 Yulan Xiong
Yulan Xiong Jianzhong Yu
Jianzhong Yu- Department of Anatomy and Physiology, Kansas State University College of Veterinary Medicine, Manhattan, KS, United States
Parkinson’s disease (PD) is recognized as the second most common neurodegenerative disorder after Alzheimer’s disease. Unfortunately, there is no cure or proven disease modifying therapy for PD. The recent discovery of a number of genes involved in both sporadic and familial forms of PD has enabled disease modeling in easily manipulable model systems. Various model systems have been developed to study the pathobiology of PD and provided tremendous insights into the molecular mechanisms underlying dopaminergic neurodegeneration. Among all the model systems, the power of Drosophila has revealed many genetic factors involved in the various pathways, and provided potential therapeutic targets. This review focuses on Drosophila models of PD, with emphasis on how Drosophila models have provided new insights into the mutations of dominant genes causing PD and what are the convergent mechanisms.
Introduction
Parkinson’s disease (PD) is recognized as the most common movement disorder and the second most common neurodegenerative disorder after Alzheimer’s disease (1). The classical motor features including akinesia, resting tremor, muscle rigidity, and postural imbalance are clinical symptoms in PD patients, and the none-motor features including cognitive impairment, psychiatric symptoms, sleep disorders, autonomic dysfunction, pain, and fatigue also frequently occur (1). The progressive degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta is the cause for the cardinal symptoms (2). Although the majority of PD cases appear to be sporadic, the identification of causative genes that cause familial forms of PD has led to important insights into the pathogenesis of this progressive neurodegenerative disease (3). To date, genes encoding α-synuclein (α-Syn), leucine-rich repeat kinase 2 (LRRK2), Parkin, phosphatase and tensin homolog deleted on chromosome 10-induced putative kinase 1 (PINK1), DJ-1, vacuolar protein sorting 35 (VPS35), and glucocerebrosidase (GBA), among others are associated with genetic forms of PD that closely resemble idiopathic PD (3–8). Among these genes, LRRK2, α-synuclein, GBA, and VPS35 are the dominant traits, and parkin, DJ-1, and PINK1 are the recessive genes. Various model systems have been developed to study the function of PD-causing genes and provided tremendous insights into the molecular mechanisms underlying DA neurodegeneration. While few of genetic models in rodents recapitulate the cardinal features of PD, the power of Drosophila has revealed many genetic factors involved in the various pathways, and provided potential therapeutic targets. Here, we focus on the dominant genes causing PD. We discuss how Drosophila models have provided new insights into the mutations of dominant genes causing PD and what are the convergent mechanisms.
Drosophila as a Model in the Study of PD
Drosophila melanogaster, as a non-mammalian animal, provides a simple, yet powerful, in vivo system to model PD pathobiology. Drosophila has well-defined nerve system. Particularly, in adult brain, Drosophila has distinct DA neuronal clusters including about 200 DA neurons and displays complicated behaviors mimicking some human behaviors which are DA dependent. Both transgenic and knockout approaches have been utilized to develop Drosophila models of PD. The Drosophila Gal4/UAS system is a powerful tool for targeted transgene expression and has been used to direct selective expression of mutant PD genes. As a simple organism, Drosophila provides great advantages in conducting genome-wide modifier screenings and high-throughput drug screenings. Modifier screenings allow analyses of genome-wide genetic interactions based on the modification of given phenotypes and further identify components of diverse signaling pathways involved in PD pathogenesis.
Several steps can be taken to establish and utilize Drosophila models to study PD:
(1) Develop Drosophila lines carrying mutant PD genes,
(2) Characterize the phenotypes of the Drosophila models and determine whether they recapitulate the pathogenesis of the disease,
(3) Explore the molecular mechanisms underlying the phenotypes,
(4) Identify genetic modifiers that suppress or enhance the disease phenotypes through genetic screenings, and dissect the signaling pathways and pathogenic mechanisms involved in pathogenesis,
(5) Screen for drug candidates,
(6) Study the impact of environmental (e.g., toxin) or aging influence in combination with genetic factors on the pathogenesis of PD.
