 Naruhiko Sahara1*
Naruhiko Sahara1* Masafumi Shimojo1Maiko Ono1
Masafumi Shimojo1Maiko Ono1 Hiroyuki Takuwa1
Hiroyuki Takuwa1 Marcelo Febo2Makoto Higuchi1Tetsuya Suhara1
Marcelo Febo2Makoto Higuchi1Tetsuya Suhara1
- 1Department of Functional Brain Imaging Research, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Science and Technology, Chiba, Japan
- 2Department of Psychiatry and Neuroscience, University of Florida College of Medicine, Gainesville, FL, United States
Association of tau deposition with neurodegeneration in Alzheimer’s disease (AD) and related tau-positive neurological disorders collectively referred to as tauopathies indicates contribution of tau aggregates to neurotoxicity. The discovery of tau gene mutations in FTDP-17-tau kindreds has provided unequivocal evidence that tau abnormalities alone can induce neurodegenerative disorders. Therefore, visualization of tau accumulation would offer a reliable, objective index to aid in the diagnosis of tauopathy and to assess the disease progression. Positron emission tomography (PET) imaging of tau lesions is currently available using several tau PET ligands. Because most tau PET ligands have the property of an extrinsic fluorescent dye, these ligands are considered to be useful for both PET and fluorescence imaging. In addition, small-animal magnetic resonance imaging (MRI) is available for both structural and functional imaging. Using these advanced imaging techniques, in vivo studies on a mouse model of tauopathy will provide significant insight into the translational research of neurodegenerative diseases. In this review, we will discuss the utilities of PET, MRI, and fluorescence imaging for evaluating the disease progression of tauopathy.
Introduction
Dementia is a leading cause of death in developed countries. Increasing age is the greatest risk factor for dementia. About 46 million people in the world are estimated to be suffering from dementia, and this figure is expected to rise to 135 million by 2050. Alzheimer’s disease (AD) is the most common type of dementia. The global impact of AD will strikingly affect social and economic costs. Distinctive features of AD are the deposited accumulations of β-amyloid (Aβ) protein fragments and intracellular neurofibrillary tangles (NFTs) (1). NTFs are closely associated with the severity of brain function loss in AD (2). Therefore, making tau protein a target in the treatment of AD has become a major therapeutic strategy.
Recent advances in positron emission tomography (PET) imaging research have led to significant breakthroughs with the application of newly developed tracers for visualizing regional tau depositions (3–8). This technology allows us to non-invasively evaluate the progression of tau pathology in living brains. PBB3 was developed as a novel PET tracer that binds with tau for the diagnosis of AD and other neurodegenerative diseases regarded as tauopathies (4, 9). PET imaging using [11C]PBB3 (radiotracer) is superior in detecting tau deposits, and it is closely aligned with disease symptoms. This was also confirmed by our tauopathy mouse models (4; Ishikawa, forthcoming1). 11C-labeled tracers, due to their short half-life, are generally unsuitable for clinical practice, but [11C]PBB3 can be used to evaluate certain tau conditions including AD (4, 10). The fluorescent quality of PBB3 makes it especially useful for multimodal imaging, allowing us to assess its binding by microscopic observation of related cells and tissues.
Animal models of tauopathy are essential for preclinical studies of AD and related dementias. The rTg4510 mouse line was developed to model aspects of tauopathy through forebrain expression of the P301L mutated human tau (11). Expression of human tau is controlled by the tetracycline transactivator transgene under the CaMKIIα promoter. This mouse line develops progressive intracellular tau aggregations in corticolimbic areas and forebrain atrophy (11). The age-dependent tau pathology of rTg4510 mice has been investigated in detail by immunohistochemical and biochemical examinations (12–14). These postmortem brain-based studies showed an extensive increase of pathological tau inclusions in the cerebral cortex and hippocampus between 4 and 6 months of age. Now, for both current and therapeutic strategies, rather than endpoint measurements, researchers are eagerly involved in the development of new in vivo protocols for tracking the progressive pathological status in living animals.
Taking advantage of the multimodality of the PBB3 ligand, in vivo monitoring of NFT formation is now available for possible in tauopathy mouse models, including the rTg4510 mouse. In addition, advanced brain magnetic resonance imaging (MRI) techniques have enabled us to visualize neuronal dysfunction and the structural changes related to neurodegenerative processes in living animals. Here, we will introduce in vivo multimodal imaging technologies, including PET, MRI, and fluorescence imaging to investigate the real-time events of tau-related neuropathology.
