 Yuhai Zhao1,2
Yuhai Zhao1,2 Walter J. Lukiw1,3,4*
Walter J. Lukiw1,3,4*
- 1LSU Neuroscience Center, Louisiana State University Health Sciences Center New Orleans, New Orleans, LA, United States
- 2Department of Anatomy and Cell Biology, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 3Department of Ophthalmology, Louisiana State University Health Sciences Center New Orleans, New Orleans, LA, United States
- 4Department of Neurology, Louisiana State University Health Sciences Center New Orleans, New Orleans, LA, United States
The first indication of a potential mechanistic link between the pathobiology of the human gastrointestinal (GI)-tract microbiome and its contribution to the pathogenetic mechanisms of sporadic Alzheimer’s disease (AD) came a scant 4 years ago (1). Ongoing research continues to strengthen the hypothesis that neurotoxic microbial-derived components of the GI tract microbiome can cross aging GI tract and blood–brain barriers and contribute to progressive proinflammatory neurodegeneration, as exemplified by the AD-process. Of central interest in these recent investigations are the pathological roles played by human GI tract resident Gram-negative anaerobic bacteria and neurotropic viruses—two prominent divisions of GI tract microbiome-derived microbiota—which harbor considerable pathogenic potential. It is noteworthy that the first two well-studied microbiota—the GI tract abundant Gram-negative bacteria Bacteroides fragilis and the neurotropic herpes simplex virus-1 both share a final common pathway of NF-κB (p50/p65) activation and microRNA-146a induction with ensuing pathogenic stimulation of innate-immune and neuroinflammatory pathways. These appear to strongly contribute to the inflammation-mediated amyloidogenic neuropathology of AD. This communication: (i) will review recent research contributions that have expanded our understanding of the nature of the translocation of microbiome-derived neurotoxins-across biophysiological barriers; (ii) will assess multiple-recent investigations of the induction of the proinflammatory pathogenic microRNA-146a by these two prominent classes of human microbiota; and (iii) will discuss the role of molecular neurobiology and mechanistic contribution of polymicrobial infections to AD-type neuropathological change.
Overview
Containing 95% of the entire human microbiome, the gastrointestinal (GI) tract is the largest reservoir of microbes in the body (2, 3). Consisting of a densely packed, genetically diverse repository of about 1014 microorganisms, the human GI tract microbiome consists mostly of anaerobic bacterial and viral species with fungi, protozoa, archaebacteria and other microorganisms making up the remainder (1, 3–11). The contribution of these microbial species to human neurobiological health, aging, and disease is becoming increasingly recognized, and the molecular genetics, epigenetics, biophysics, and signaling mechanisms of the species involved and their abundance, speciation, and complexity in health and disease are becoming increasingly understood. One recently described benefit of GI tract microbiota is that they may help protect against inflammatory neurodegeneration, such as those encountered in Alzheimer’s disease (AD) brain, in part by supporting the generation of select short chain fatty acids which interfere with the formation and aggregation of toxic soluble amyloid beta (Aβ) peptides (12). Indeed, while normally confined within the healthy human GI tract microbiome, with aging and disease microbiota and/or their exuded neurotoxins may leak across normally protective biophysiological barriers inducing a persistent systemic inflammatory condition that may be an early indicator and biomarker for the onset of chronic inflammatory neurodegenerative disorders that include AD (2, 13–17). This article (i) will focus on recent advances in our understanding of the neurotropic herpes simplex virus-1 (HSV-1) and the GI tract abundant Gram-negative bacillus Bacteroides fragilis to AD-type neurological change; (ii) will evaluate several recent findings on the involvement of the inducible microRNA-146a (miRNA-146a) by these two prominent classes of human microbiota; and (iii) consider the possibility that polymicrobial infections involving both bacterial- and viral-derived neurotoxins may make a significant pathogenic contribution to chronic, insidious and fatal neurological diseases of the human central and peripheral nervous system (CNS and PNS).
HSV-1 and Sporadic AD
The icosahedral capsid-enveloped HSV-1 is a neurotropic, neuroinvasive group 1 member of the herpes virus family Herpesviridae (18–20). As a 155,000 base pair double-stranded DNA (dsDNA) virus, HSV-1 contains at least 74 genes and is known to be capable of establishing a persistent and lifelong latency in human CNS and PNS tissues (21–28). Interestingly, human populations are infected with at least 8 different types of herpes viruses, including HSV1 and HSV2 (also termed HHV1 and HHV2, involved in oral and genital herpes, respectively), varicella zoster virus (human herpesvirus HHV3), Epstein-Barr virus (HHV4), cytomegalovirus (HHV5), herpes lymphotropic virus (HHV6), human myeloradiculoneuropathy/encephalopathy virus (HHV7), and Kaposi sarcoma-associated herpesvirus (termed KSV or HHV8). In general herpes viruses: (i) are detectable in human nervous tissue and their presence is not related to either age or gender (27, 29); (ii) have variable patterns of activation that can be separated into low-reactivation and high-reactivation phenotypes (24, 30); (iii) are highly neuroinvasive, establishing themselves as a “persistent infection” of neurons and neuronal ganglia of both the CNS and PNS (27); (iv) are adapted to lifelong “latent” infection of their human hosts (4, 5, 23, 31); (v) are activated by physiological stimuli that involve stress, mediated in part by reactive nitrogen and oxygen species (4, 5, 18, 23, 27, 29, 32, 33); (vi) make extensive use of multiple immune-evasion strategies to shield themselves from the host innate-immune system (4, 5); (vii) when neuroactive are highly proinflammatory, and progressively and irreversibly incapacitate neurons (34); and (viii) induce proinflammatory microRNAs such as miRNA-146a in the host and induce AD-type inflammatory gene signaling immediately after HSV-1 infection of human neuronal-glial (HNG) cells in primary culture including the rounding up of cell bodies and retraction of neurites [(4, 5, 18, 35–37); Figure 1].