Modeling LRRK2-Associated PD in Drosophila
Mutations in the LRRK2 gene (PARK8, dardarin) is the most common known genetic cause of PD and cause late-onset, autosomal dominant PD with age-related penetrance and clinical features identical to late-onset sporadic PD (9, 10). LRRK2 is a large multifunctional protein about 280 kD. It includes two important enzymatic domains, which are a GTPase domain and a kinase domain, and several protein interaction domains including a LRRK2-specific repeat domain, a leucine-rich repeat, and a WD40 repeat (11–13). Disease causing mutations are found in both enzymatic domains, indicating their importance in disease pathogenesis (11, 12). The most prevalent LRRK2 mutation, G2019S, within the kinase domain, accounts for ~1% of sporadic late-onset PD and 5–6% of familial PD worldwide (14). In North African Arabs and Ashkenazi Jews, the frequency of LRRK2–G2019S mutation can be as high as 30–40% in PD patients (15, 16). Patients with the G2019S mutation exhibit Lewy bodies (LBs) in most cases and incomplete penetrance even at advanced ages (1). However, mutations in the GTPase domain such as R1441 C/G often vary on LB pathology and exhibit nearly complete penetrance (9, 10). This suggests that these pathogenic mutations may cause disease via distinct pathogenic pathways/mechanisms. Tremendous work suggests LRRK2 GTPase and kinase enzyme activities may reciprocally regulate each other to direct LRRK2 functions in diverse cellular signaling pathways (17, 18). LRRK2 is demonstrated to be involved in protein translation, vesicle trafficking, mitochondrial function, lysosomal–autophagy, and cytoskeletal dynamics (13, 18–22). However, how LRRK2 mutations cause neurodegeneration in PD still need to be defined. To this end, various animal models of LRRK2-associated PD have been generated (23–25). Among these models, LRRK2 Drosophila models have provided unique and critical insights on LRRK2 functions (Table 1).