MRI Studies for a Mouse Model of Tauopathy
Magnetic resonance imaging-based volumetry is a valuable tool for assessing disease progression in humans (15–19). As translational research of neurodegenerative diseases, several groups have reported studies using volumetric MRI in mouse models (20–25). Age-dependent volume reduction in both cerebral cortex and hippocampus of the rTg4510 mouse line was clearly demonstrated on MRI (24, 25). The volume (size) of the cerebral cortex from 5 to 8-month-old rTg4510 mice was significantly less (by approximately 20%) than that of age-matched non-transgenic (non-tg) mice (24). Brain atrophy in rTg4510 mice as examined by MRI was in agreement with previous histopathology findings (13, 26). A gender difference showing more severe phenotype in female rTg4510 mice compared with males (27) was confirmed by volumetric MRI study (24).
In addition to volumetric MRI, MR-based in vivo imaging techniques, including MR spectroscopy (MRS), manganese-enhanced MRI (MEMRI), arterial spin labeling, amide proton transfer imaging, and diffusion tensor imaging (DTI), have been tested to evaluate brain functions and microstructural changes in living rTg4510 mice (24, 25, 28–32). MEMRI is a technique for measuring neuronal function, because the manganese ion (Mn2+) accumulates in actively firing neurons through voltage-gated Ca2+ channels and serves as a positive contrast agent in T1-weighted brain images (33, 34). Using this technique, Perez et al. observed a reduction in both neuronal activity and volume in the hippocampus of 6-month-old rTg4510 mice (25). Fontaine et al. demonstrated neuronal dysfunction in the CA3 and CA1 regions of 3-month-old rTg4510 mice (32). Together, these studies illustrate the importance of the MEMRI method as a diagnostic research tool for the early detection of neuronal dysfunction in rTg4510 mice.
Diffusion tensor imaging is another MR-based imaging technique, which is applied to assess the integrity and organization of myelinated structures such as axons in white matter (WM) and also in gray matter regions (35). For this purpose, we used DTI to examine age-related alterations in WM diffusion anisotropy in rTg4510 and non-tg control mice (29). Our results indicated that 8-month-old rTg4510 mice show significant reductions in fractional anisotropy (FA) in WM structures compared with control non-tg and young (2.5-month-old) rTg4510 mice. The microstructural changes contributing to these age- and tauopathy-associated WM changes were supported by electron microscopic evidence of disorganized axonal processes and the presence of interprocess spaces, which could explain the reduced FA in aged rTg4510 mice (29). Reduced FA at a later age (over 7.5 months) was confirmed by another research group (30, 31). Since, reduced FA values in WM were mostly observed at an age that included the presence of significant atrophy and tau pathology, this standard method may be useful for assessing the therapeutic efficacy of treatments that may be used to reduce or prevent neurodegenerative progression. On the other hand, examination with additional diffusion anisotropy indices such as the mode of anisotropy allowed us to detect early signs (at 2.5 months old) of WM disorganization (29). Although disorganization of myelin morphology at this age was not determined, WM degeneration may be one of the early signs of tauopathy. Nevertheless, reduced WM integrity associated with tau pathology was confirmed by in vivo DTI studies.
Tau PET Tracers
Current tau PET tracers are mostly designed by β-sheet binding properties. In principle, filamentous tau aggregates (e.g., NFTs, neuropile threads, tufted astrocytes, astrocytic plaques, and coiled bodies) will be labeled by these tracers with distinct specificity and selectivity. Up to date, three types of radiotracers have been widely tested for the clinical assessment of patients with tauopathy [reviewed in Ref. (36)]. These tracers include the arcyquinoline derivative THK5351 (37), the pyrido-indole derivative AV-1451 (38, 39), and the phenyl/pyridinyl-butadienyl-benzothiazole/benzothiazolium derivative PBB3 (4). Screening of potential tracers was performed by in vitro binding assays in AD brain homogenates and autoradiographies of AD brain sections. [18F]THK5351, [18F]AV-1451, and [11C]PBB3 showed good affinity for tau deposits and no selectivity of amyloid plaques. Accumulating evidence has shown that tau PET using these tracers might not detect pretangles in tauopathy brains (9, 40). Comparative in vitro binding assay of [18F]AV-1451 and [11C]PBB3 revealed that the binding of [11C]PBB3 to non-AD-type tau pathology (e.g., tufted astrocytes, astrocytic plaques, and pick bodies) was higher than that of [18F]AV-1451 (9). Another comparative study of [3H]AV-1451 and [3H]THK523 showed a distinct binding affinity for NFTs (41). Potential off-target bindings of [18F]AV-1451 and [18F]THK5351 to monoamine oxidase A (MAO-A) and MAO-B, respectively, were also observed (36, 42, 43). Although binding properties of tau PET tracers have been vigorously investigated for the past decade, the specificity and selectivity of PET signals to tau pathology are still to be fully understood.