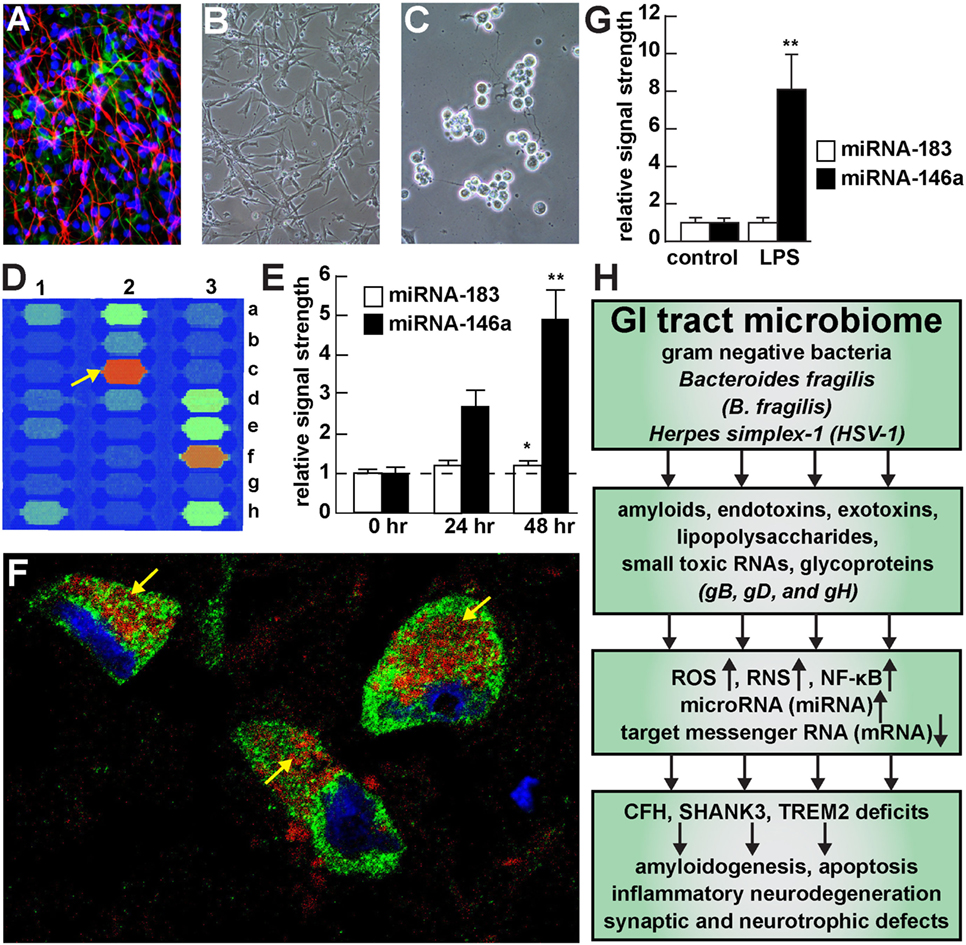
Figure 1. Viral and bacterial contribution to neuroinflammatory signaling in sporadic Alzheimer’s disease (AD) brain—(A) primary human neuronal-glial (HNG) cells after ~2 weeks in primary coculture; the cell density is approximately 75% neurons and 25% astroglia at ~60% confluency; human primary neuronal and glial “support” cell cocultures are utilized because human neuronal cells do not culture well by themselves (11); neuronal cells are stained with neuron-specific β-tubulin (red; λmax = 690 nm), glial cells are stained with glial-specific glial fibrillary acidic protein (GFAP; green; λmax = 525 nm), and nuclei are stained with DAPI/Hoechst 33258 stain (blue; λmax = 470 nm); photo magnification 30×; (B) control HNG cells in primary culture; as in previous panel; light microscopy 30×; (C) HNG cells exposed to HSV-1 for 48 h; note “rounding up of cell bodies and retraction of neurites”; (D) miRNA array analysis of an overexpressed proinflammatory miRNA-146a in HSV-1 treated HNG cells; on this miRNA array an upregulated miRNA-146a is indicated at position 2c (33); (E) time course of induction (0, 24, and 48 h) of a pathogenic miRNA-146a in HNG cells by HSV-1; at 48 h the abundance of miRNA-146a was almost fivefold over controls; miRNA-183 represents an unchanging miRNA control; (F) association of lipopolysaccharide (LPS) with the neuronal cytoplasm and the periphery of neuronal nuclei in AD neocortex—NeuN (neuron-specific green stain; λmax = 520 nm), LPS (red stain; λmax = 690 nm), and DAPI (blue stain; λmax = 470 nm); human superior temporal lobe AD neocortex (Brodmann A22); note organization of LPS into a “clathrin-like” lattice or “net” within the neuronal cell cytoplasm (yellow arrows); (G) induction of the proinflammatory miRNA-146a in LPS-stressed HNG cells is almost eightfold over controls after 48 h; (H) flow chart of the potential contribution of the GI tract-resident microbiome-abundant Gram-negative bacilli Bacteroides fragilis and herpes simplex-1 (HSV-1) to neuropathological pathways; stressed B. fragilis secrete a remarkable quantity of neurotoxins that include amyloids, endotoxins, exotoxins, LPS, and small toxic microRNA-like RNAs; upon HSV-1 invasion of human neurons cell surface proteins serve as receptors for viral entry and the HSV-1 glycoproteins gB, gD, and gH are required for infection of, and the maintenance of latency in human neurons; in both cases B. fragilis and HSV-1 in turn induce the evolution of reactive oxygen species (ROS), reactive nitrogen species (RNS), and the proinflammatory transcription factor NF-κB and upregulate a small family of NF-κB-sensitive microRNAs (miRNAs) including miRNA-146a that interact with the 3′-untranslated region (3′-UTR) of messenger RNA (mRNA) targets (1, 36, 37). In the case of B. fragilis signaling this drives the downregulation of complement factor H (CFH), the postsynaptic cytoskeletal SHANK3 and the triggering receptor expressed in myeloid/microglial cells TREM2; deficits in CFH, SHANK3, and TREM2 resulting in the inability to clear excessive amyloid from brain cells, amyloidogenesis, apoptosis, inflammatory neurodegeneration and synaptic and neurotropic deficits. Because all miRNA fractions were obtained from short postmortem interval (PMI) human tissue samples and miRNAs have been reported to have a high rate of depolymerization (degradation) under physiological, and especially pathophysiological conditions, only upregulated miRNAs were studied here; in fact miRNA upregulation and mRNA downregulation appears to be a very common posttranslational genetic regulatory mechanism in the human and murine CNS [see text (38, 39)]. Note that parts of Figure 1 have been considerably updated from a previous version (37, 40–42).