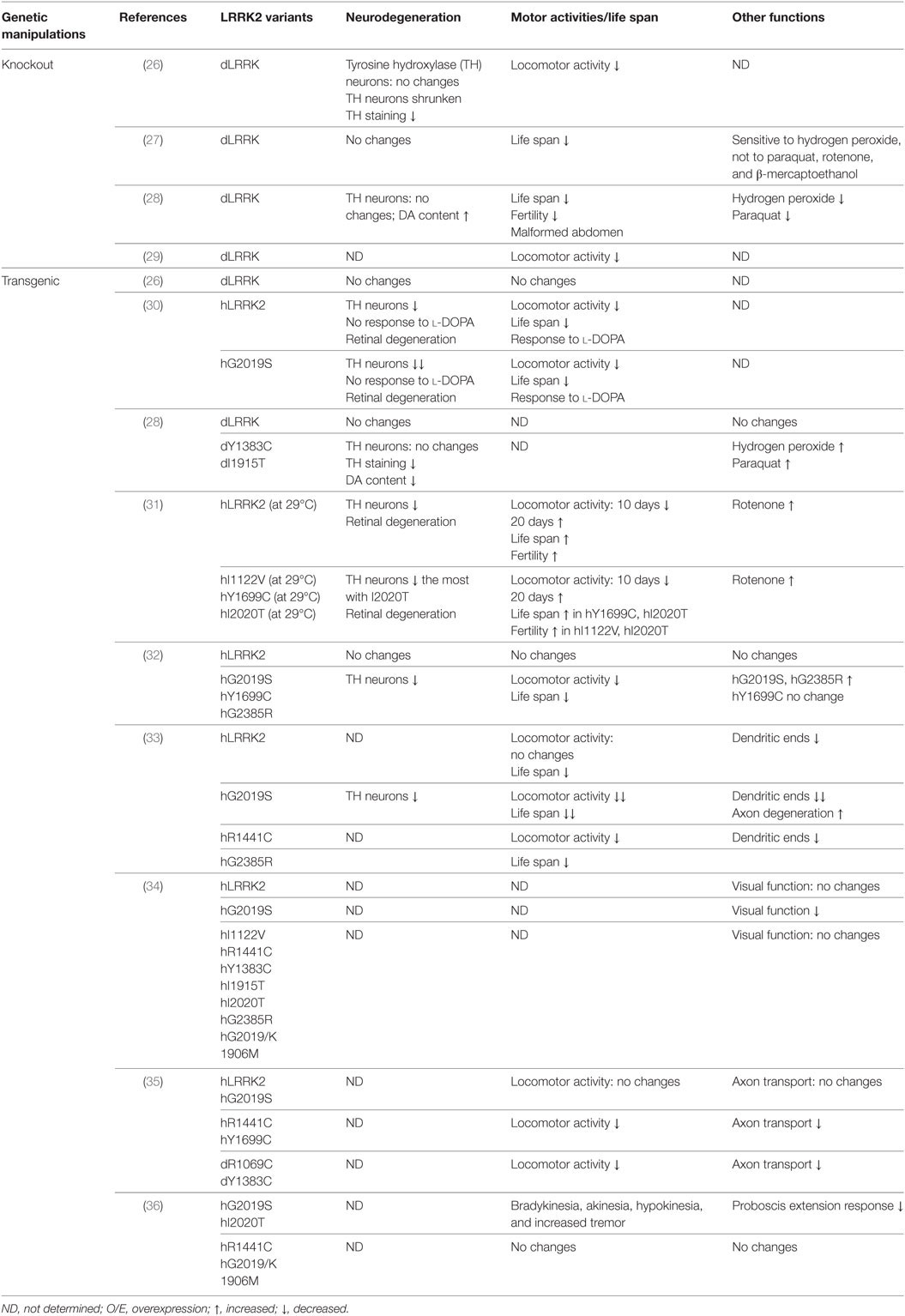
Table 1. Drosophila models for leucine-rich repeat kinase 2 (LRRK2)-associated Parkinson’s disease.
LRRK2 Drosophila Models
LRRK2 Knockout Drosophila Models
Drosophila has a single human LRRK2 homolog, dLRRK. Residues changed by PD-causing mutations in human LRRK2 are highly conserved in dLRRK (Table 1). To study the physiological function of endogenous LRRK2, dLRRK loss-of-function mutant fly lines have been generated. One major line used in the studies is LRRKe03680 from the Exelixis Collection at the Harvard Medical School. It was generated with piggyBac element insertion in the intron between exon 5 and exon 6 of dLRRK gene. In characterization of dLRRK knockout Drosophila model on PD related pathogenesis, one study reported knockout of dLRRK exhibited a decrease in tyrosine hydroxylase (TH) immunostaining, shrunken DA neurons, and locomotor activity deficits (26), while three studies reported that the homozygous mutant flies developed unchanged number and pattern of DA neurons as well as a normal life span (27–29). Furthermore, the sensitivity of those dLRRK2 knockout flies response to oxidative stress have been examined. Wang et al. showed that dLRRK mutant flies are selectively sensitive to H2O2 (27). By contrast, a report by Imai et al. demonstrated that dLRRK knockout flies are relatively resistant to paraquat and H2O2 treatment (28). Thus, the exact role of dLRRK in PD-related pathogenesis remains elusive. As the majority of studies supported that dLRRK is not required for DA neurons survival and this is consistent with the results from LRRK2 knockout rodents (mice or rats), the general agreement is that LRRK2-induced neuronal toxicity is from a gain-of-function but not a loss-of-function mechanism.