Tau PET Imaging Studies of rTg4510 Mice
Micro-PET imaging of tau pathology in tg mouse models of tauopathy has contributed to the characterization of novel PET tracers. Using [18F]THK523, micro-PET imaging in 6-month-old rTg4510 mice successfully showed significantly higher radiotracer retentions in brains compared with non-tg or PS1/APP mice (44). [11C]PBB3-PET imaging in another tauopathy mouse model, PS19 tg (expressing P301S mutant human tau) (4), and [18F]THK5117-PET imaging in P301S tg and biGT (bigenic GSK-3β × P301L tau) tg mice (45) were reported to show higher tracer uptake in tg than in non-tg mice. On the other hand, there was no significant difference in retention of [18F]AV-1451 between TAPP (bigenic APPswe × P301L tau) tg and non-tg mice (39). Inconsistency of these micro-PET analyses was mostly due to the use of different tg mouse models. Technical limitations also stem from the diversity of pathological characteristics in different mouse models. The distribution of tau deposits and the time course of pathological tau accumulation in tg mice differed from each other. It is very important to compare in vivo bindings of different tracers with reproducible mouse models. To initiate the development of a screening platform for tau PET tracers, our group has recently demonstrated longitudinal micro-PET imaging in rTg4510 mice (see text footnote 1). Consistent with neuropathological and biochemical observations, our [11C]PBB3 PET imaging of rTg4510 mice showed an age-dependent increase in [11C]PBB3 signal and that [11C]PBB3 retention was inversely correlated with neocortical volumes. The increasing [11C]PBB3 signal reached a plateau by 7 months of age. The correlation between [11C]PBB3 levels and brain atrophy disappeared in rTg4510 mice over 7 months old. Since, tau pathology is tightly linked to neuronal loss, the disappearance rate of [11C]PBB3-positive neurons may increase at age over 7 months. It should be noted that microglial activation in rTg4510 brains examined using translocator protein (TSPO) (18-kDa TSPO)-PET imaging showed significant correlation with both [11C]PBB3 level and brain atrophy (see text footnote 1). rTg4510 will be a useful model for investigating the mechanisms of tau-induced neuroinflammation. Nevertheless, in combination with tau PET imaging, the imaging of neuroinflammation will offer an additional diagnostic parameter for tauopathy.
In Vivo Fluorescence Imaging for Detecting Tau Pathology
The invention of two-photon excitation laser scanning microscopy has given new impetus to the research field of investigating in vivo brain cell dynamics (46). In combination with newly developed fluorescence imaging techniques, cellular and molecular mechanisms underlying brain functions and impairments can be examined using in vivo animal models. In theory, the spatial scales of two-photon microscopy are from a micron to a millimeter. The side length of field-of-views can reach a few 100 μm. The imaging depth reached has been up to ~1 mm depending on the properties of the tissue (47). Using this technology, AD mouse models have been investigated with regard to monitoring the time course of disease progression [reviewed in Ref. (48)]. Hyman’s group first reported in vivo visualization of amyloid plaques in APP tg mouse lines (PDAPP mice and tg2576 mice) (49, 50). Clearance of plaques by anti-Aβ antibody was monitored by two-photon microscopy after Thioflavin S injection into the brain. Fluorescence imaging using Pittsburgh Compound B (PiB), which is used for amyloid PET imaging, was also examined to visualize amyloid plaques in APP tg mouse lines (51). As a result, the multimodality of PiB ligand for both fluorescence and PET imaging was clearly demonstrated. Because the PBB3 ligand was derived from the same tracer family as the PiB ligand, a similar approach was used to visualize the tau pathology in PS19 