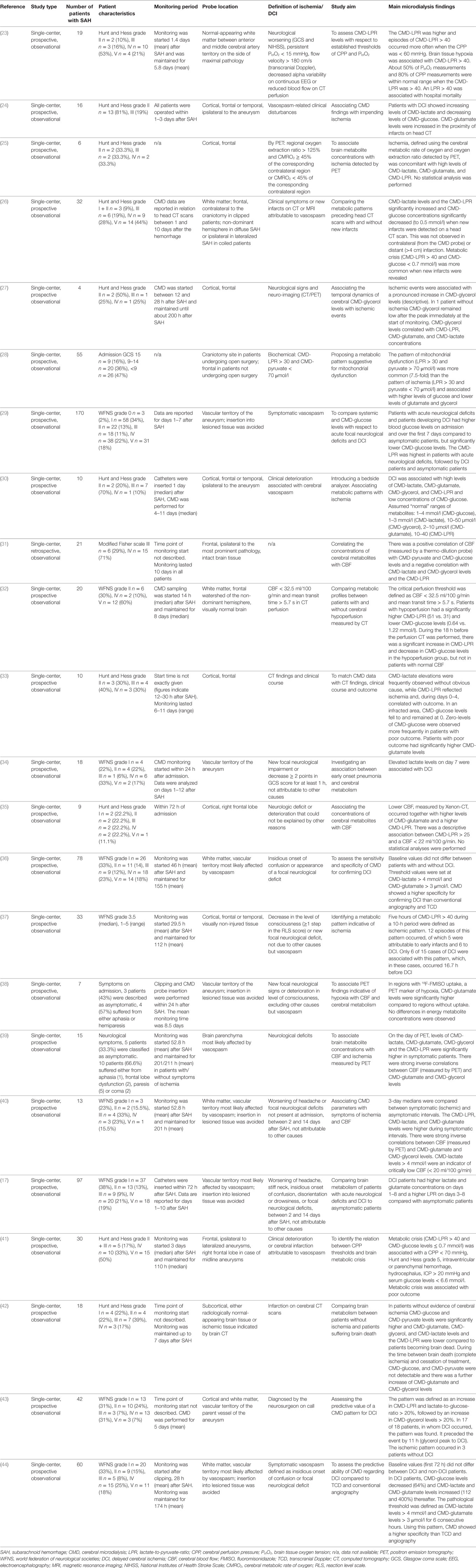
Table 4. The clinical use of CMD as a marker of cerebral hypoperfuison and DCI in SAH patients.
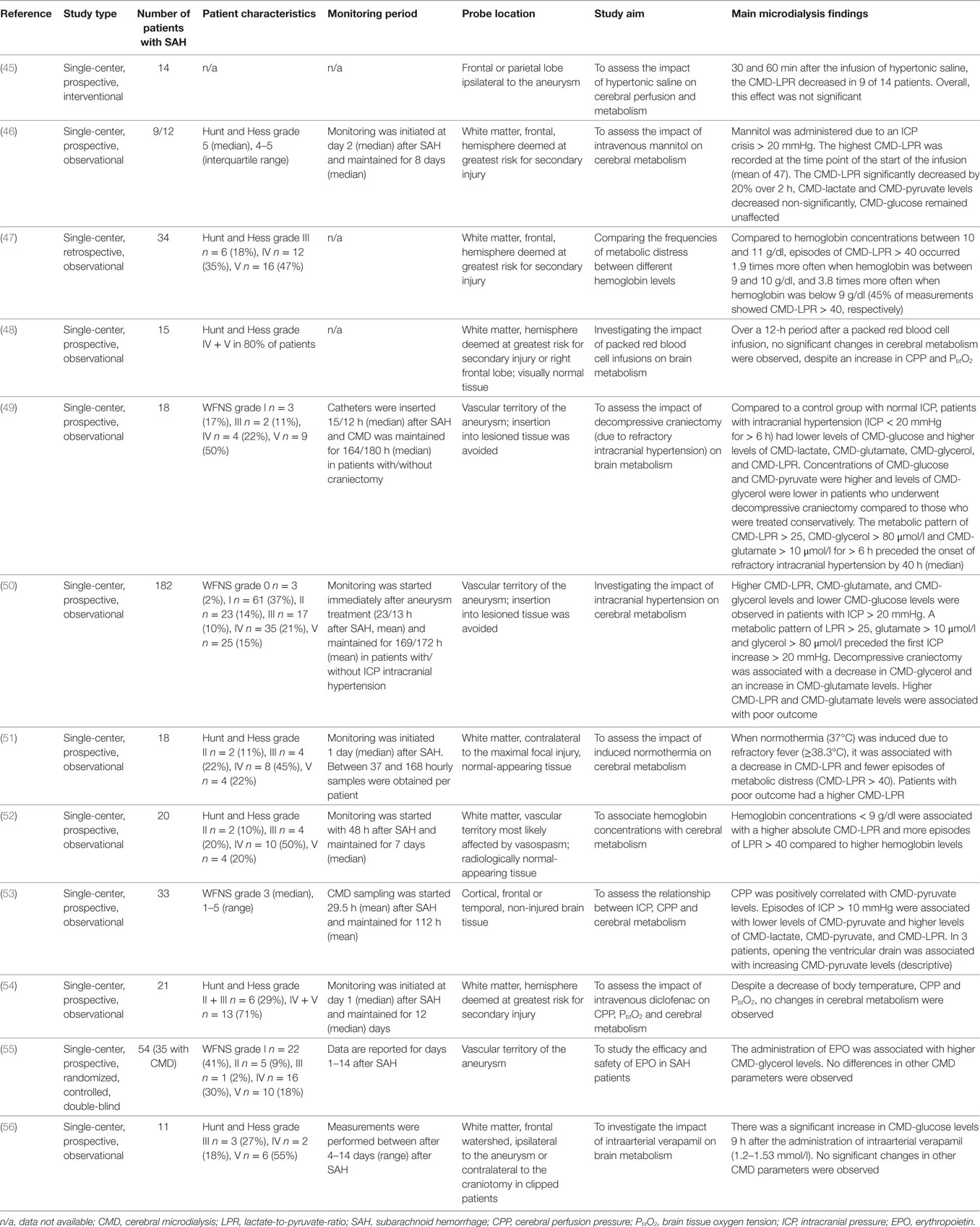
Table 5. CMD in monitoring treatment effects in SAH patients.