Herpes viruses in general, and HSV-1 in particular, are significant components of the human GI tract microbiome and also occupy a prominent role in human illnesses triggered by primary herpetic infection or reactivation of HSV from the latent state (30, 36, 43). Indeed HSV-1 presence is common in neuronal ganglia innervating the human GI tract, HSV-1-derived signaling molecules can act on enteric neurons to influence GI tract motility, and HSV-1 reactivating from these sites play a role in recurrent GI tract disorders, especially in immune-compromised or immune-incompetent humans (1, 30, 36, 43). Although an epidemiological link between HSV-1 infection and Alzheimer’s disease (AD) was first suggested almost 30 years ago (44), the molecular-genetic mechanism of this pathogenic association is yet to be fully elucidated. A preferential association of HSV-1 with trigeminal sensory ganglion and the incubation of HSV-1 with HNG cells in primary coculture results in a number of morphological, neurochemical, biophysical, and genetic changes to the neurons favorable to the propagation of the infecting agent and detrimental to the function of the host cells, enabling the latent occupation of the neuronal cell cytoplasm and/or genome by HSV-1. Reactivation of latent herpesviruses can directly alter host cytokine profiles through both viral expression of cytokine-like proteins and upregulation in the host expression of members of the arachidonic acid signaling cascade including interleukin-6 (IL-6), cytoplasmic phospholipase A2 (cPLA2), the inducible prostaglandin synthase cyclooxygenase-2, the neuroinflammatory cytokine interleukin-1beta (IL-1β), specific viral encoded and secreted small non-coding RNA (sncRNA) and microRNAs, and the modification and modulation of expression of host gene transcription pathways, such as that for nuclear factor κB [NF-κB (1, 45, 46)]. Interestingly, a generalized upregulation of inflammatory signaling has been associated with both HSV-1 infection of stressed brain cells in primary culture and in AD, where there occurs increased expression of NF-κB-regulated proinflammatory microRNAs such as miRNA-146a (1, 25, 26, 46). In turn an upregulated miRNA-146a (i) is known to be important in the significant downregulation of complement factor H (CFH) and a chronic stimulation of an atypical and pathogenic innate-immune response (35, 36, 47–49); (ii) decreases the expression of the proline-rich postsynapse-associated synaptogenic glycoprotein SHANK3 with resulting synaptic disorganization and functional loss (36, 50–52); and (iii) induces a downregulation in the expression of the triggering receptor expressed in myeloid/microglial cells (TREM2) with an ensuing stimulation of tau neuropathology and deficits in the phagocytosis and clearance of amyloid from the neuronal parenchyma (36, 53, 54). These actions combined indicate a prominent role for HSV-1-induced miRNA-146a in the activation of key elements of the arachidonic acid cascade and proinflammatory pathways known to contribute to Alzheimer-type neuropathological change and evasion of HSV-1 from the host complement system (Figure 1). Interestingly, for infectivity to be attained and sustained, the dsDNA HSV genome must enter the host cell through means of fusion of its envelope with the host cellular membrane and/or via endocytosis involving the HSV-1 viral entry glycoproteins gB, gD, and gH and other elements of the HSV-1 secretome. Of interest is that many of these same glycoproteins are utilized by HSV-1 to promote HSV-1 infectivity and survival, and counteract host antiviral innate-immune responses—many of these later responses involve miRNA-146a signaling (18, 19, 55).