tg mice (4). Moreover, the current chronic cranial window setting enables the use of long-period two-photon imaging for more than 2 months (52). As for our study, longitudinal monitoring of PBB3-positive neurons has been performed in living rTg4510 mice (Takuwa et al., manuscript in preparation). Our data showed that PBB3-positive inclusions were visualized with two-photon imaging at an age of as early as 4 months, with fluorescence signals then reaching a plateau at 6 months. These data are in agreement with both [11C]PBB3 PET imaging and volumetric MRI, suggesting multimodal imaging utilities of the PBB3 ligand. On the other hand, Hyman’s group has investigated the mechanism of neuronal loss in rTg4510 mouse brains using the fluorescence indicator of caspase activation and β-sheet ligand X-34 (a Congo red derivative) (53). Their data indicate that NFT formation has a neuroprotective effect against caspase-mediated neuronal death. Synaptic dysfunction is another key event taking place during the pathogenesis of tauopathy. Previous reports showed decreased dendritic spine and synapse density in cortical slices prepared from 9 to 10-month-old rTg4510 mice (54, 55). Most recently, Jackson et al. demonstrated the visualization of synapses in the somatosensory cortex of rTg4510 mice by two-photon microscopy after the injection of adeno-associated virus that drove neuronal expression of either GFP or GCaMP6m (56). For longitudinal imaging, GFP-expressing axonal and dendritic regions were imaged weekly to investigate the turnover of axonal terminal boutons and dendritic spines. For functional imaging, GCaMP6-expressing neurons were imaged in lightly anesthetized animals to measure neuronal activity in response to whisker stimulation. The authors observed mismatched abnormalities in pre-and post-synaptic turnover coinciding with disrupted neuronal activity at 5 months of age. Their data suggests that synaptic dysfunction precedes tangle-associated neurodegeneration. Although linkage between aggregated tau formation and synaptic dysfunction remains unclear, in vivo monitoring of tau pathology at cellular levels provides an advantage for dissecting the mechanisms of tau toxicity.
Design for a Diagnostic Platform of Tauopathy
The rTg4510 mouse is one of the widely used models of tauopathy. As described above, several groups have conducted in vivo imaging studies on rTg4510 mice (Table 1). Because the time course of tau pathology and forebrain atrophy have been well examined using postmortem materials, experimental designs for drug intervention can be easily designed (Figure 1). Since, this mouse model allows the tetracycline-repressible overexpression of human tau, doxycycline treatment for the suppression of tau expression will provide a positive control in an experimental design of the evaluation of therapeutic candidates. Previous study showed that suppression of tau from 5.5 months of age reversed memory deficits, while tangles persisted and continued to accumulate (11). Therefore, early intervention starting at 2 months of age could be more effective to prevent both NFT formation and brain atrophy (Figure 1B). Although effects will be limited, late intervention would be worthwhile if aggravation of tau-induced brain atrophy can be slowed down (Figure 1C). In our study, we developed a unique diagnostic platform of in vivo imaging of volumetric MRI, tau-PET, and TSPO-PET (Figure 1D). Brains of living rTg4510 mice can be used to monitor their volume, pathological tau accumulation, and neuroinflammation. Moreover, live cell imaging with two-photon microscopy allows us to capture NFT formation and neuronal death in rTg4510 mice. Using these platforms, we expect to be able to validate several drug candidates in the foreseeable future.
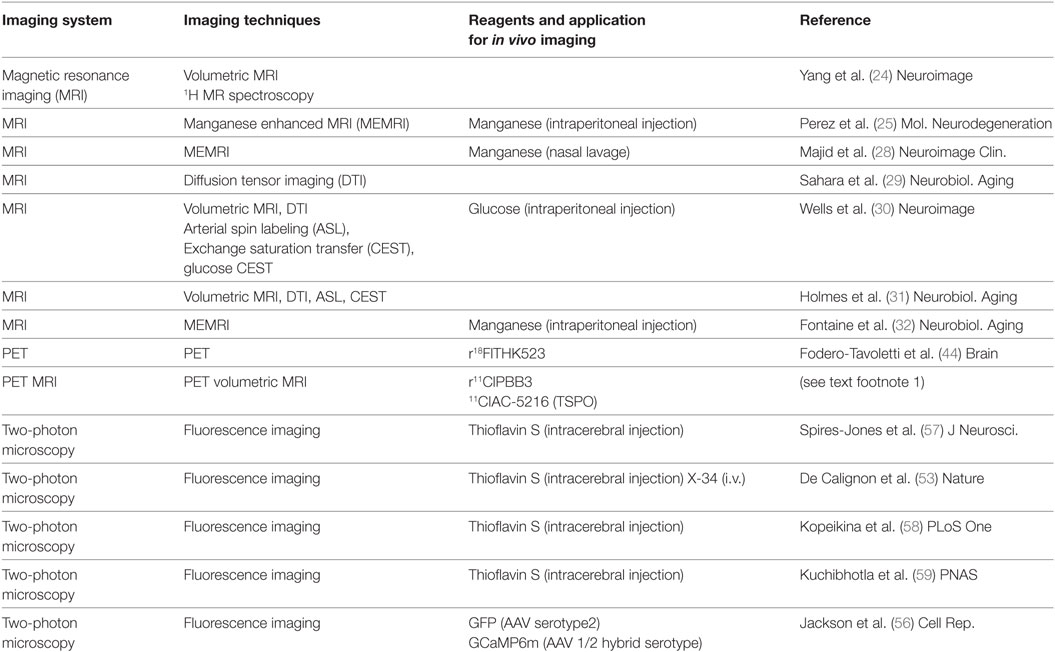
Table 1. Reference list for in vivo imaging in living rTg4510 mice.