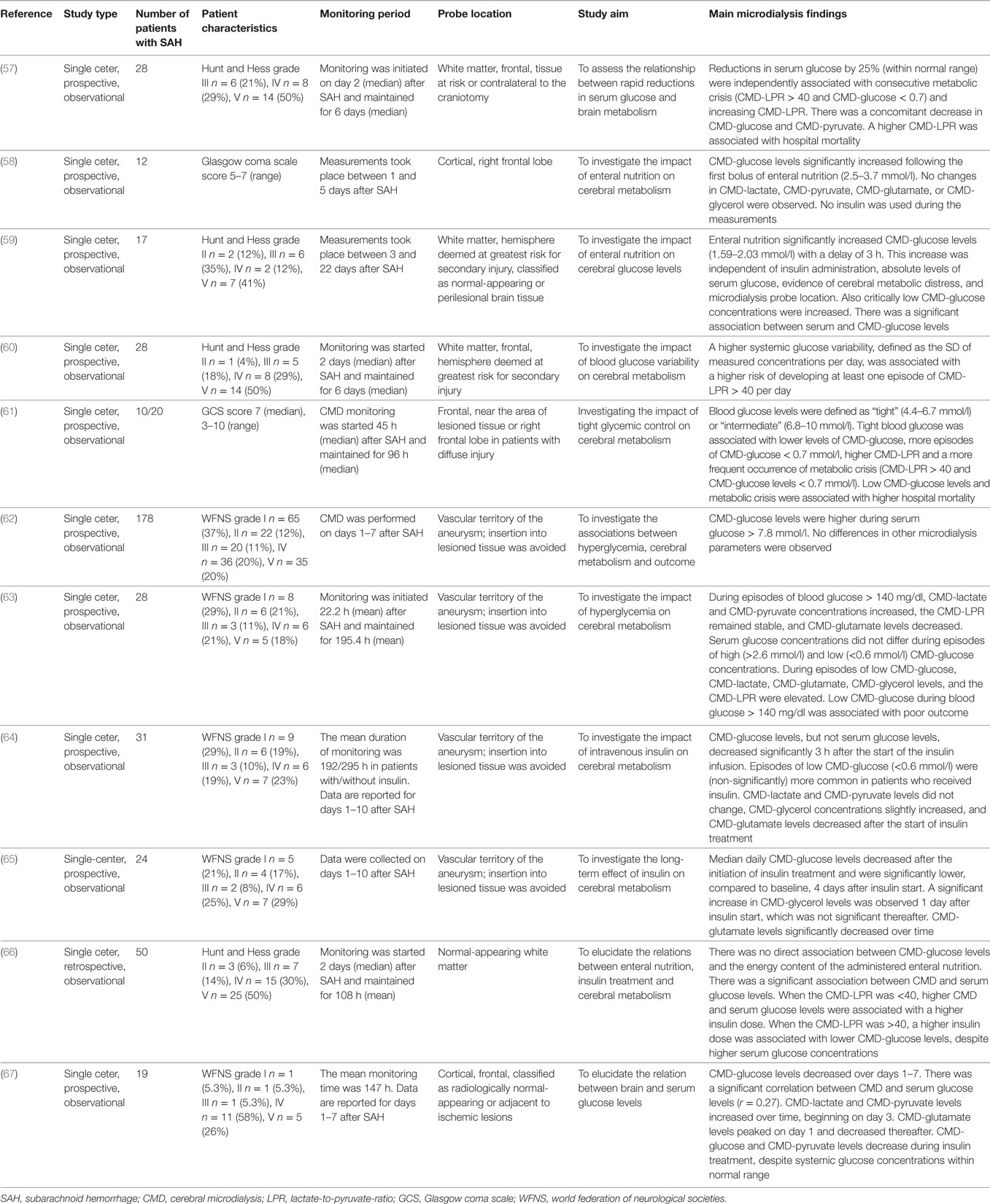
Table 6. CMD and systemic glucose management in SAH patients.
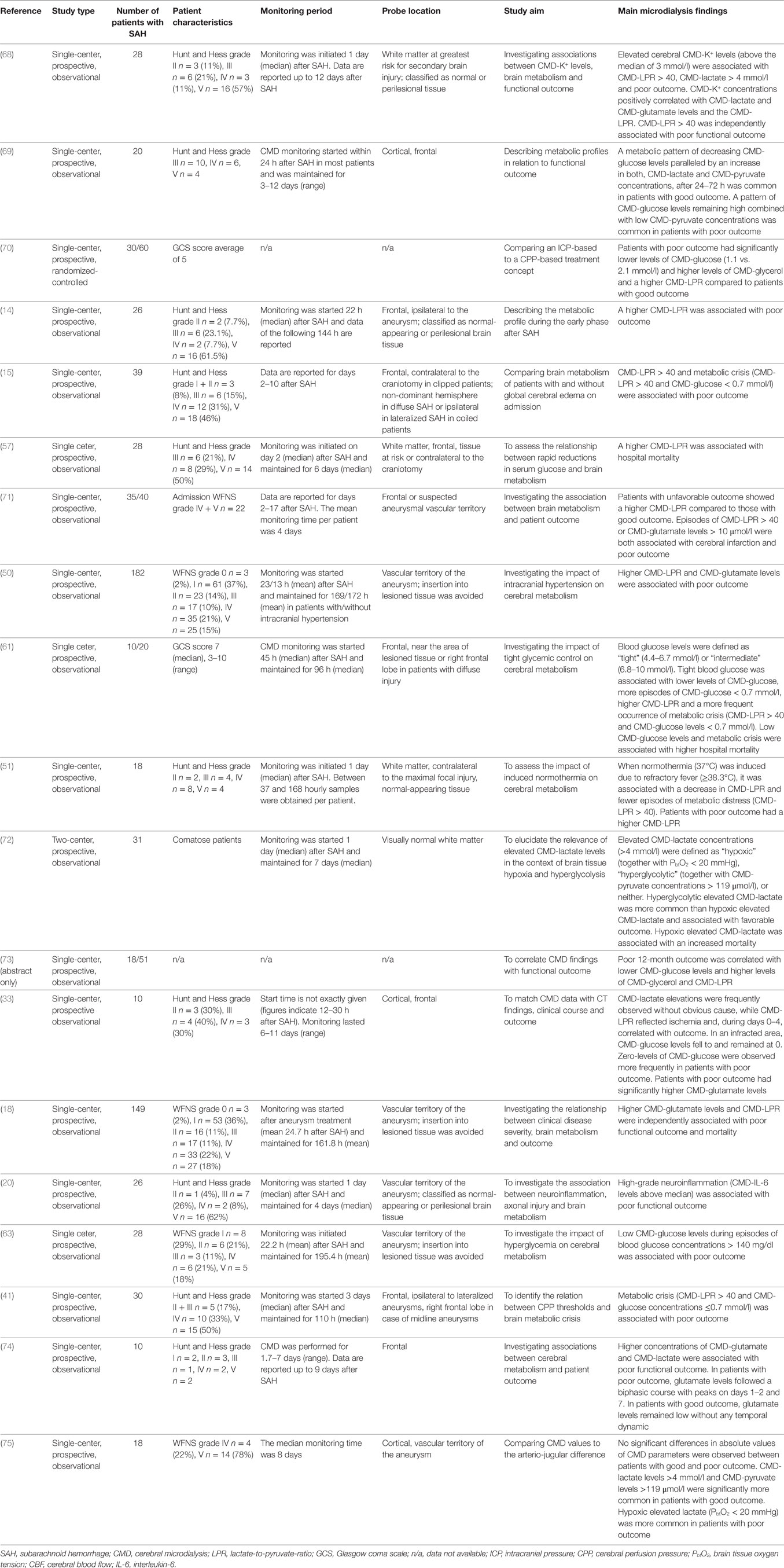
Table 7. Brain metabolism and outcome after SAH.