LRRK2 Transgenic Drosophila Models
Patients carrying heterozygou s or homozygous LRRK2 pathogenic mutations have similar disease risk and progression, supporting LRRK2 dominant nature (1) (Table 1). Indeed, in contrast to dLRRK loss-of-function mutant, overexpression of either human LRRK2 (hLRRK2) or dLRRK pathogenic mutations in flies leads to an age-dependent DA neuronal loss and DA-responsive locomotor deficits (28, 30–33, 36, 37). Notably, different LRRK2 mutations cause different phenotypes of the degeneration. One study demonstrated that dopaminergic expression of LRRK2 G2019S led to non-autonomous neurodegeneration in visual system (34). This degeneration is specific to G2019S mutation and dependent on kinase activity. Another report showed that GTPase-COR domain mutations R1441C or Y1699C, but not G2019S, preferentially inhibits axonal transport in Drosophila and causes locomotor deficits (35). This suggests that the defects depend on LRRK2 GTPase activity (35). Recently, Cording et al. reported that expressing either the G2019S or I2020T but not R1441C, or kinase dead LRRK2 in DA neurons reduces proboscis extension response, with bradykinesia, akinesia, and tremor (36). These studies support the possibility that different LRRK2 pathogenic mutations act at distinct pathways and cause disease via distinct pathogenic mechanisms. The LRRK2 transgenic fly models support the gain-of-function of LRRK2 in PD pathogenesis.
LRRK2 Drosophila Models Reveal LRRK2 Functions in PD
LRRK2 Functions in Vesicular Trafficking
Vesicular trafficking has been implicated to play crucial roles in neurodegeneration (38). LRRK2 Drosophila models have provided extensive evidence of potential roles for LRRK2 in various vesicle trafficking processes including endocytosis, ER-Golgi and retromer trafficking, and autophagy–lysosomal pathways (39). dLRRK was reported to localize to the membranes of late endosomes and lysosomes, physically and functionally interacts with Rab7, a key mediator of late endosomal transport and lysosome biogenesis (40). Nonsense alleles in dLRRK induced by ethyl methanesulfonate causes striking defects in the autophagy–lysosomal pathways (41). Furthermore, LRRK2 has been shown to interact with clathrin-light chains to limit Rac1 activation on endosomes (42). Importantly, studies in Drosophila show that LRRK2 phosphorylates endophilin A (EndoA), a central component of synaptic endocytosis, and synaptojanin 1 (SJ1), a synaptic vesicle protein which was recently linked to recessive PD (37, 43–45). LRRK2 regulates EndoA and SJ1 by phosphorylation at synapses, which facilitates synaptic endocytosis through clathrin uncoating at the synaptic terminals. In addition, LRRK2’s role in retromer and ER–Golgi trafficking was highlighted by genetic interactions between LRRK2 and VPS35, Rab7L1, ArfGAP1 in Drosophila (46–49). Moreover, dLRRK has been demonstrated to regulate axonal transport and Golgi outpost dynamics in dendrites through the golgin Lava lamp (35, 50). Taken together, these studies strongly support the roles of LRRK2 in vesicular trafficking processes, which may provide potential mechanisms for α-Syn accumulation in LRRK2-associated PD.
LRRK2 Functions in Protein Translation Machinery
The first evidence of LRRK2 function in protein translation was demonstrated in Drosophila (28). In this study, both dLRRK and human LRRK2 can phosphorylate eukaryotic initiation factor 4E-binding protein (4E-BP), a negative regulator of eukaryotic initiation factor 4E-mediated protein translation and a key mediator downstream of mTOR signaling to various stress responses (28). The notion that LRRK2 functions in protein translation was further strengthened by the study from the same group that LRRK2 interacts with the microRNA pathway to regulate protein synthesis (51). However, these studies were done in Drosophila system and need to be extended to mammalian systems. Subsequently, Martin and colleagues, using LRRK2 Drosophila model and human DA neurons, demonstrated that LRRK2 phosphorylates ribosome protein s15 to regulate protein translation and mediate LRRK2-induced neurodegeneration (52). Recently, Penney et al. showed that LRRK2 targets Furin1 to promote cap-dependent translation, which is required for LRRK2 synaptic function (53). Taken together, these findings support convergent evidence that LRRK2 regulates protein translation machinery directly or indirectly, which could be a potential therapeutic avenue for LRRK2-associated PD.