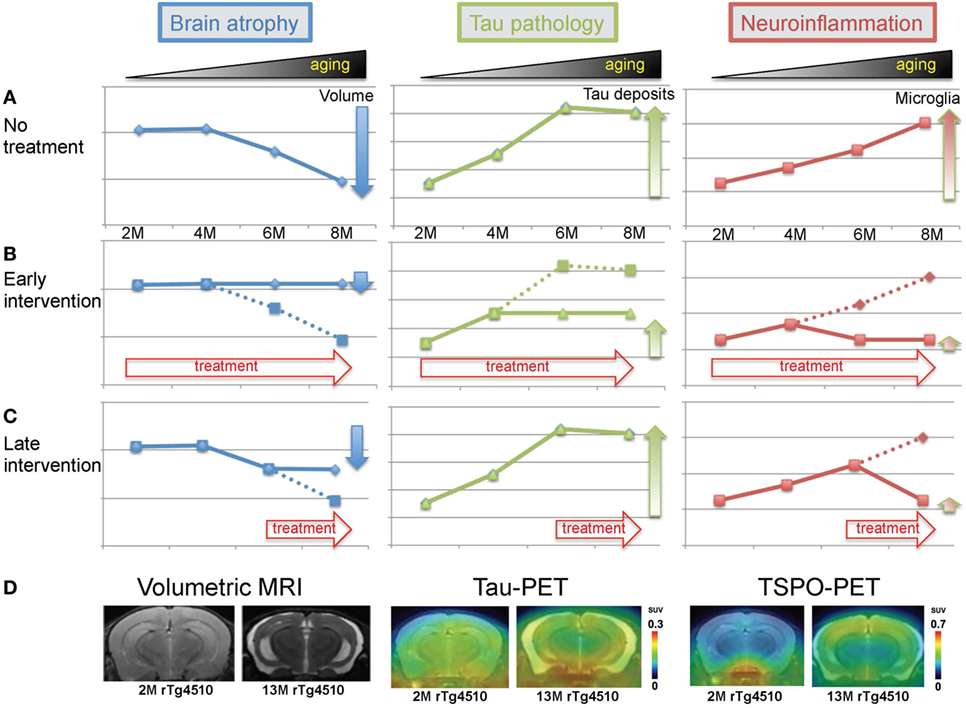
Figure 1. Imaging modalities for investigating tau-induced brain atrophy and neuroinflammation. (A) Theoretical processes of volume change, pathological tau accumulation, and microglial activation in rTg4510 mice. (B) Effects of early intervention. Treatment was started from 2 months of age. (C) Effects of late intervention. Treatment was started from 6 months of age. (D) Representative images of volumetric magnetic resonance imaging (MRI), tau-positron emission tomography (PET), and translocator protein-PET in 2- and 13-month-old rTg4510 mice.
Conclusion
For more effective therapies, the pre-clinical evaluation of drugs for tauopathy is needed. The use of animal models that recapitulate the critical features of the disease, such as NFTs, cognitive impairment, brain atrophy, and neuronal loss, is essential. The rTg4510 mouse model of tauopathy fulfills the required features, despite the fact that tau protein was expressed at a non-physiologically higher level over the total life span. In vivo, brain imaging offers reliable approaches to validating the pathological status and to determining the efficacy of drugs for exploring disease-modifying therapies.
Author Contributions
Authors wrote and proofed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported in part by grants from Grant-in-Aid for Science Research on Innovation Area (“Brain Protein Aging” 26117001 to NS) and Scientific Research (C) (15K06793 to NS) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and from the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development, AMED. MH holds a patent on compounds including PBB3 (JP 5422782/EP 12 844 742.3).