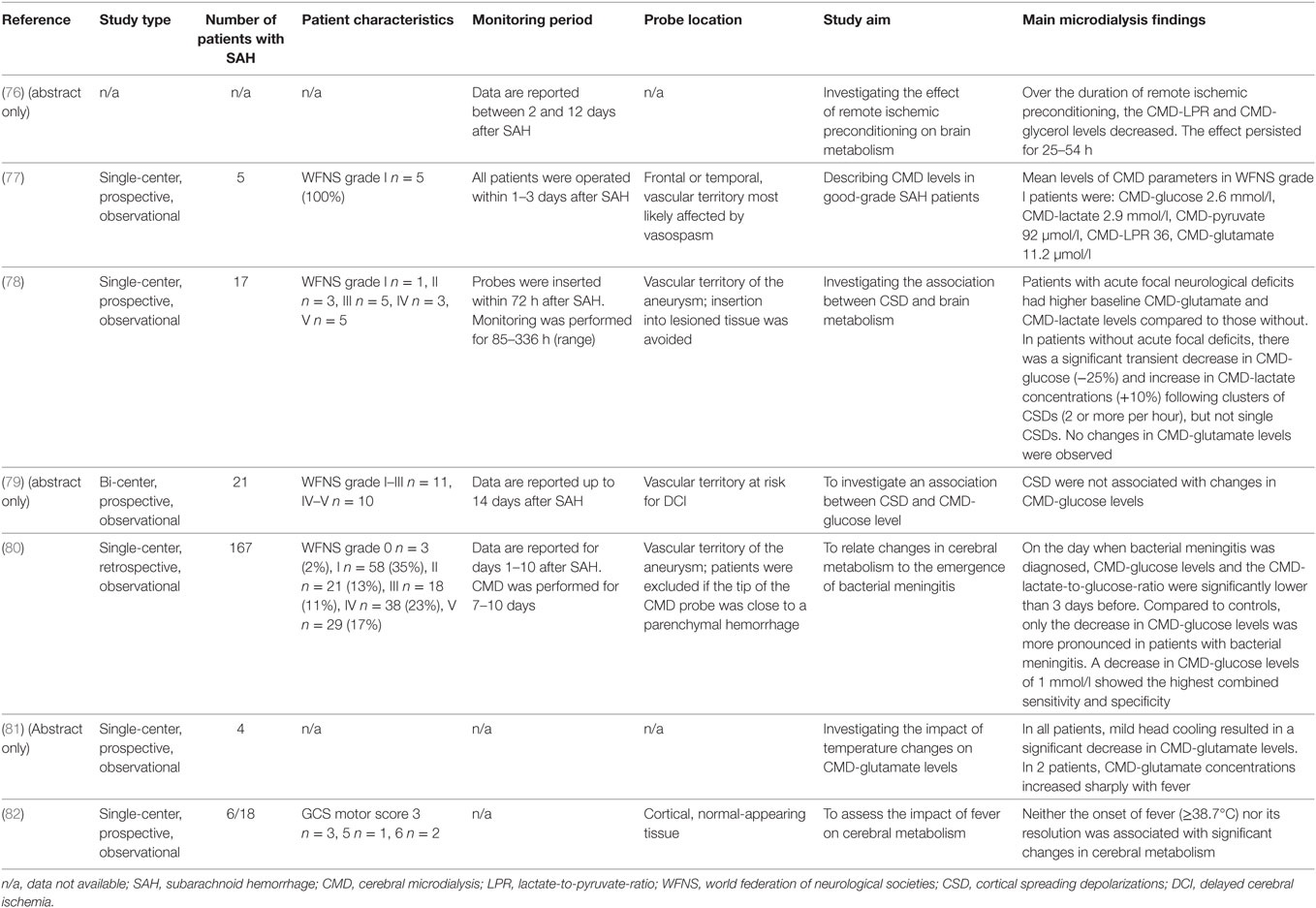
Table 8. CMD findings not directly related to the discussed topics.
Post-Procedural Monitoring
We identified nine studies focusing on the early phase after aneurysm treatment (Table 3) (14–22). In summary, CMD-LPR > 40 during the early phase is a sensitive marker for poor clinical grade on admission (18), radiological evidence of global cerebral edema (15), intracranial hypertension (19), and poor 3-month outcome (14). Critically, low levels of CMD-glucose were associated with acute neurological deficits (16). Trend analysis may indicate the clinical course with “normalization of CMD-parameters” being associated with clinical improvement and pathological evolution of brain metabolism with permanent neurological deficits (16).
CMD and Acute Focal Neurological Deficits after SAH
Most patients underwent aneurysm clipping. In a study including 26 poor-grade SAH patients (68% Hunt and Hess grade IV–V), CMD-LPR and CMD-glutamate were highest at the start of neuromonitoring, indicating metabolic distress (mean CMD-LPR > 40) and excitotoxicity, and significantly decreased thereafter (14). A higher CMD-LPR was associated with poor 3-month outcome (modified Rankin scale 4–6). CMD-glucose significantly decreased, however, did not reach critically low levels in this cohort with systemic glucose levels of 135–150 mg/dl (7.5–8.3 mmol/l) (14). A study including 149 SAH patients, of whom 89 (60%) were admitted with good clinical grades (WFNS grades ≤ 3), reported higher CMD-LPR and CMD-lactate levels in poor-grade patients, already at the start of neuromonitoring, compared to good-grade patients (18). Moreover, higher CMD-glycerol levels were reported in poor-grade patients, significantly decreasing over the first days (18).
In another study including 97 SAH patients, the authors compared patients with and without acute focal neurological deficits immediately after SAH due to SAH-related parenchymal hematoma and/or perioperative/periinterventional ischemia. In patients with focal deficits, CMD-lactate, CMD-LPR, CMD-glutamate, and CMD-glycerol were pathologically increased throughout the first week, whereas these parameters remained within normal range in patients without acute neurological deficits (16, 17). CMD-glucose levels were significantly lower in patients with acute focal deficits (16, 17), despite higher blood glucose concentrations (overall mean blood glucose levels were approximately 7.2–8.3 mmol/l = 130–150 mg/dl) (29). Regarding temporal dynamics, a trend toward normalization of CMD values was associated with clinical improvement, whereas further deterioration was associated with permanent neurological deficits in patients with acute focal deficits (16).
CMD and Admission Global Cerebral Edema
Two studies compared cerebral metabolic changes in patients with and without global cerebral edema (GCE) diagnosed by CT-imaging of the brain. Helbok et al. found an association between a higher frequency of metabolic crisis (significantly higher LPR, lower brain glucose levels) and GCE (15). Zetterling et al. described a pattern of cerebral hypermetabolism (higher lactate and pyruvate levels, no significant differences in LPR and glucose levels) in GCE patients (21). Despite this discrepancy, these findings indicate altered brain energy metabolism in SAH patients with GCE. Intracranial hypertension (ICP > 20 mmHg), often a result of brain edema or focal lesions, was associated with a pathologically elevated LPR (>40) and significantly lower CMD-glucose levels (19).
The Clinical Use of CMD as a Marker of Cerebral Hypoperfuison and DCI in SAH Patients
Delayed cerebral ischemia occurs in up to 30% of SAH patients, mostly between 4 and 10 days after the hemorrhage, and was defined by an international group of experts as either clinical deterioration or cerebral infarction not attributable to other causes (83). In the literature published before 2010, we found a considerable heterogeneity in the definition of delayed ischemia after SAH. Detailed information on the definition used in individual trials is given in Table 4.
Several studies investigated metabolic changes associated with parameters of cerebral perfusion, including CBF, CPP, and imaging surrogates. In summary, a negative correlation between CBF and CMD-lactate, CMD-LPR, CMD-glutamate, and CMD-glycerol has been described, while CMD-pyruvate and CMD-glucose are commonly positively correlated with CBF (25, 31, 32, 35, 38–40). Other studies focused on DCI and found increases in CMD-lactate and CMD-glutamate as early sensitive markers (17). Metabolic derangement with increasing CMD-LPR (> 40) and decreasing CMD-glucose (< 0.7 mmol/l) may occur up to 16 h before DCI onset (26, 32). In the following, we give detailed information on some studies, all studies are listed in Table 4.
Schmidt et al. found an association of CPP < 70 mmHg with a higher incidence of metabolic crisis (CMD-LPR > 40 and CMD-glucose levels < 0.7 mmol/l) and further worsening of brain metabolism at lower CPP values (41). Similarly, a higher CMD-LPR and increased episodes of metabolic distress (CMD-LPR > 40) were observed at a CPP < 60 mmHg in another study including 19 SAH patients (23). However, it is important to elaborate that a high CMD-LPR may also indicate metabolic distress in the absence of ischemia. In this regard, Jacobsen et al. defined an elevated LPR (>30), together with pyruvate levels within normal range (>70 μmol/l) as mitochondrial dysfunction and found this pattern to be 7.5-fold more common than metabolic changes indicative for cerebral ischemia (LPR > 30 and CMD-pyruvate < 70 μmol/l) (28). Mitochondrial dysfunction was moreover associated with higher levels of CMD-glucose and lower levels of CMD-glutamate and CMD-glycerol compared to ischemic episodes.