LRRK2 Functions in Dendritic Degeneration and Synaptic Dysfunction
Mutant LRRK2 functions in dendritic degeneration were first revealed by the evidence that LRRK2 G2019S induces mislocalization of the axonal protein tau in dendrites and in turn causes dendrite degeneration (33). This may act through tau phosphorylation by the glycogen synthase kinase 3β, which is promoted by LRRK2 G2019S (33). In addition, LRRK2 regulates synaptic morphology through phosphorylation of Futsch at the presynaptic compartments and interaction with 4E-BP at the postsynaptic site of the Drosophila neuromuscular junctions (NMJs) (54). Furthermore, a recent finding indicates that loss of dLRRK disrupts the retrograde synaptic compensation while overexpression of either dLRRK or hLRRK2 can induce a retrograde enhancement of presynaptic release (53). This regulation of synaptic homeostasis might act through a mechanism that LRRK2 promotes cap-dependent translation (53). These studies suggest that LRRK2 might regulate synaptic function in neural circuits.
LRRK2 Drosophila Models as Platforms to Identify Potential Therapeutic Compounds
The genetic LRRK2 Drosophila models provide promising in vivo platforms for inhibitor identification and validation, and drug development. It has shown that sorafenib, curcumin, or GW5074 significantly suppressed LRRK2 PD-like phenotypes in Drosophila (55, 56). Melatonin attenuates hLRRK2-induced synaptic dysfunction and sleep disorders (57). Although candidate compounds have been used in these studies, they open the possibility of performing compound screens. Recently, Lin et al. identified compounds from the FDA-approved licensed drug library that could rescue LRRK2-induced neurite degeneration, motor disability, and DA neuron loss (58). Of 640 compounds, lovastatin had the highest lipophilicity, which facilitates crossing the blood–brain barrier (58). These studies provide significant steps toward the development of new drugs for treatment of LRRK2-associated PD.
Modeling α-Syn-Associated PD in Drosophila
The discovery of the first missense mutation A53T in the SNCA gene in 1997 (59) and the insoluble aggregated α-Syn forms as the major component of LBs, a pathological hallmark of PD (60), heralded a new era in PD research. Since then, more SNCA pathogenic mutations as well as multiplications of SNCA have been identified as genetic causes of PD [review in Ref (61).]. In addition, multiple genome-wide association studies have identified SNCA as a risk factor for sporadic PD (62, 63). These findings revealed a central role of SNCA in PD. SNCA encodes α-Syn protein, a small protein with 140 amino acid residues. It is highly soluble and enriched at presynaptic terminals, where it binds lipids and regulates synaptic vesicle release and it has a propensity of self-aggregate to form oligomeric species and LB-like fibrils (64, 65). Multiple evidence suggest that oligomeric species of α-Syn, which are precursors for higher-order fibrillar aggregates in LBs, are pathogenically toxic and the culprits for neuronal degeneration (66). Recently, prion-like behavior of α-Syn has attracted a lot of attention and been debated in playing an important role in the pathogenesis of PD (67, 68). Although Drosophila have no homolog of SNCA, pathogenic mutations and multiplication of SNCA causing PD with dominant inheritance pattern implicates a toxic gain-of-function mechanism, which led to suitable transgenic modeling in fly by overexpressing wild-type or mutant α-Syn (69) (Table 2).