B. fragilis and Sporadic AD
Bacteroides fragilis is a Gram-negative anaerobic bacterium and major component of the human GI tract microbiome (1, 56, 57). The absolute abundance of B. fragilis in the human GI tract appears to be regulated in large part by the intake of dietary fiber such that diets low in soluble fiber tend to proliferate anaerobic Gram-negative bacterial species (6, 7, 57, 58). B. fragilis secretes a remarkably varied array of highly proinflammatory neurotoxins which, when released from the confines of the healthy GI tract into the systemic circulation and neurovasculature are highly toxic to nervous tissues of the CNS (7, 58). One important aspect of this process is the transfer of these B. fragilis toxins (BFTs) through the GI tract and the blood–brain barrier (BBB), dynamic structures which are known to become considerably more “leaky” with aging and disease. Multiple reviews on GI tract and BBB structure and function have been recently published including (i) the role of BBB breakdown and dysfunction in neurodegenerative process and how targeting the BBB can influence the course of AD (59); (ii) the role of human ATP-binding cassette transporters across lipid membranes of the BBB in AD (60); (iii) the design, development and use of polymer-based, lipid-based, and inorganic-based nanocarriers to aid in biophysiological barrier research, and the design of drugs which can cross these barriers (61); and (iv) in depth studies on brain capillary endothelial cells, pericytes, astrocytes, platelets, and basement membranes and their interactions that form the basis for the neurovascular unit and the BBB and GI tract barriers (62, 63).
When stressed, overpopulated or pathogenically stimulated, B. fragilis releases a remarkably complex array of endotoxins and exotoxins (such as fagilysin), lipooligosaccahrides (LOS), lipopolysaccharide (LPS), including an extremely proinflammatory B. fragilis LPS (BF-LPS), microRNA-like sncRNA, and a wide variety of bacterial-derived amyloids (9–11, 56, 57, 64–66). These neurotoxins may have both PNS and CNS effects. For example, Bacteroides fragilis endotoxins are a leading cause of anaerobic bacteremia, sepsis and/or systemic inflammatory distress in the PNS through their generation of the highly proinflammatory zinc metalloprotease metalloproteinase BFT fragilysin (67, 68). BFT has recently been shown to disrupt epithelial cells of GI tract barriers via cleavage of the synaptic adhesion zonula adherens protein E-cadherin (67, 69, 70). BFT also has strong CNS effects in the induction of NF-κB signaling and miRNA-146a upregulation in HNG cells in primary culture, cells originally derived from human CNS tissues (9–11, 65, 68). Similarly BF-LPS, a characteristic component of the outer leaflet of the outer membrane of Gram-negative bacteria B. fragilis shed into the extracellular space plays a key role in host–pathogen interaction of the innate-immune system in part via the induction of NF-κB (33, 41, 42, 65, 71–73). Of related interest is that while microbiome-derived, secreted LPS, proteolytic endotoxins, and amyloid monomers are generally soluble as monomers, over time they gradually form into insoluble fibrous protein aggregates that are microglial cell activating and characteristic of several common, age-related disorders of the human systemic circulation, PNS and CNS including systemic inflammatory response syndrome, multiple sclerosis, prion disease, and AD (36, 53, 74–77). Again, the one common denominator regarding the pathogenic actions of these neurotoxins is their ability to upregulate NF-κB and NF-κB-sensitive genes, including the significant transcriptional upregulation of a small family of NF-κB-sensitive proinflammatory miRNAs such as miRNA-146a (18, 35–37, 53, 78–83). Interestingly, upregulated NF-κB-miRNA-146a circuits have also been implicated in other progressive neurodegenerative diseases that include Down’s syndrome (Trisomy 21) and the human prion diseases sporadic Creutzfeldt–Jakob disease and Gerstmann–Straussler–Scheinker syndrome (41, 42, 81, 82, 84).
Unanswered Questions
Many unanswered questions remain concerning the role of microbiome-derived neurotoxins and their contribution to the progressive inflammatory neurodegeneration of sporadic AD—and these include, prominently: Does life-long exposure to specific infectious agents predispose one to develop AD at a later age? How do the secreted toxins from B. fragilis and HSV-1, and other microbiota, progressively leak across the GI tract barrier into the systemic circulation and on through the BBB to CNS compartments? Are other transcription factors besides NF-κB and other proinflammatory microRNAs besides miRNA-146a involved in driving AD-type neurodegeneration? What combinations of bacterial- and viral-based neurotoxins and perhaps other microbiome-derived toxins are the most efficient in inducing a progressive proinflammatory neurodegeneration? Do microbial-derived neurotoxins or polymicrobial infections exhibit synergism in their toxicities toward neural cells of the PNS and CNS? Do prebiotics, carbohydrates and specialized plant-based dietary fibers that nourish beneficial microbes already in the GI tract, or probiotics, consisting of beneficial “health-promoting” microbes have any role in AD onset or progression? Would it possible to tailor a life-long dietary intake that minimizes the risk of CNS-based age-related neuroinflammatory diseases such as AD? Is it possible to devise prebiotic, probiotic, anti-neurotoxin, anti-NF-κB, anti-microRNA, or combinations of these approaches for therapeutic benefit in the clinical management of AD?