In poor-grade SAH patients requiring sedation and mechanical ventilation, neurological deterioration may not be detected. In these patients, CMD provides useful information on the metabolic state of the injured brain and may even indicate metabolic changes before DCI occurs. Sarrafzadeh et al. reported higher levels of CMD-lactate and CMD-glutamate in patients with DCI compared to those who did not develop DCI already on day 1 and throughout the first week after the bleeding (17). A higher CMD-LPR was observed from day 3 on (17). This is in line with findings by Nilsson et al., who concluded that CMD-lactate and CMD-glutamate may be the earliest and most sensitive markers of ischemia, followed by the CMD-LPR and CMD-glycerol (30). Furthermore, lower brain glucose levels during the first 3 days after SAH were reported in DCI patients, despite higher blood glucose concentrations when compared to patients who did not develop DCI (29).
Changes in CMD parameters were observed even 12–16 h before the occurrence of DCI and included a significant increase in CMD-LPR to values >40 and decreases in CMD-glucose to levels <0.7 mmol/l (26, 32, 37). At a comparable time point, also elevations of CMD-lactate, CMD-glutamate, and CMD-glycerol were reported (43, 44). The sensitivity of the method is limited due to the local measurement. Therefore, ischemia occurring in brain tissue distant to the monitoring catheter may not be detected (26, 37). Lack of specificity results from metabolic distress secondary to non-ischemic CMD-LPR elevations and hyperglycolytic (non-ischemic) lactate increase (28, 72). Nevertheless, CMD had a higher specificity for predicting DCI than transcranial ultrasound and conventional angiography (44). Unfortunately, most studies do not report on the effect of current treatment strategies for DCI (induced hypertension, intraarterial spasmolysis). Sarrafzadeh et al. reported a decrease in CMD-glutamate levels, but no change in other parameters, associated with “triple-H therapy” (17). Further research focusing on metabolic changes following such interventions is surely warranted.
In summary, studies assessing CBF directly in the region around the CMD probe revealed a highly consistent metabolic pattern of increased CMD-lactate, CMD-glutamate, CMD-glycerol levels, and CMD-LPR during episodes of hypoperfusion, whereas CMD-glucose and CMD-pyruvate levels were positively correlated with CBF. CMD-lactate and CMD-glutamate are early and sensitive markers of impending DCI, but lack of specificity. However, CMD parameters showed a higher specificity for predicting DCI than transcranial ultrasound and conventional angiography (37, 43, 44). Regarding trend analyses, a CMD-LPR increase above 40 and decreasing CMD-glucose concentrations preceded the occurrence of delayed ischemia by several hours (26, 32); however, ischemia that occurred remote from the CMD probe or in the contralateral hemisphere was not detected (26).
CMD in Monitoring Treatment Effects
Several studies have investigated the effect of pharmacological and non-pharmacological interventions on cerebral metabolism as summarized in Table 5. Studied interventions included the treatment of intracranial hypertension, either with osmotherapy (45, 46), by cerebrospinal fluid drainage via external ventriculostomy (53), or by decompressive craniectomy (49, 50), fever treatment with diclofenac or targeted temperature management (51, 52, 54), the management of anemia and administration of packed red blood cells (47, 48, 52), and the administration of erythropoietin or verapamil (55, 56). The impact of enteral nutrition and insulin on brain metabolism will be discussed in the next chapter.
CMD and Systemic Glucose Management
There is still an ongoing debate on the optimal systemic glucose target in critically ill patients (84, 85). CMD-glucose levels represent the net effect of delivered glucose and glucose consumption. Little is known about the impact of glucose transport and diffusion in acutely brain injured patients.
Severe hyperglycemia (>200 mg/dl = 11.1 mmol/l) is associated with poor outcome in SAH patients (86). Some studies investigated the association between systemic and brain interstitial glucose levels (Table 6). Oddo et al. described the brain metabolic profile during episodes of “low” (<4.4 mmol/), “tight” (4.4–6.7 mmol/l), “intermediate” (6.8–10 mmol/l), and high blood glucose levels in a mixed population of neurocritical care patients, including 10 patients with SAH. Compared to intermediate systemic glucose levels, tight glycemic control was associated with lower CMD-glucose levels and more episodes of CMD-glucose < 0.7 mmol/l, as well as a higher CMD-LPR and more episodes of metabolic crisis (CMD-LPR > 40 and CMD-glucose concentrations < 0.7mmol/l). Metabolic crisis and CMD-glucose < 0.7 mmol/l were associated with higher hospital mortality (61). The significant association of systemic- and CMD-glucose is supported by the results of other groups (59, 66); however, some studies indicate a poor correlation (63, 67), especially in the injured brain. Decreased CMD-glucose is moreover observed when delivery is reduced (i.e., reduction in CBF) or under conditions of increased consumption (i.e., higher body temperature, seizures and the occurrence of cortical spreading depolarizations) (66, 78, 82).
Pathologically low CMD-glucose levels (<0.7 or <0.6 mmol/l) were associated with poor outcome in SAH patients (41, 61, 63). In the recent consensus statement on the use of CMD, clinical experts suggest to intervene when pathologically low CMD-glucose levels are detected (2). This attempt should only be made with respect to probe location (in healthy-appearing brain tissue on head computed tomography), baseline blood glucose levels, and in the absence of brain ischemia. Proposed interventions targeting higher systemic glucose levels include intravenous or enteral glucose administration and the reevaluation of insulin treatment (2).
While no data on intravenous glucose administration are available, three studies sought to investigate the impact of enteral feeding on cerebral metabolism. Schmidt et al. did not observe a direct relation between CMD-glucose levels and the energy content of the administered enteral nutrition (66). Other groups investigating the temporal association of enteral feeding and the metabolic profile revealed time-related increases in CMD-glucose levels not affecting other CMD parameters (58, 59). CMD-glucose levels even increased from critically low (<0.7 mmol/l) levels at baseline independent of probe location (59).
Insulin treatment was associated with a decrease of CMD-glucose independent of serum glucose levels, resulting in a higher incidence of critically low CMD-glucose levels (<0.6 mmol/l) (64, 67), especially under conditions of cerebral metabolic distress (LPR > 40) (66). Moreover, rapid reductions in serum glucose concentrations were associated with a decrease in CMD-glucose (57). In line with this, a higher serum glucose variability was associated with a higher rate of cerebral metabolic distress (60). In summary, tight glycemic control may be associated with pathologically low CMD-glucose levels (neuroglucopenia) in critically ill patients with acute neurologic injury. If brain metabolic monitoring indicates critically low glucose levels (i.e., <0.2 mmol/l) targeting a more liberal glucose regimen (110–150 mg/dl or up to 180 mg/dl) may be indicated.
CMD and Outcome
Studies reporting an association of CMD parameters with patient outcome are summarized in Table 7. The largest study includes 182 SAH patients and found higher CMD-LPR and CMD-glutamate values during the first week after SAH being significantly and independently associated with poor functional outcome after 12 months (50). Patients with poor functional 3–6 months outcome had significantly more episodes of CMD-LPR > 40 and CMD-glutamate levels > 10 μmol/l compared to those with favorable outcome (71). An elevated CMD-LPR was also associated with hospital mortality (57). Pathologically low CMD-glucose levels < 0.7 mmol/l were observed more often in patients with poor outcome and in those who died (41, 61, 63). Elevated CMD-lactate is a less specific marker for neuroprognostication, as both, associations with good (in a hyperglycolytic context) and poor (due to hypoxia) outcome were reported (72).
Trend analysis is important as a shift to hypermetabolism, indicated by an increase in both, CMD-lactate and CMD-pyruvate levels, was observed in patients with favorable outcome. Persistent low CMD-pyruvate levels without increase to normal values were associated with poor outcome (69). Persistent low CMD-glutamate levels were associated with good functional outcome, whereas increases at day 1 and day 7 were associated with poor outcome (74).