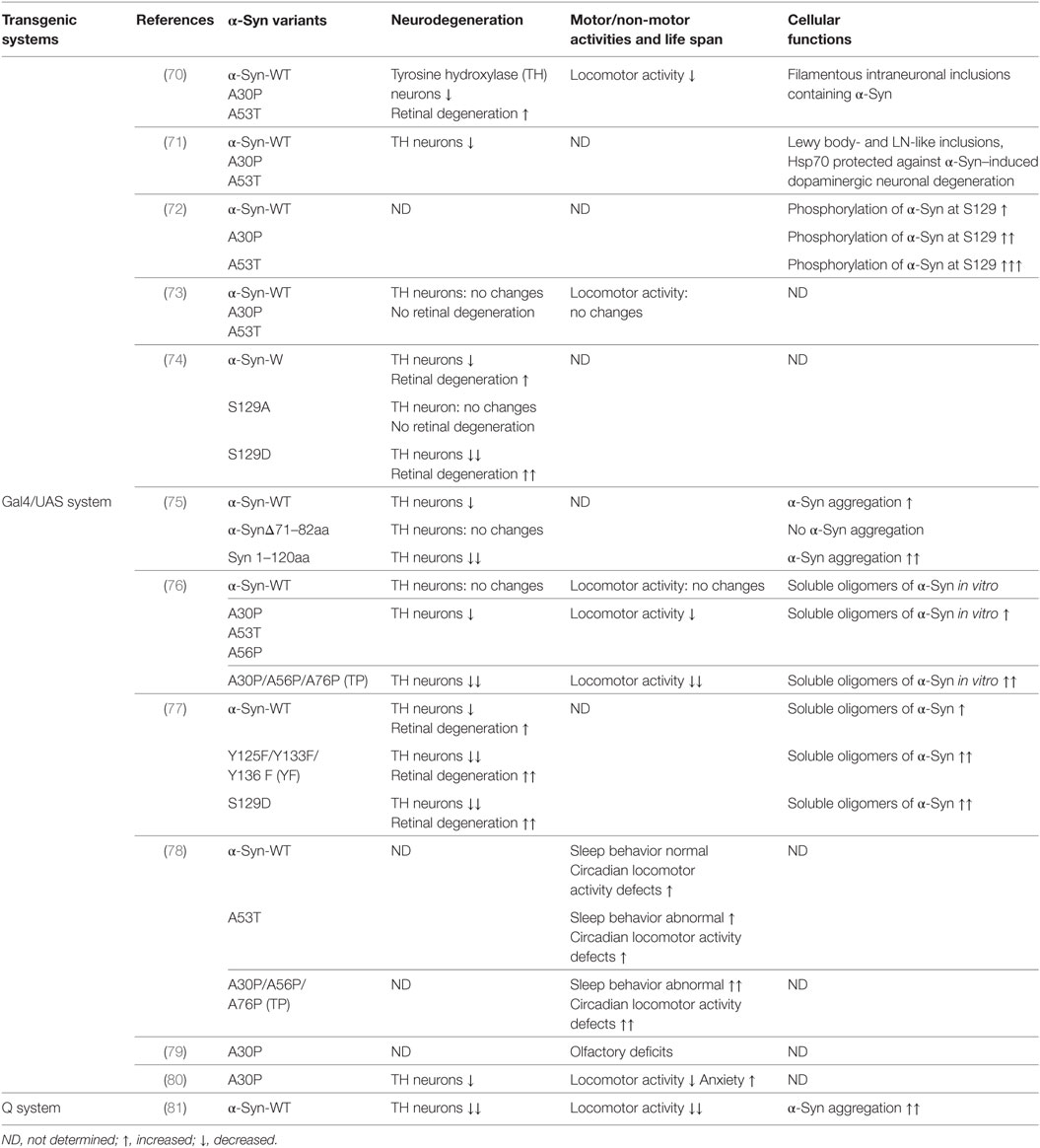
Table 2. Transgenic Drosophila models of α-Syn-associated Parkinson’s disease.