Footnote
- ^Ishikawa I, Tokunaga M, Maeda J, Minamihisamatsu T, Shimojo M, Takuwa H, et al. In vivo visualization of tau accumulation, microglial activation and brain atrophy in a mouse model of tauopathy rTg4510. J Alzheimers Dis (2018, Forthcoming).
References
1. Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat (1995) 8:429–31. doi:10.1002/ca.980080612
2. Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol (1997) 41:17–24. doi:10.1002/ana.410410106
3. Harada R, Okamura N, Furumoto S, Yoshikawa T, Arai H, Yanai K, et al. Use of a benzimidazole derivative BF-188 in fluorescence multispectral imaging for selective visualization of Tau protein fibrils in the Alzheimer’s disease brain. Mol Imaging Biol (2014) 16:19–27. doi:10.1007/s11307-013-0667-2
4. Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron (2013) 79:1094–108. doi:10.1016/j.neuron.2013.07.037
5. Okamura N, Furumoto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M, et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med (2013) 54:1420–7. doi:10.2967/jnumed.112.117341
6. Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, et al. Early clinical PET imaging results with the novel PHF-Tau radioligand [F18]-T808. J Alzheimers Dis (2014) 38:171–84. doi:10.3233/JAD-130098
7. Ariza M, Kolb HC, Moechars D, Rombouts F, Andres JI. Tau positron emission tomography (PET) imaging: past, present, and future. J Med Chem (2015) 58:4365–82. doi:10.1021/jm5017544
8. Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol (2015) 14:114–24. doi:10.1016/S1474-4422(14)70252-2
9. Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain (2017) 140(3):764–80. doi:10.1093/brain/aww339
10. Kimura Y, Ichise M, Ito H, Shimada H, Ikoma Y, Seki C, et al. PET quantification of Tau pathology in human brain with 11C-PBB3. J Nucl Med (2015) 56:1359–65. doi:10.2967/jnumed.115.160127
11. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science (2005) 309:476–81. doi:10.1126/science.1113694
12. Ramsden M, Kotilinek L, Forster C, Paulson J, Mcgowan E, Santacruz K, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci (2005) 25:10637–47. doi:10.1523/JNEUROSCI.3279-05.2005
13. Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, et al. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol (2009) 174:228–38. doi:10.2353/ajpath.2009.080764
14. Sahara N, Deture M, Ren Y, Ebrahim AS, Kang D, Knight J, et al. Characteristics of TBS-extractable hyperphosphorylated tau species: aggregation intermediates in rTg4510 mouse brain. J Alzheimers Dis (2013) 33:249–63. doi:10.3233/JAD-2012-121093
15. Du AT, Schuff N, Amend D, Laakso MP, Hsu YY, Jagust WJ, et al. Magnetic resonance imaging of the entorhinal cortex and hippocampus in mild cognitive impairment and Alzheimer’s disease. J Neurol Neurosurg Psychiatry (2001) 71:441–7. doi:10.1136/jnnp.71.4.441
16. Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, et al. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch Neurol (2006) 63:693–9. doi:10.1001/archneur.63.5.693
17. Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, Segal S, et al. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer disease. Neurology (2007) 68:828–36. doi:10.1212/01.wnl.0000256697.20968.d7
18. Fan Y, Batmanghelich N, Clark CM, Davatzikos C, Alzheimer’s Disease Neuroimaging Initiative. Spatial patterns of brain atrophy in MCI patients, identified via high-dimensional pattern classification, predict subsequent cognitive decline. Neuroimage (2008) 39:1731–43. doi:10.1016/j.neuroimage.2007.10.031
19. Whitwell JL, Shiung MM, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, et al. MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology (2008) 70:512–20. doi:10.1212/01.wnl.0000280575.77437.a2
20. McDaniel B, Sheng H, Warner DS, Hedlund LW, Benveniste H. Tracking brain volume changes in C57BL/6J and ApoE-deficient mice in a model of neurodegeneration: a 5-week longitudinal micro-MRI study. Neuroimage (2001) 14:1244–55. doi:10.1006/nimg.2001.0934
21. Weiss C, Venkatasubramanian PN, Aguado AS, Power JM, Tom BC, Li L, et al. Impaired eyeblink conditioning and decreased hippocampal volume in PDAPP V717F mice. Neurobiol Dis (2002) 11:425–33. doi:10.1006/nbdi.2002.0555