Conclusion
The potential contribution of neurotoxic components of the human GI tract microbiome to the initiation, development and/or progression of the sporadic AD process appears to be both complex and significant. We still remain in the early stages of understanding the GI tract microbiome-brain axis in sporadic AD, and the biophysics, molecular mechanics, genetics and epigenetics of just how this is accomplished is becoming increasingly understood. Major bacterial and viral species of the human microbiome such as the Gram-negative bacillus B. fragilis and the neurotropic HSV-1 secrete a remarkably complex array of highly pathogenic proinflammatory neurotoxins which, when released from the confines of a healthy GI tract, are highly toxic to neurons of the CNS and PNS. Interestingly, while an environmental cause for sporadic AD has often been suggested, a strong source of powerful neurotoxins already resides within us. For example, BF-LPS represents an internally generated GI tract microbiome-derived neurotoxin capable of driving AD-type change and has enormous potential to initiate and/or propagate inflammatory neurodegeneration along the gut–brain axis. Some understudied aspects of the bioavailability of GI tract generated neurotoxins are (i) their translocation through the GI tract and BBB that involves dynamic structures which are known to become more “leaky” with aging and disease; (ii) the direct influence of these endotoxins, such as fragilysin, which targets zonula adherens protein E-cadherin and cell-cell adhesion; and (iii) the molecular exchanges between the GI tract, the systemic circulation and parenchyma of the central CNS (59, 61, 67, 69, 70). To cite another important example, BF-LPS (i) represents an internally generated GI tract microbiome-derived neurotoxin capable of driving AD-type change and (ii) has enormous potential to initiate and/or propagate inflammatory neurodegeneration along the GI tract–CNS axis (9–11, 33, 37, 41, 42). It is remarkable that of the few GI tract-derived microbes so far studied that all appear to be employing an NF-κB-miRNA-146a signaling pathway that promotes amyloidogenesis, apoptosis, inflammatory neurodegeneration, synaptic and neurotropic defects—all of which are characteristic aspects of AD-type neuropathology (Figure 1) (85–87). Furthering our molecular and mechanistic understanding of how individual secreted components of the GI tract microbiome—including potentially neurotoxic exudates consisting of endotoxins and exotoxins, fragilysin, select lipoglycans, LOS and LPS, specific LPS such as BF-LPS, amyloids and sncRNAs—affect the PNS and CNS may uncover potential and novel strategies for GI tract-based modulation of neural function and the more efficacious clinical treatment of terminal neurological disease.
Ethics Statement
All procedures involving murine and postmortem human tissues were followed in strict accordance with the ethics review board policies at donor institutions, and the Institutional Biosafety Committee/Institutional Review Board ethical guidelines at the LSU Health Sciences Center, LA 70112, USA.
Author Contributions
YZ and WL analyzed and discussed the review literature; YZ performed the experiments; and WL wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in this article was presented in part at the Vavilov Institute of General Genetics Autumn 2017 Seminar Series (Институт общей генетики имени Вавилова Осень 2017 Семинар серии) in Moscow, Russia, October 2017 and at the Society for Neuroscience (SFN) Annual Meeting, Washington, DC, USA, November 2017. Sincere thanks are extended to Drs. L. Carver, L. Cong, J. G. Cui, F. Culicchia, C. Eicken, V. Jaber, K. Navel, A. I. Pogue, W. Poon, and the late Drs. J. M. Hill and T. P. Kruck for helpful discussions in this research area, for short postmortem interval (PMI) human brain and retinal tissues or extracts, for initial bioinformatics and data interpretation, and to D. Guillot for expert technical assistance and medical artwork. Thanks are also extended to the University of California at Irvine Brain Bank, the University of Maryland Brain Bank, the Harvard Brain Tissue Resource Center, the LSU School of Medicine and the many neuropathologists, physicians, and researchers in the US and Canada who have provided high quality, short PMI human brain tissue fractions for scientific analysis. Research on microRNAs, proinflammatory and pathogenic signaling in the Lukiw laboratory involving the microbiome, the innate-immune response, amyloidogenesis, synaptogenesis, and neuroinflammation in AD, prion and in other neurological diseases was supported through an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN) and NIH grants NEI EY006311, NIA AG18031, and NIA AG038834 (WL). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging, the National Center for Research Resources, or the National Institutes of Health.
References
1. Bhattacharjee S, Lukiw WJ. Alzheimer’s disease and the microbiome. Front Cell Neurosci (2013) 7:153. doi:10.3389/fncel.2013.00153
2. Friedland RP, Chapman MR. The role of microbial amyloid in neurodegeneration. PLoS Pathog (2017) 13(12):e1006654. doi:10.1371/journal.ppat.1006654
3. Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep (2017) 7(1):13537. doi:10.1038/s41598-017-13601-y
4. Hill JM, Bhattacharjee S, Pogue AI, Lukiw WJ. The gastrointestinal tract microbiome and potential link to Alzheimer’s disease. Front Neurol (2014) 5:43. doi:10.3389/fneur.2014.00043
5. Hill JM, Clement C, Pogue AI, Bhattacharjee S, Zhao Y, Lukiw WJ. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front Aging Neurosci (2014) 6:127. doi:10.3389/fnagi.2014.00127
6. Alkasir R, Li J, Li X, Jin M, Zhu B. Human gut microbiota: the links with dementia development. Protein Cell (2017) 8:90–102. doi:10.1007/s13238-016-0338-6
7. Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci (2017) 20:145–55. doi:10.1038/nn.4476