Discussion
This review demonstrates that CMD is a powerful tool providing almost continuous brain metabolic information at bedside. Based on the published literature, pathological changes in brain metabolism are associated with disease severity, mechanisms of primary and secondary brain injury, and poor long-term outcome after SAH.
As summarized in this review, the major limitation of the published literature is that CMD monitoring was reported mostly in small single-center trials of neurological and neurosurgical ICUs with extensive experience in the use of CMD as adjunct to other multimodal neuromonitoring techniques. In many centers, implementation of CMD as a clinical tool is still limited due to financial constraints and the lack of evidence to improve patient’s outcome. Future trials may address this gap in the literature by targeting brain metabolic parameters as endpoints to improve brain homeostasis as no monitoring tool can improve patient’s outcome unless coupled with a therapeutic intervention. On the other hand, there is a need to re-define commonly used outcome parameters and to move from a simplistic functional outcome score to a more sophisticated approach including neuropsychological testing, quality of life measures, and brain tissue outcome.
The invasiveness of CMD limits its application to poor-grade SAH patients. This implicates that this review merely summarizes brain metabolic changes of unconscious patients and results are not generalizable to all clinical grades. Based on the recently published consensus statement, the use of CMD is recommended in poor-grade SAH patients with prolonged ventilation and patients with secondary neurologic deterioration requiring mechanical ventilation.
Despite these limitations metabolic monitoring in the early and subacute phase after SAH provides insight into pathophysiological mechanisms of primary and secondary brain injury on the brain tissue level. As shown in our review, the detection of impeding ischemia is the most extensively studied application of CMD in SAH patients. CMD has been shown to have the potential to be used as early warning tool of brain tissue ischemia hours before the insult (17, 43, 44), even if clinically silent (26). In this regard, it is important to mention that we recognized a large variability in the definition of DCI throughout the published literature. Although more homogeneity was detected after the year 2010 when Vergouwen et al. defined DCI based on clinical and/or radiographic criteria (83), earlier studies are difficult to compare as information on radiographic evidence of new infarctions related to vasospasm were commonly not reported.
Another limitation is that microdialysis probe location differed in individual studies by means of targeting either brain tissue ipsilateral to the bleeding aneurysm or the contralateral hemisphere and monitoring the cortical gray matter vs. subcortical white matter. Furthermore, probe location was rarely adequately addressed as covariate in multivariate models, which limits the interpretation of results and conclusions. It is important to mention that brain chemistry can only be interpreted correctly based on the relation to focal injury on brain imaging (“normal appearing brain tissue” vs. “perilesional” vs. “intralesional”) (2). As recommended by clinical experts, future trials reporting microdialysis data should at least include information on catheter location, the catheter type used, perfusion fluid composition, the perfusion fluid flow rate, and time from ictus to monitoring (2).
The interpretation of CMD-derived information requires profound knowledge of the complex underlying pathophysiology. As shown in Table 1, metabolic changes may be ambiguous, as similar patterns may indicate different underlying pathophysiological processes. However, taking into account, the occurrence of specific patterns in relation to the bleeding onset may help to discriminate between early and late onset ischemic patterns or patterns of mitochondrial dysfunction. A clinical guidance is given in Table 1 and may be combined with other neuromonitoring parameters such as brain tissue oxygenation, ICP, and CBF.
Clinical implication of microdialysis monitoring besides the detection of secondary ischemic insults include guidance of systemic glucose management, CPP optimization, defining individual transfusion thresholds and monitoring of brain chemistry during pharmacological and non-pharmacological interventions (41, 49, 51, 52, 57, 61, 63, 67).
The only treatment decision based on changes in brain metabolism currently recommended by clinical experts is the treatment of low cerebral glucose taking into account baseline systemic glucose concentration, catheter location, and the etiology of neuroglucopenia. In the knowledge of its potential, there is a need to integrate brain metabolic changes and to define CMD-based endpoints in future clinical trials.
Conclusion
Cerebral microdialysis is used in the clinical management of severe SAH together with ICP, PbtO2, and other neuromonitoring parameters. In the knowledge of its limitations, this method provides a novel insight into pathophysiological processes of primary and secondary brain injury. Recent consensus on microdialysis monitoring paves the way for improved protocols and targeted interventions. The major task for future research integrates the prospective evaluation of predefined interventions to improve brain tissue physiology aiming toward a personalized management of SAH patients in the future.
Author Contributions
RH was involved in the idea, design, data acquisition, article selection, writing, interpretation of data, and final revision of the manuscript. MK was involved in article selection, writing, interpretation of data, and final revision of the manuscript. AS, MG, and VR were involved in the data acquisition, interpretation of data, and final revision of the manuscript. BP, RB, and ES were involved in article selection, writing, interpretation of data, and final revision of the manuscript. All authors read and approved the final version of the manuscript and are accountable for the content, data interpretation, and data accuracy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Persson L, Hillered L. Chemical monitoring of neurosurgical intensive care patients using intracerebral microdialysis. J Neurosurg (1992) 76(1):72–80. doi:10.3171/jns.1992.76.1.0072
2. Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med (2015) 41(9):1517–28. doi:10.1007/s00134-015-3930-y
3. Ungerstedt U. Microdialysis – principles and applications for studies in animals and man. J Intern Med (1991) 230(4):365–73. doi:10.1111/j.1365-2796.1991.tb00459.x
4. Hutchinson PJ, O’Connell MT, Nortje J, Smith P, Al-Rawi PG, Gupta AK, et al. Cerebral microdialysis methodology – evaluation of 20 kDa and 100 kDa catheters. Physiol Meas (2005) 26(4):423–8. doi:10.1088/0967-3334/26/4/008
5. Hutchinson PJ, O’Connell MT, Al-Rawi PG, Maskell LB, Kett-White R, Gupta AK, et al. Clinical cerebral microdialysis: a methodological study. J Neurosurg (2000) 93(1):37–43. doi:10.3171/jns.2000.93.1.0037
6. Bellander BM, Cantais E, Enblad P, Hutchinson P, Nordstrom CH, Robertson C, et al. Consensus meeting on microdialysis in neurointensive care. Intensive Care Med (2004) 30(12):2166–9. doi:10.1007/s00134-004-2461-8
7. Sehba FA, Pluta RM, Zhang JH. Metamorphosis of subarachnoid hemorrhage research: from delayed vasospasm to early brain injury. Mol Neurobiol (2011) 43(1):27–40. doi:10.1007/s12035-010-8155-z
8. Starke RM, Connolly ES Jr, Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid, Hemorrhage. Rebleeding after aneurysmal subarachnoid hemorrhage. Neurocrit Care (2011) 15(2):241–6. doi:10.1007/s12028-011-9581-0
9. Hutchinson PJ, Al-Rawi PG, O’Connell MT, Gupta AK, Maskell LB, Hutchinson DB, et al. Monitoring of brain metabolism during aneurysm surgery using microdialysis and brain multiparameter sensors. Neurol Res (1999) 21(4):352–8. doi:10.1080/01616412.1999.11740943
10. Hutchinson PJ, O’Connell MT, Al-Rawi PG, Kett-White CR, Gupta AK, Maskell LB, et al. Increases in GABA concentrations during cerebral ischaemia: a microdialysis study of extracellular amino acids. J Neurol Neurosurg Psychiatry (2002) 72(1):99–105. doi:10.1136/jnnp.72.1.99
11. Kett-White R, Hutchinson PJ, Al-Rawi PG, Czosnyka M, Gupta AK, Pickard JD, et al. Cerebral oxygen and microdialysis monitoring during aneurysm surgery: effects of blood pressure, cerebrospinal fluid drainage, and temporary clipping on infarction. J Neurosurg (2002) 96(6):1013–9. doi:10.3171/jns.2002.96.6.1013
12. Kett-White R, Hutchinson PJ, al-Rawi PG, Gupta AK, O’Connell MT, Pickard JD, et al. Extracellular lactate/pyruvate and glutamate changes in patients during per-operative episodes of cerebral ischaemia. Acta Neurochir Suppl (2002) 81:363–5.