22. Lau JC, Lerch JP, Sled JG, Henkelman RM, Evans AC, Bedell BJ. Longitudinal neuroanatomical changes determined by deformation-based morphometry in a mouse model of Alzheimer’s disease. Neuroimage (2008) 42:19–27. doi:10.1016/j.neuroimage.2008.04.252
23. Maheswaran S, Barjat H, Rueckert D, Bate ST, Howlett DR, Tilling L, et al. Longitudinal regional brain volume changes quantified in normal aging and Alzheimer’s APP x PS1 mice using MRI. Brain Res (2009) 1270:19–32. doi:10.1016/j.brainres.2009.02.045
24. Yang D, Xie Z, Stephenson D, Morton D, Hicks CD, Brown TM, et al. Volumetric MRI and MRS provide sensitive measures of Alzheimer’s disease neuropathology in inducible tau transgenic mice (rTg4510). Neuroimage (2011) 54:2652–8. doi:10.1016/j.neuroimage.2010.10.067
25. Perez PD, Hall G, Kimura T, Ren Y, Bailey RM, Lewis J, et al. In vivo functional brain mapping in a conditional mouse model of human tauopathy (taup301l) reveals reduced neural activity in memory formation structures. Mol Neurodegener (2013) 8:9. doi:10.1186/1750-1326-8-9
26. Spires TL, Orne JD, Santacruz K, Pitstick R, Carlson GA, Ashe KH, et al. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol (2006) 168:1598–607. doi:10.2353/ajpath.2006.050840
27. Yue M, Hanna A, Wilson J, Roder H, Janus C. Sex difference in pathology and memory decline in rTg4510 mouse model of tauopathy. Neurobiol Aging (2011) 32:590–603. doi:10.1016/j.neurobiolaging.2009.04.006
28. Majid T, Ali YO, Venkitaramani DV, Jang MK, Lu HC, Pautler RG. In vivo axonal transport deficits in a mouse model of fronto-temporal dementia. Neuroimage Clin (2014) 4:711–7. doi:10.1016/j.nicl.2014.02.005
29. Sahara N, Perez PD, Lin WL, Dickson DW, Ren Y, Zeng H, et al. Age-related decline in white matter integrity in a mouse model of tauopathy: an in vivo diffusion tensor magnetic resonance imaging study. Neurobiol Aging (2014) 35:1364–74. doi:10.1016/j.neurobiolaging.2013.12.009
30. Wells JA, O’callaghan JM, Holmes HE, Powell NM, Johnson RA, Siow B, et al. In vivo imaging of tau pathology using multi-parametric quantitative MRI. Neuroimage (2015) 111:369–78. doi:10.1016/j.neuroimage.2015.02.023
31. Holmes HE, Colgan N, Ismail O, Ma D, Powell NM, O’callaghan JM, et al. Imaging the accumulation and suppression of tau pathology using multiparametric MRI. Neurobiol Aging (2016) 39:184–94. doi:10.1016/j.neurobiolaging.2015.12.001
32. Fontaine SN, Ingram A, Cloyd RA, Meier SE, Miller E, Lyons D, et al. Identification of changes in neuronal function as a consequence of aging and tauopathic neurodegeneration using a novel and sensitive magnetic resonance imaging approach. Neurobiol Aging (2017) 56:78–86. doi:10.1016/j.neurobiolaging.2017.04.007
33. Pautler RG, Mongeau R, Jacobs RE. In vivo trans-synaptic tract tracing from the murine striatum and amygdala utilizing manganese enhanced MRI (MEMRI). Magn Reson Med (2003) 50:33–9. doi:10.1002/mrm.10498
34. Lee JH, Silva AC, Merkle H, Koretsky AP. Manganese-enhanced magnetic resonance imaging of mouse brain after systemic administration of MnCl2: dose-dependent and temporal evolution of T1 contrast. Magn Reson Med (2005) 53:640–8. doi:10.1002/mrm.20368
35. Mori S, Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron (2006) 51:527–39. doi:10.1016/j.neuron.2006.08.012
36. Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener (2017) 12:19. doi:10.1186/s13024-017-0162-3
37. Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, et al. 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med (2016) 57:208–14. doi:10.2967/jnumed.115.164848
38. Zhang W, Arteaga J, Cashion DK, Chen G, Gangadharmath U, Gomez LF, et al. A highly selective and specific PET tracer for imaging of tau pathologies. J Alzheimers Dis (2012) 31:601–12. doi:10.3233/JAD-2012-120712
39. Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement (2013) 9:666–76. doi:10.1016/j.jalz.2012.11.008
40. Harada R, Okamura N, Furumoto S, Tago T, Yanai K, Arai H, et al. Characteristics of Tau and its ligands in PET imaging. Biomolecules (2016) 6:7. doi:10.3390/biom6010007