α-Syn Transgenic Drosophila Models
Feany and Bender first developed α-Syn transgenic Drosophila models by overexpressing either wild-type or familial mutants A53T and A30P of human α-Syn using the conventional Gal4/USA expression system (70). These models recapitulate the essential features of PD: adult-onset loss of DA neurons, filamentous intraneuronal inclusions containing α-Syn and locomotor dysfunction (70). In an independent study, Auluck et al. confirmed the phenotypes reported by Feany and Bender (71). In addition to these phenotypes, Chen et al. demonstrated olfactory deficits and elevated anxiety in a α-Syn transgenic Drosophila model expressing A30P (79, 80). There are deficits in two out of three olfactory modalities, odor discrimination and tested-olfactory acuity. A30P expression in dopamine neurons is necessary for both acuity and discrimination deficits. Gajula Balija et al. showed the non-motor symptoms such as an abnormal sleep-like behavior, locomotor deficits, and abnormal circadian periodicity when targeted expression of pre-fibrillar α-Syn mutants in a subset of serotonergic and DA neurons (78). In 2017, the Feany group expended the scope of their previous α-Syn transgenic Drosophila models using a binary expression system, the Q system, which relies on the transcriptional activation by the Neurospora protein QF2 to activate transgene expression (81). This new α-Syn Drosophila model shows robust neurodegeneration, early-onset locomotor deficits, and abundant α-Syn aggregation (81). Although there is some discrepancy over the strength of the phenotypes (73, 82), the α-Syn transgenic Drosophila models are widely used to delineate underlying pathogenic mechanisms and identify novel proteins mediating α-Syn toxicity.
α-Syn Drosophila Models Reveal α-Syn Functions in PD
α-Syn Aggregation and Misfolding in α-Syn-Induced Neurotoxicity
Accumulating evidence revealed α-Syn aggregation and misfolding plays a central role in the pathogenesis of PD and synucleinopathies. Wild type or mutant α-Syn has been demonstrated to be aggregated as inclusions when overexpressing in flies (70, 71, 81). Structurally engineered α-Syn mutants with an increased oligomerization propensity increase neurotoxicity in Drosophila (76). Truncation of α-Syn contributes to aggregation and LB formation. Expression of α-Syn with a deletion of NAC domain (α-Syn Δ71–82) did not show evidence of α-Syn aggregation and any DA neurodegeneration, suggesting the essential role of NAC domain of α-Syn in aggregation and toxicity (75). By contrast, a C-terminal truncated α-Syn has ability to promote aggregation and enhance neurotoxicity (75). Interestingly, calpain-cleaved α-Syn fragments with similar molecular weight to truncated α-Syn have been identified in the PD/DLB patient brains and α-Syn-expressing flies (83). This suggests the physiological and pathological importance of the truncated α-Syn.
Protein quality control systems including molecular chaperones and protein degradation function as a defense mechanism against protein misfolding and aggregation. Identification of suppressors in these systems further supports a critical role of toxic oligomers and aggregation in α-Syn-induced neurotoxicity. Histone deacetylase 6 suppresses α-Syn-induced DA neurodegeneration and promotes the formation of α-Syn inclusions by reducing α-Syn oligomers (84). Auluck et al. demonstrated that increasing the level of chaperone Hsp70 ameliorated the toxicity of α-Syn to DA neurons while a reduction in chaperone activity enhanced α-Syn-induced DA neuronal loss in Drosophila system (71). In addition, decreased level of the mitochondrial chaperone protein tumor necrosis factor receptor associated protein-1 enhanced A53T-α-Syn-induced age-dependent DA neuron loss in fly (85). Molecular chaperones assist proper protein folding and thus protect against α-Syn misfolding and aggregation. If proteins have been misfolded and aggregated, they are cleared by degradation. The ubiquitin-protease system and the autophagy-lysosome systems are two major protein degradation systems. Using Drosophila and cell culture systems, Lee et al. demonstrated that co-expression of ubiquitin can rescue α-Syn-induced neurotoxicity. This neuroprotection is dependent on the formation of lysine 48 polyubiquitin linkage, which is known to target protein degradation (86). This observation is further strengthened by evidence that overexpression of the ubiquitin ligase Nedd4 can rescue α-Syn-induced degenerative phenotype in fly (87). Furthermore, the deubiquitinase Usp8 interacted and partly colocalized with α-Syn, and deubiquitinated K63-linked chains on α-Syn. Knockdown of Usp8 in fly reduced α-Syn levels and α-Syn-induced toxicity (88). In addition, Cullen et al. showed that the defect of cathepsin D, a major lysosomal aspartyl protease, enhanced α-Syn-induced neurodegeneration in vivo in Drosophila (89). Taken together, these results confirmed that protein quality control systems function as a protection mechanism against α-Syn aggregation and misfolding.