13. Bhatia R, Hashemi P, Razzaq A, Parkin MC, Hopwood SE, Boutelle MG, et al. Application of rapid-sampling, online microdialysis to the monitoring of brain metabolism during aneurysm surgery. Neurosurgery (2006) 58(4 Suppl 2):ONS-313–20; discussion ONS-321. doi:10.1227/01.NEU.0000208963.42378.83
14. Helbok R, Schiefecker AJ, Beer R, Dietmann A, Antunes AP, Sohm F, et al. Early brain injury after aneurysmal subarachnoid hemorrhage: a multimodal neuromonitoring study. Crit Care (2015) 19:75. doi:10.1186/s13054-015-0809-9
15. Helbok R, Ko SB, Schmidt JM, Kurtz P, Fernandez L, Choi HA, et al. Global cerebral edema and brain metabolism after subarachnoid hemorrhage. Stroke (2011) 42(6):1534–9. doi:10.1161/STROKEAHA.110.604488
16. Sarrafzadeh A, Haux D, Sakowitz O, Benndorf G, Herzog H, Kuechler I, et al. Acute focal neurological deficits in aneurysmal subarachnoid hemorrhage: relation of clinical course, CT findings, and metabolite abnormalities monitored with bedside microdialysis. Stroke (2003) 34(6):1382–8. doi:10.1161/01.STR.0000074036.97859.02
17. Sarrafzadeh AS, Sakowitz OW, Kiening KL, Benndorf G, Lanksch WR, Unterberg AW. Bedside microdialysis: a tool to monitor cerebral metabolism in subarachnoid hemorrhage patients? Crit Care Med (2002) 30(5):1062–70. doi:10.1097/00003246-200205000-00018
18. Sarrafzadeh A, Haux D, Kuchler I, Lanksch WR, Unterberg AW. Poor-grade aneurysmal subarachnoid hemorrhage: relationship of cerebral metabolism to outcome. J Neurosurg (2004) 100(3):400–6. doi:10.3171/jns.2004.100.3.0400
19. Sarrafzadeh AS, Thomale UW, Haux D, Unterberg AW. Cerebral metabolism and intracranial hypertension in high grade aneurysmal subarachnoid haemorrhage patients. Acta Neurochir Suppl (2005) 95:89–92. doi:10.1007/3-211-32318-X_19
20. Schiefecker AJ, Dietmann A, Beer R, Pfausler B, Lackner P, Kofler M, et al. Neuroinflammation is associated with brain extracellular TAU-protein release after spontaneous subarachnoid hemorrhage. Curr Drug Targets (2017) 18(12):1408–16. doi:10.2174/1389450117666160201111804
21. Zetterling M, Hallberg L, Hillered L, Karlsson T, Enblad P, Ronne Engstrom E. Brain energy metabolism in patients with spontaneous subarachnoid hemorrhage and global cerebral edema. Neurosurgery (2010) 66(6):1102–10. doi:10.1227/01.NEU.0000370893.04586.73
22. Zetterling M, Hillered L, Samuelsson C, Karlsson T, Enblad P, Ronne-Engstrom E. Temporal patterns of interstitial pyruvate and amino acids after subarachnoid haemorrhage are related to the level of consciousness – a clinical microdialysis study. Acta Neurochir (Wien) (2009) 151(7):771–80; discussion 780. doi:10.1007/s00701-009-0384-4
23. Chen HI, Stiefel MF, Oddo M, Milby AH, Maloney-Wilensky E, Frangos S, et al. Detection of cerebral compromise with multimodality monitoring in patients with subarachnoid hemorrhage. Neurosurgery (2011) 69(1):53–63; discussion 63. doi:10.1227/NEU.0b013e3182191451
24. De Micheli E, Pinna G, Piovan E, Prisco R, Hillered L, Persson L, et al. Monitoring subtle neurometabolic changes in subarachnoid hemorrhage patients using microdialysis: a study on 16 cases. Acta Neurochir Suppl (2001) 77:149–53.
25. Enblad P, Valtysson J, Andersson J, Lilja A, Valind S, Antoni G, et al. Simultaneous intracerebral microdialysis and positron emission tomography in the detection of ischemia in patients with subarachnoid hemorrhage. J Cereb Blood Flow Metab (1996) 16(4):637–44. doi:10.1097/00004647-199607000-00014
26. Helbok R, Madineni RC, Schmidt MJ, Kurtz P, Fernandez L, Ko SB, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care (2011) 14(2):162–7. doi:10.1007/s12028-010-9472-9
27. Hillered L, Valtysson J, Enblad P, Persson L. Interstitial glycerol as a marker for membrane phospholipid degradation in the acutely injured human brain. J Neurol Neurosurg Psychiatry (1998) 64(4):486–91. doi:10.1136/jnnp.64.4.486
28. Jacobsen A, Nielsen TH, Nilsson O, Schalen W, Nordstrom CH. Bedside diagnosis of mitochondrial dysfunction in aneurysmal subarachnoid hemorrhage. Acta Neurol Scand (2014) 130(3):156–63. doi:10.1111/ane.12258
29. Kerner A, Schlenk F, Sakowitz O, Haux D, Sarrafzadeh A. Impact of hyperglycemia on neurological deficits and extracellular glucose levels in aneurysmal subarachnoid hemorrhage patients. Neurol Res (2007) 29(7):647–53. doi:10.1179/016164107X248983
30. Nilsson OG, Brandt L, Ungerstedt U, Saveland H. Bedside detection of brain ischemia using intracerebral microdialysis: subarachnoid hemorrhage and delayed ischemic deterioration. Neurosurgery (1999) 45(5):1176–84; discussion 1184–5. doi:10.1097/00006123-199911000-00032
31. Papadopoulos D, Filippidis A, Krommidas G, Vretzakis G, Paterakis K, Komnos A, et al. Regional cerebral blood flow and cellular environment in subarachnoid hemorrhage: a thermal Doppler flowmetry and microdialysis study. Neurol Neurochir Pol (2017) 51(1):66–71. doi:10.1016/j.pjnns.2016.11.002
32. Patet C, Quintard H, Zerlauth JB, Maibach T, Carteron L, Suys T, et al. Bedside cerebral microdialysis monitoring of delayed cerebral hypoperfusion in comatose patients with poor grade aneurysmal subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry (2017) 88(4):332–8. doi:10.1136/jnnp-2016-313766
33. Persson L, Valtysson J, Enblad P, Warme PE, Cesarini K, Lewen A, et al. Neurochemical monitoring using intracerebral microdialysis in patients with subarachnoid hemorrhage. J Neurosurg (1996) 84(4):606–16. doi:10.3171/jns.1996.84.4.0606
34. Radolf S, Smoll N, Drenckhahn C, Dreier JP, Vajkoczy P, Sarrafzadeh AS. Cerebral lactate correlates with early onset pneumonia after aneurysmal SAH. Transl Stroke Res (2014) 5(2):278–85. doi:10.1007/s12975-013-0292-z
35. Rostami E, Engquist H, Johnson U, Howells T, Ronne-Engstrom E, Nilsson P, et al. Monitoring of cerebral blood flow and metabolism bedside in patients with subarachnoid hemorrhage – a Xenon-CT and Microdialysis Study. Front Neurol (2014) 5:89. doi:10.3389/fneur.2014.00089
36. Sakowitz OW, Sarrafzadeh AS, Benndorf G, Lanksch WR, Unterberg AW. On-line microdialysis following aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl (2001) 77:141–4.