8. Westfall S, Lomis N, Kahouli I, Dia SY, Singh SP, Prakash S. Microbiome, probiotics and neurodegenerative diseases. Cell Mol Life Sci (2017) 74:3769–87. doi:10.1007/s00018-017-2550-9
9. Zhao Y, Cong L, Jaber V, Lukiw WJ. Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front Immunol (2017) 8:1064. doi:10.3389/fimmu.2017.01064
10. Zhao Y, Jaber V, Lukiw WJ. Secretory products of the human GI tract microbiome and their potential impact on AD: detection of LPS in AD hippocampus. Front Cell Infect Microbiol (2017) 7:318. doi:10.3389/fcimb.2017.00318
11. Zhao Y, Cong L, Lukiw WJ. Lipopolysaccharide (LPS) accumulates in neocortical neurons of alzheimer’s disease (AD) brain and impairs transcription in human neuronal-glial primary co-cultures. Front Aging Neurosci (2017) 9:407. doi:10.3389/fnagi.2017.00407
12. Ho L, Ono K, Tsuji M, Mazzola P, Singh R, Pasinetti GM. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev Neurother (2018) 18(1):83–90. doi:10.1080/14737175.2018.1400909
13. Lim SL, Rodriguez-Ortiz CJ, Kitazawa M. Infection, systemic inflammation, and Alzheimer’s disease. Microbes Infect (2015) 17:549–56. doi:10.1016/j.micinf.2015.04.004
14. Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement (2016) 12:719–32. doi:10.1016/j.jalz.2016.02.010
15. Lukiw WJ. The microbiome, microbial-generated proinflammatory neurotoxins, and Alzheimer’s disease. J Port Health Sci (2017) 5(4):393–6. doi:10.1016/j.jshs.2016.08.008
16. Magalhães TNC, Weiler M, Teixeira CVL, Hayata T, Moraes AS, Boldrini VO, et al. Systemic inflammation and multimodal biomarkers in amnestic mild cognitive impairment and Alzheimer’s disease. Mol Neurobiol (2017). doi:10.1007/s12035-017-0795-9
17. Walker KA, Hoogeveen RC, Folsom AR, Ballantyne CM, Knopman DS, Windham BG, et al. Midlife systemic inflammatory markers are associated with late-life brain volume: the ARIC study. Neurology (2017) 89:2262–70. doi:10.1212/WNL.0000000000004688
18. Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport (2009) 20:1500–5. doi:10.1097/WNR.0b013e3283329c05
19. Su C, Zhan G, Zheng C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J (2016) 13:38. doi:10.1186/s12985-016-0495-5
20. Rechenchoski DZ, Faccin-Galhardi LC, Linhares REC, Nozawa C. Herpesvirus: an underestimated virus. Folia Microbiol (Praha) (2017) 62:151–6. doi:10.1007/s12223-016-0482-7
21. McGeoch DJ. The genome of herpes simplex virus: structure, replication and evolution. J Cell Sci Suppl (1987) 7:67–94. doi:10.1242/jcs.1987.Supplement_7.6
22. Lehtinen M, Koivisto V, Lahtinen P, Lehtinen T, Aaran RK, Leinikki P. Phospholipase A2 activity is copurified together with herpes simplex virus-specified Fc receptor proteins. Intervirology (1988) 29:50–6. doi:10.1159/000150028
23. Hill JM, Lukiw WJ, Gebhardt BM, Higaki S, Loutsch JM, Myles ME, et al. Gene expression analyzed by microarrays in HSV-1 latent mouse trigeminal ganglion following heat stress. Virus Genes (2001) 23:273–80. doi:10.1023/A:1012517221937
24. Toma HS, Murina AT, Areaux RG, Neumann DM, Bhattacharjee PS, Foster TP, et al. Ocular HSV-1 latency, reactivation and recurrent disease. Semin Ophthalmol (2008) 23:249–73. doi:10.1080/08820530802111085
25. Lam MM, Mapletoft JP, Miller MS. Abnormal regulation of the antiviral response in neurological/neurodegenerative diseases. Cytokine (2016) 88:251–8. doi:10.1016/j.cyto.2016.09.002
26. McNamara J, Murray TA. Connections between herpes simplex virus type 1 and Alzheimer’s disease pathogenesis. Curr Alzheimer Res (2016) 13:996–1005. doi:10.2174/1567205013666160314150136
27. Sochocka M, Zwolińska K, Leszek J. The infectious etiology of Alzheimer’s disease. Curr Neuropharmacol (2017) 15:996–1009. doi:10.2174/1570159X15666170313122937
28. Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, Waubant E. The gut microbiome in human neurological disease. Ann Neurol (2017) 81:369–82. doi:10.1002/ana.24901
29. Hill JM, Ball MJ, Neumann DM, Azcuy AM, Bhattacharjee PS, Bouhanik S, et al. The high prevalence of herpes simplex virus type 1 DNA in human trigeminal ganglia is not a function of age or gender. J Virol (2008) 82:8230–4. doi:10.1128/JVI.00686-08
30. Dreyfus DH. Herpesviruses and the microbiome. J Allergy Clin Immunol (2013) 132:1278–86. doi:10.1016/j.jaci.2013.02.039
31. Higaki S, Gebhardt B, Lukiw W, Thompson H, Hill J. Gene expression profiling in the HSV-1 latently infected mouse trigeminal ganglia following hyperthermic stress. Curr Eye Res (2003) 26:231–8. doi:10.1076/ceyr.26.3.231.14892
32. Hill JM, Nolan NM, McFerrin HE, Clement C, Foster TP, Halford WP, et al. HSV-1 latent rabbits shed viral DNA into their saliva. Virol J (2012) 9:221. doi:10.1186/1743-422X-9-221
33. Hill JM, Lukiw WJ. Microbial-generated amyloids and Alzheimer’s disease (AD). Front Aging Neurosci (2015) 7:9. doi:10.3389/fnagi.2015.00009
34. Harris SA, Harris EA. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer’s disease. J Alzheimers Dis (2015) 48:319–533. doi:10.3233/JAD-142853
35. Lukiw WJ, Zhao Y, Cui JG. An NF-kB-sensitive miRNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem (2008) 283:31315–22. doi:10.1074/jbc.M805371200
36. Jaber V, Zhao Y, Lukiw WJ. Alterations in micro RNA-messenger RNA (miRNA-mRNA) coupled signaling networks in sporadic AD. J Alzheimers Dis Parkinsonism (2017) 7:312. doi:10.4172/2161-0460.1000312
37. Pogue AI, Lukiw WJ. Up-regulated pro-inflammatory microRNAs (miRNAs) in Alzheimer’s disease (AD) and age-related macular degeneration (AMD). Cell Mol Neurobiol (2018). doi:10.1007/s10571-017-0572-3
38. Clement C, Hill JM, Dua P, Culicchia F, Lukiw WJ. Analysis of RNA from Alzheimer’s disease post-mortem brain tissues. Mol Neurobiol (2016) 53:1322–8. doi:10.1007/s12035-015-9105-6
39. Rissland OS, Subtelny AO, Wang M, Lugowski A, Nicholson B, Laver JD, et al. The influence of microRNAs and poly(A) tail length on endogenous mRNA-protein complexes. Genome Biol (2017) 18:211. doi:10.1186/s13059-017-1330-z