41. Cai L, Qu B, Hurtle BT, Dadiboyena S, Diaz-Arrastia R, Pike VW. Candidate PET radioligand development for neurofibrillary tangles: two distinct radioligand binding sites identified in postmortem Alzheimer’s disease brain. ACS Chem Neurosci (2016) 7:897–911. doi:10.1021/acschemneuro.6b00051
42. Harada R, Ishiki A, Kai H, Sato N, Furukawa K, Furumoto S, et al. Correlations of 18F-THK5351 PET with post-mortem burden of tau and astrogliosis in Alzheimer’s disease. J Nucl Med (2017). doi:10.2967/jnumed.117.197426
43. Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M, et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res Ther (2017) 9:25. doi:10.1186/s13195-017-0253-y
44. Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, Mclean CA, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain (2011) 134:1089–100. doi:10.1093/brain/awr038
45. Brendel M, Jaworska A, Probst F, Overhoff F, Korzhova V, Lindner S, et al. Small-animal PET imaging of tau pathology with 18F-THK5117 in 2 transgenic mouse models. J Nucl Med (2016) 57:792–8. doi:10.2967/jnumed.115.163493
46. Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron (2006) 50:823–39. doi:10.1016/j.neuron.2006.05.019
47. Theer P, Denk W. On the fundamental imaging-depth limit in two-photon microscopy. J Opt Soc Am A Opt Image Sci Vis (2006) 23:3139–49. doi:10.1364/JOSAA.23.003139
48. Shimojo M, Higuchi M, Suhara T, Sahara N. Imaging multimodalities for dissecting Alzheimer’s disease: advanced technologies of positron emission tomography and fluorescence imaging. Front Neurosci (2015) 9:482. doi:10.3389/fnins.2015.00482
49. Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, et al. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med (2001) 7:369–72. doi:10.1038/85525
50. Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, et al. Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J Neurosci (2001) 21:858–64.
51. Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, et al. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci U S A (2003) 100:12462–7. doi:10.1073/pnas.2034101100
52. Tomita Y, Kubis N, Calando Y, Tran Dinh A, Meric P, Seylaz J, et al. Long-term in vivo investigation of mouse cerebral microcirculation by fluorescence confocal microscopy in the area of focal ischemia. J Cereb Blood Flow Metab (2005) 25:858–67. doi:10.1038/sj.jcbfm.9600077
53. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature (2010) 464:1201–4. doi:10.1038/nature08890
54. Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, et al. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol (2010) 223:385–93. doi:10.1016/j.expneurol.2009.07.029
55. Crimins JL, Rocher AB, Peters A, Shultz P, Lewis J, Luebke JI. Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol (2011) 122:551–64. doi:10.1007/s00401-011-0877-0
56. Jackson JS, Witton J, Johnson JD, Ahmed Z, Ward M, Randall AD, et al. Altered synapse stability in the early stages of tauopathy. Cell Rep (2017) 18:3063–8. doi:10.1016/j.celrep.2017.03.013
57. Spires-Jones TL, De Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, et al. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci (2008) 28:862–7. doi:10.1523/JNEUROSCI.3072-08.2008
58. Kopeikina KJ, Wegmann S, Pitstick R, Carlson GA, Bacskai BJ, Betensky RA, et al. Tau causes synapse loss without disrupting calcium homeostasis in the rTg4510 model of tauopathy. PLoS One (2008) 8:e80834. doi:10.1371/journal.pone.0080834
Keywords: tau protein, transgenic mouse, positron emission tomography, magnetic resonance imaging, two-photon microscopy
Citation: Sahara N, Shimojo M, Ono M, Takuwa H, Febo M, Higuchi M and Suhara T (2017) In Vivo Tau Imaging for a Diagnostic Platform of Tauopathy Using the rTg4510 Mouse Line. Front. Neurol. 8:663. doi: 10.3389/fneur.2017.00663
Received: 16 October 2017; Accepted: 23 November 2017;
Published: 07 December 2017
Edited by:
Jesus Avila, Universidad Autonoma de Madrid, SpainReviewed by:
Felix Hernandez, Consejo Superior de Investigaciones Científicas (CSIC), SpainMar Perez, Universidad Autonoma de Madrid, Spain
Copyright: © 2017 Sahara, Shimojo, Ono, Takuwa, Febo, Higuchi and Suhara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naruhiko Sahara, c2FoYXJhLm5hcnVoaWtvQHFzdC5nby5qcA==