37. Samuelsson C, Hillered L, Enblad P, Ronne-Engstrom E. Microdialysis patterns in subarachnoid hemorrhage patients with focus on ischemic events and brain interstitial glutamine levels. Acta Neurochir (Wien) (2009) 151(5):437–46; discussion 446. doi:10.1007/s00701-009-0265-x
38. Sarrafzadeh AS, Nagel A, Czabanka M, Denecke T, Vajkoczy P, Plotkin M. Imaging of hypoxic-ischemic penumbra with (18)F-fluoromisonidazole PET/CT and measurement of related cerebral metabolism in aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab (2010) 30(1):36–45. doi:10.1038/jcbfm.2009.199
39. Sarrafzadeh A, Haux D, Plotkin M, Ludemann L, Amthauer H, Unterberg A. Bedside microdialysis reflects dysfunction of cerebral energy metabolism in patients with aneurysmal subarachnoid hemorrhage as confirmed by 15 O-H2 O-PET and 18 F-FDG-PET. J Neuroradiol (2005) 32(5):348–51. doi:10.1016/S0150-9861(05)83168-2
40. Sarrafzadeh AS, Haux D, Ludemann L, Amthauer H, Plotkin M, Kuchler I, et al. Cerebral ischemia in aneurysmal subarachnoid hemorrhage: a correlative microdialysis-PET study. Stroke (2004) 35(3):638–43. doi:10.1161/01.STR.0000116101.66624.F1
41. Schmidt JM, Ko SB, Helbok R, Kurtz P, Stuart RM, Presciutti M, et al. Cerebral perfusion pressure thresholds for brain tissue hypoxia and metabolic crisis after poor-grade subarachnoid hemorrhage. Stroke (2011) 42(5):1351–6. doi:10.1161/STROKEAHA.110.596874
42. Schulz MK, Wang LP, Tange M, Bjerre P. Cerebral microdialysis monitoring: determination of normal and ischemic cerebral metabolisms in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg (2000) 93(5):808–14. doi:10.3171/jns.2000.93.5.0808
43. Skjoth-Rasmussen J, Schulz M, Kristensen SR, Bjerre P. Delayed neurological deficits detected by an ischemic pattern in the extracellular cerebral metabolites in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg (2004) 100(1):8–15. doi:10.3171/jns.2004.100.1.0008
44. Unterberg AW, Sakowitz OW, Sarrafzadeh AS, Benndorf G, Lanksch WR. Role of bedside microdialysis in the diagnosis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg (2001) 94(5):740–9. doi:10.3171/jns.2001.94.5.0740
45. Al-Rawi PG, Zygun D, Tseng MY, Hutchinson PJ, Matta BF, Kirkpatrick PJ. Cerebral blood flow augmentation in patients with severe subarachnoid haemorrhage. Acta Neurochir Suppl (2005) 95:123–7. doi:10.1007/3-211-32318-X_27
46. Helbok R, Kurtz P, Schmidt JM, Stuart RM, Fernandez L, Malhotra R, et al. Effect of mannitol on brain metabolism and tissue oxygenation in severe haemorrhagic stroke. J Neurol Neurosurg Psychiatry (2011) 82(4):378–83. doi:10.1136/jnnp.2009.198754
47. Kurtz P, Schmidt JM, Claassen J, Carrera E, Fernandez L, Helbok R, et al. Anemia is associated with metabolic distress and brain tissue hypoxia after subarachnoid hemorrhage. Neurocrit Care (2010) 13(1):10–6. doi:10.1007/s12028-010-9357-y
48. Kurtz P, Helbok R, Claassen J, Schmidt JM, Fernandez L, Stuart RM, et al. The effect of packed red blood cell transfusion on cerebral oxygenation and metabolism after subarachnoid hemorrhage. Neurocrit Care (2016) 24(1):118–21. doi:10.1007/s12028-015-0180-3
49. Nagel A, Graetz D, Vajkoczy P, Sarrafzadeh AS. Decompressive craniectomy in aneurysmal subarachnoid hemorrhage: relation to cerebral perfusion pressure and metabolism. Neurocrit Care (2009) 11(3):384–94. doi:10.1007/s12028-009-9269-x
50. Nagel A, Graetz D, Schink T, Frieler K, Sakowitz O, Vajkoczy P, et al. Relevance of intracranial hypertension for cerebral metabolism in aneurysmal subarachnoid hemorrhage. Clinical article. J Neurosurg (2009) 111(1):94–101. doi:10.3171/2009.1.JNS08587
51. Oddo M, Frangos S, Milby A, Chen I, Maloney-Wilensky E, Murtrie EM, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke (2009) 40(5):1913–6. doi:10.1161/STROKEAHA.108.534115
52. Oddo M, Milby A, Chen I, Frangos S, MacMurtrie E, Maloney-Wilensky E, et al. Hemoglobin concentration and cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke (2009) 40(4):1275–81. doi:10.1161/STROKEAHA.108.527911
53. Samuelsson C, Howells T, Kumlien E, Enblad P, Hillered L, Ronne-Engstrom E. Relationship between intracranial hemodynamics and microdialysis markers of energy metabolism and glutamate-glutamine turnover in patients with subarachnoid hemorrhage. Clinical article. J Neurosurg (2009) 111(5):910–5. doi:10.3171/2008.8.JNS0889
54. Schiefecker AJ, Pfausler B, Beer R, Sohm F, Sabo J, Knauseder V, et al. Parenteral diclofenac infusion significantly decreases brain-tissue oxygen tension in patients with poor-grade aneurysmal subarachnoid hemorrhage. Crit Care (2013) 17(3):R88. doi:10.1186/cc12714
55. Springborg JB, Moller C, Gideon P, Jorgensen OS, Juhler M, Olsen NV. Erythropoietin in patients with aneurysmal subarachnoid haemorrhage: a double blind randomised clinical trial. Acta Neurochir (Wien) (2007) 149(11):1089–101; discussion 1101. doi:10.1007/s00701-007-1284-z
56. Stuart RM, Helbok R, Kurtz P, Schmidt M, Fernandez L, Lee K, et al. High-dose intra-arterial verapamil for the treatment of cerebral vasospasm after subarachnoid hemorrhage: prolonged effects on hemodynamic parameters and brain metabolism. Neurosurgery (2011) 68(2):337–45; discussion 345. doi:10.1227/NEU.0b013e318201be47
57. Helbok R, Schmidt JM, Kurtz P, Hanafy KA, Fernandez L, Stuart RM, et al. Systemic glucose and brain energy metabolism after subarachnoid hemorrhage. Neurocrit Care (2010) 12(3):317–23. doi:10.1007/s12028-009-9327-4
58. Kinoshita K, Moriya T, Utagawa A, Sakurai A, Mukoyama T, Furukawa M, et al. Change in brain glucose after enteral nutrition in subarachnoid hemorrhage. J Surg Res (2010) 162(2):221–4. doi:10.1016/j.jss.2009.06.009
59. Kofler M, Schiefecker AJ, Beer R, Gaasch M, Rhomberg P, Stover J, et al. Enteral nutrition increases interstitial brain glucose levels in poor-grade subarachnoid hemorrhage patients. J Cereb Blood Flow Metab (2017). doi:10.1177/0271678X17700434
60. Kurtz P, Claassen J, Helbok R, Schmidt J, Fernandez L, Presciutti M, et al. Systemic glucose variability predicts cerebral metabolic distress and mortality after subarachnoid hemorrhage: a retrospective observational study. Crit Care (2014) 18(3):R89. doi:10.1186/cc13857
61. Oddo M, Schmidt JM, Carrera E, Badjatia N, Connolly ES, Presciutti M, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med (2008) 36(12):3233–8. doi:10.1097/CCM.0b013e31818f4026