40. Zhao Y, Lukiw WJ. Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J Nat Sci (2015) 1(7):e138.
41. Zhao Y, Dua P, Lukiw WJ. Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s disease (AD). J Alzheimers Dis Parkinsonism (2015) 5(1):177.
42. Zhao Y, Pogue AI, Lukiw WJ. MicroRNA (miRNA) signaling in the human CNS in sporadic Alzheimer’s disease (AD)-novel and unique pathological features. Int J Mol Sci (2015) 16:30105–16. doi:10.3390/ijms161226223
43. Lavery EA, Coyle WJ. Herpes simplex virus and the alimentary tract. Curr Gastroenterol Rep (2008) 10:417–23. doi:10.1007/s11894-008-0078-8
44. Walker DG, O’Kusky JR, McGeer PL. In situ hybridization analysis for herpes simplex virus nucleic acids in Alzheimer disease. Alzheimer Dis Assoc Disord (1989) 3:123–31. doi:10.1097/00002093-198903030-00001
45. Hill JM, Clement C. Herpes simplex virus type 1 DNA in human corneas: what are the virological and clinical implications? J Infect Dis (2009) 200(1):1–4. doi:10.1086/599330
46. Panday A, Inda ME, Bagam P, Sahoo MK, Osorio D, Batra S. Transcription factor NF-κB: an update on intervention strategies. Arch Immunol Ther Exp (Warsz) (2016) 64:463–83. doi:10.1007/s00005-016-0405-y
47. Lukiw WJ, Alexandrov PN. Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol Neurobiol (2012) 46:11–9. doi:10.1007/s12035-012-8234-4
48. Lukiw WJ, Surjyadipta B, Dua P, Alexandrov PN. Common micro RNAs (miRNAs) target complement factor H (CFH) regulation in Alzheimer’s disease (AD) and in age-related macular degeneration (AMD). Int J Biochem Mol Biol (2012) 3(1):105–16.
49. Shaw PX, Stiles T, Douglas C, Ho D, Fan W, Du H, et al. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci (2016) 3(2):196–221.
50. Gong Y, Lippa CF, Zhu J, Lin Q, Rosso AL. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer’s disease. Brain Res (2009) 1292:191–8. doi:10.1016/j.brainres.2009.07.056
51. Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J (2010) 277:3051–67. doi:10.1111/j.1742-4658.2010.07719.x
52. Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol (2014) 74:113–22. doi:10.1002/dneu.22128
53. Zhao Y, Jaber V, Lukiw WJ. Over-expressed pathogenic miRNAs in Alzheimer’s disease (AD) and prion disease (PrD) drive deficits in TREM2-mediated Aβ42 peptide clearance. Fron Aging Neurosci (2016) 8:140. doi:10.3389/fnagi.2016.00140
54. Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener (2017) 12:56. doi:10.1186/s13024-017-0197-5
55. Miettinen JJ, Matikainen S, Nyman TA. Global secretome characterization of herpes simplex virus 1-infected human primary macrophages. J Virol (2012) 86:12770–8. doi:10.1128/JVI.01545-12
56. Hofer U. Microbiome: B. fragilis and the brain. Nat Rev Microbiol (2014) 12:76–7. doi:10.1038/nrmicro3197
57. Yang NJ, Chiu IM. Bacterial signaling to the nervous system through toxins and metabolites. J Mol Biol (2017) 429:587–605. doi:10.1016/j.jmb.2016.12.023
58. Heinritz SN, Weiss E, Eklund M, Aumiller T, Heyer CM, Messner S, et al. Impact of a high-fat or high-fiber diet on intestinal microbiota and metabolic markers in a pig model. Nutrients (2016) 8(5):E317. doi:10.3390/nu8050317
59. Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med (2017) 214:3151–69. doi:10.1084/jem.20171406
60. Pereira CD, Martins F, Wiltfang J, da Cruz E, Silva OAB, Rebelo S. ABC transporters are key players in Alzheimer’s disease. J Alzheimers Dis (2018) 61(2):463–85. doi:10.3233/JAD-170639
61. Tsou YH, Zhang XQ, Zhu H, Syed S, Xu X. Drug delivery to the brain across the blood-brain barrier using nanomaterials. Small (2017) 13(43). doi:10.1002/smll.201701921
62. Mancuso ME, Santagostino E. Platelets: much more than bricks in a breached wall. Br J Haematol (2017) 178:209–19. doi:10.1111/bjh.14653
63. Yamazaki Y, Kanekiyo T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int J Mol Sci (2017) 18:E1965. doi:10.3390/ijms18091965
64. Foster JA, Lyte M, Meyer E, Cryan JF. Gut microbiota and brain function: an evolving field in neuroscience. Int J Neuropsychopharmacol (2016) 19:yv114. doi:10.1093/ijnp/pyv114
65. Lukiw WJ. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front Microbiol (2016) 7:1544. doi:10.3389/fmicb.2016.01544
66. Mancuso C, Santangelo R. Alzheimer’s disease and gut microbiota modifications: the long way between preclinical studies and clinical evidence. Pharmacol Res (2018) 129:329–36. doi:10.1016/j.phrs.2017.12.009
67. Choi VM, Herrou J, Hecht AL, Teoh WP, Turner JR, Crosson S, et al. Activation of Bacteroides fragilis toxin by a novel protease contributes to anaerobic sepsis in mice. Nat Med (2016) 22:563–7. doi:10.1038/nm.4077
68. Fathi P, Wu S. Isolation, detection, and characterization of enterotoxigenic Bacteroides fragilis in clinical samples. Open Microbiol J (2016) 10:57–63. doi:10.2174/1874285801610010057
69. Seong E, Yuan L, Arikkath J. Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh Migr (2015) 9:202–13. doi:10.4161/19336918.2014.994919
70. Zhan LS, Davies SS. Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med (2016) 8:46. doi:10.1186/s13073-016-0296-x
71. Jiang Q, Jin S, Jiang Y, Liao M, Feng R, Zhang L, et al. Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol Neurobiol (2017) 54(1):594–600. doi:10.1007/s12035-015-9670-8
72. Maldonado RF, Sá-Correia I, Valvano MA. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol Rev (2016) 40:480–93. doi:10.1093/femsre/fuw007
73. Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem (2016) 136:457–74. doi:10.1111/jnc.13411
74. Asti A, Gioglio L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J Alzheimers Dis (2014) 39:169–79. doi:10.3233/JAD-131394
75. Clark IA, Vissel B. Amyloid β: one of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate AD. Br J Pharmacol (2015) 172:3714–27. doi:10.1111/bph.13181
76. Devier DJ, Lovera JF, Lukiw WJ. Increase in NF-κB-sensitive miRNA-146a and miRNA-155 in multiple sclerosis (MS) and pro-inflammatory neurodegeneration. Front Mol Neurosci (2015) 8:5. doi:10.3389/fnmol.2015.00005
77. Richards RI, Robertson SA, O’Keefe LV, Fornarino D, Scott A, Lardelli M, et al. The enemy within: innate surveillance-mediated cell death, the common mechanism of neurodegenerative disease. Front Neurosci (2016) 10:193. doi:10.3389/fnins.2016.00193
78. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103:12481–6. doi:10.1073/pnas.0605298103
79. Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TP, Percy ME, et al. Characterization of an NF-kB-regulated, miRNA-146a mediated down-regulation of CFH in metal-sulfate-stressed human brain cells. J Inorg Biochem (2009) 103:1591–5. doi:10.1016/j.jinorgbio.2009.05.012
80. Lukiw WJ. NF-kB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp Neurol (2012) 235:484–90. doi:10.1016/j.expneurol.2011.11.022
81. Boese AS, Saba R, Campbell K, Majer A, Medina S, Burton L, et al. MicroRNA abundance is altered in synaptoneurosomes during prion disease. Mol Cell Neurosci (2016) 71:13–24. doi:10.1016/j.mcn.2015.12.001
82. Arena A, Iyer AM, Milenkovic I, Kovacs GG, Ferrer I, Perluigi M, et al. Developmental expression and dysregulation of miRNA-146a and miRNA-155 in Down’s syndrome and mouse models of Down’s syndrome and Alzheimer’s disease. Curr Alzheimer Res (2017) 14:1305–17. doi:10.2174/1567205014666170706112701
83. Gupta P, Bhattacharjee S, Sharma AR, Sharma G, Lee SS, Chakraborty C. miRNAs in AD – a therapeutic perspective. Curr Alzheimer Res (2017) 14:1198–206. doi:10.2174/1567205014666170829101016
84. Lukiw WJ, Dua P, Pogue AI, Eicken C, Hill JM. Upregulation of micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt-Jakob disease (sCJD) and Gerstmann-Straussler-Scheinker (GSS) syndrome. J Toxicol Environ Health (2011) 74:1460–8. doi:10.1080/15287394.2011.618973
85. Lukiw WJ. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem Res (2004) 29:1287–97. doi:10.1023/B:NERE.0000023615.89699.63
86. Lukiw WJ, Cui JG, Yuan LY, Bhattacharjee PS, Corkern M, Clement C, et al. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport (2010) 21:922–7. doi:10.1097/WNR.0b013e32833da51a
Keywords: Alzheimer’s disease, Bacteroides fragilis, herpes simplex virus-1, microRNA-146a, polymicrobial infections
Citation: Zhao Y and Lukiw WJ (2018) Microbiome-Mediated Upregulation of MicroRNA-146a in Sporadic Alzheimer’s Disease. Front. Neurol. 9:145. doi: 10.3389/fneur.2018.00145
Received: 26 December 2017; Accepted: 27 February 2018;
Published: 19 March 2018
Edited by:
Karen-Anne McVey Neufeld, McMaster University, CanadaReviewed by:
Karen Ann Scott, University College Cork, IrelandRichard O’Connor, Icahn School of Medicine at Mount Sinai, United States
Copyright: © 2018 Zhao and Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter J. Lukiw, d2x1a2l3QGxzdWhzYy5lZHU=