62. Schlenk F, Vajkoczy P, Sarrafzadeh A. Inpatient hyperglycemia following aneurysmal subarachnoid hemorrhage: relation to cerebral metabolism and outcome. Neurocrit Care (2009) 11(1):56–63. doi:10.1007/s12028-009-9222-z
63. Schlenk F, Nagel A, Graetz D, Sarrafzadeh AS. Hyperglycemia and cerebral glucose in aneurysmal subarachnoid hemorrhage. Intensive Care Med (2008) 34(7):1200–7. doi:10.1007/s00134-008-1044-5
64. Schlenk F, Graetz D, Nagel A, Schmidt M, Sarrafzadeh AS. Insulin-related decrease in cerebral glucose despite normoglycemia in aneurysmal subarachnoid hemorrhage. Crit Care (2008) 12(1):R9. doi:10.1186/cc6776
65. Schlenk F, Sarrafzadeh AS. Is continuous insulin treatment safe in aneurysmal subarachnoid hemorrhage? Vasc Health Risk Manag (2008) 4(4):885–91. doi:10.2147/VHRM.S1924
66. Schmidt JM, Claassen J, Ko SB, Lantigua H, Presciutti M, Lee K, et al. Nutritional support and brain tissue glucose metabolism in poor-grade SAH: a retrospective observational study. Crit Care (2012) 16(1):R15. doi:10.1186/cc11160
67. Zetterling M, Hillered L, Enblad P, Karlsson T, Ronne-Engstrom E. Relation between brain interstitial and systemic glucose concentrations after subarachnoid hemorrhage. J Neurosurg (2011) 115(1):66–74. doi:10.3171/2011.3.JNS10899
68. Antunes AP, Schiefecker AJ, Beer R, Pfausler B, Sohm F, Fischer M, et al. Higher brain extracellular potassium is associated with brain metabolic distress and poor outcome after aneurysmal subarachnoid hemorrhage. Crit Care (2014) 18(3):R119. doi:10.1186/cc13916
69. Cesarini KG, Enblad P, Ronne-Engstrom E, Marklund N, Salci K, Nilsson P, et al. Early cerebral hyperglycolysis after subarachnoid haemorrhage correlates with favourable outcome. Acta Neurochir (Wien) (2002) 144(11):1121–31. doi:10.1007/s00701-002-1011-9
70. Dizdarevic K, Hamdan A, Omerhodzic I, Kominlija-Smajic E. Modified Lund concept versus cerebral perfusion pressure-targeted therapy: a randomised controlled study in patients with secondary brain ischaemia. Clin Neurol Neurosurg (2012) 114(2):142–8. doi:10.1016/j.clineuro.2011.10.005
71. Kett-White R, Hutchinson PJ, Al-Rawi PG, Gupta AK, Pickard JD, Kirkpatrick PJ. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery (2002) 50(6):1213–21; discussion 1221–2. doi:10.1097/00006123-200206000-00008
72. Oddo M, Levine JM, Frangos S, Maloney-Wilensky E, Carrera E, Daniel RT, et al. Brain lactate metabolism in humans with subarachnoid hemorrhage. Stroke (2012) 43(5):1418–21. doi:10.1161/STROKEAHA.111.648568
73. Omerhodzic I, Dizdarevic K, Rotim K, Hajdarpasic E, Niksic M, Bejtic-Custovic E, et al. Cerebral microdialysis: perioperative monitoring and treatment of severe neurosurgical patient. Acta Clin Croat (2011) 50(1):13–20.
74. Staub F, Graf R, Gabel P, Kochling M, Klug N, Heiss WD. Multiple interstitial substances measured by microdialysis in patients with subarachnoid hemorrhage. Neurosurgery (2000) 47(5):1106–15; discussion 1115–6. doi:10.1097/00006123-200011000-00016
75. Tholance Y, Barcelos GK, Dailler F, Renaud B, Marinesco S, Perret-Liaudet A. Biochemical neuromonitoring of poor-grade aneurysmal subarachnoid hemorrhage: comparative analysis of metabolic events detected by cerebral microdialysis and by retrograde jugular vein catheterization. Neurol Res (2015) 37(7):578–87. doi:10.1179/1743132815Y.0000000012
76. Gonzalez NR, Hamilton R, Bilgin-Freiert A, Dusick J, Vespa P, Hu X, et al. Cerebral hemodynamic and metabolic effects of remote ischemic preconditioning in patients with subarachnoid hemorrhage. Acta Neurochir Suppl (2013) 115:193–8. doi:10.1007/978-3-7091-1192-5_36
77. Noske DP, Peerdeman SM, Comans EF, Dirven CM, Knol DL, Girbes AR, et al. Cerebral microdialysis and positron emission tomography after surgery for aneurysmal subarachnoid hemorrhage in grade I patients. Surg Neurol (2005) 64(2):109–15; discussion 115. doi:10.1016/j.surneu.2004.09.036
78. Sakowitz OW, Santos E, Nagel A, Krajewski KL, Hertle DN, Vajkoczy P, et al. Clusters of spreading depolarizations are associated with disturbed cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke (2013) 44(1):220–3. doi:10.1161/STROKEAHA.112.672352
79. Sarrafzadeh A, Santos E, Wiesenthal D, Martus P, Vajkoczy P, Oehmchen M, et al. Cerebral glucose and spreading depolarization in patients with aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl (2013) 115:143–7. doi:10.1007/978-3-7091-1192-5_28
80. Schlenk F, Frieler K, Nagel A, Vajkoczy P, Sarrafzadeh AS. Cerebral microdialysis for detection of bacterial meningitis in aneurysmal subarachnoid hemorrhage patients: a cohort study. Crit Care (2009) 13(1):R2. doi:10.1186/cc7689
81. Shuaib A, Kanthan R, Goplen G, Griebel R, el-Azzouni H, Miyashita H, et al. In-vivo microdialysis study of extracellular glutamate response to temperature variance in subarachnoid hemorrhage. Acta Neurochir Suppl (1996) 67:53–8.
82. Stocchetti N, Protti A, Lattuada M, Magnoni S, Longhi L, Ghisoni L, et al. Impact of pyrexia on neurochemistry and cerebral oxygenation after acute brain injury. J Neurol Neurosurg Psychiatry (2005) 76(8):1135–9. doi:10.1136/jnnp.2004.041269
83. Vergouwen MD, Vermeulen M, van Gijn J, Rinkel GJ, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke (2010) 41(10):2391–5. doi:10.1161/STROKEAHA.110.589275
84. van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, et al. Intensive insulin therapy in critically ill patients. N Engl J Med (2001) 345(19):1359–67. doi:10.1056/NEJMoa011300
85. Investigators N-SS, Finfer S, Chittock DR, Su SY, Blair D, Foster D, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med (2009) 360(13):1283–97. doi:10.1056/NEJMoa0810625
Keywords: subarachnoid hemorrhage, neuromonitoring, cerebral microdialysis, brain metabolism, treatment
Citation: Helbok R, Kofler M, Schiefecker AJ, Gaasch M, Rass V, Pfausler B, Beer R and Schmutzhard E (2017) Clinical Use of Cerebral Microdialysis in Patients with Aneurysmal Subarachnoid Hemorrhage—State of the Art. Front. Neurol. 8:565. doi: 10.3389/fneur.2017.00565
Received: 20 May 2017; Accepted: 09 October 2017;
Published: 03 November 2017
Edited by:
Adel Helmy, University of Cambridge, United KingdomReviewed by:
Diederik Bulters, University Hospital Southampton NHS Foundation Trust, United KingdomMartin Hunn, The Alfred Hospital, Australia
Copyright: © 2017 Helbok, Kofler, Schiefecker, Gaasch, Rass, Pfausler, Beer and Schmutzhard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raimund Helbok, cmFpbXVuZC5oZWxib2tAaS1tZWQuYWMuYXQ=
†These authors have contributed equally to this work.