α-Syn Phosphorylation Controls Neurotoxicity Inclusion Formation
Phosphorylation of α-Syn plays a key role in the PD pathogenesis. Phosphorylation at Ser129 is the one extensively phosphorylated in brain tissues from PD patients and related disorders, suggesting a role for Ser129 phosphorylation in disease pathogenesis (90, 91). In transgenic flies, it has been demonstrated that human α-Syn is phosphorylated at Ser129, and phosphorylation increases with age as DA neurons degenerate, mimicking the pathogenic phenomena in PD patient (72). Later on, Chen and Feany generated transgenic flies carrying the mutations at S129 of α-Syn (S129A to block phosphorylation and S129D to mimic phosphorylation) (74). Using these transgenic lines, they demonstrated phosphorylation of S129 is critical for α-Syn-induced DA neuron degeneration, and blocking S129 phosphorylation increases inclusion formation (74). As increased number of inclusion bodies correlates with reduced toxicity, this study suggested inclusion bodies might have protective function. Recently, the Feany group reported that tyrosine and serine phosphorylation of α-Syn have opposing effects: levels of soluble oligomeric species of α-Syn were increased by serine 129 phosphorylation and decreased by tyrosine 125 phosphorylation, suggesting detrimental effects of S129 phosphorylation and a neuroprotective action of T125 phosphorylation (77). These studies reveal that phosphorylation of α-Syn plays an important role in α-Syn-induced inclusion body formation and DA neurodegeneration.
α-Syn Functions in Vesicular Trafficking
The first evidence of α-Syn functions in trafficking in animal models has been reported by Cooper et al. using a combination of a genetic screening in yeast and validation in α-Syn transgenic Drosophila models (92). In this study, Rab1 rescues the neuron loss in the flies (92). Recently, using Drosophila models of α-Syn toxicity, several reports have implicated α-Syn functions in vesicular trafficking particularly through the small GTPase Rab proteins. The endosomal recycling factor Rab11 was demonstrated to modulate synaptic vesicle size, decrease α-Syn aggregation and ameliorate several α-Syn-dependent phenotypes (93). Rab7, regulating trafficking of late endosomes and autophagosomes, and Rab8, modulating post-Golgi vesicle trafficking, rescue the locomotor deficit in α-Syn flies (94, 95). Notably, other PD genes such as LRRK2 and PINK1 have also recently been linked to Rab proteins (48, 96–98). Thus, determination of the precise mechanisms of Rabs-mediated functions in PD pathogenesis is warranted.
α-Syn Functions in Mitochondrial Dysfunction and Oxidative Stress
Oxidative stress and mitochondrial dysfunction have been proposed as important causative factors for the progression of PD. Botella et al. found that DA neurons are specifically sensitive to an environmental oxidative insult (hyperoxia) induced oxidative stress. The mutant forms of α-Syn enhanced the toxicity under this stress in the Drosophila model (99). In addition, the co-expression of Cu/Zn superoxide dismutase protects against mutant α-Syn-induced DA neuronal loss (99). The same group also demonstrated that dopamine, which produces reactive oxygen species, might be involved in the α-Syn-induced neurotoxicity through oxidative stress (100). Furthermore, α-Syn-induced neuronal death in Drosophila is enhanced by the mutants that promote glutathione synthesis and conjugation (101). Natural antioxidants attenuate locomotor deficits of α-Syn transgenic flies (102). GPI, an enzyme in glucose metabolism, acts as neuroprotection from α-Syn proteotoxicity in flies (103). Recently, Feany group provided evidence that the interaction of α-Syn with spectrin initiates pathological alteration of the actin cytoskeleton and downstream neurotoxicity, and consequent mitochondrial dysfunction through altered Drp1 localization (81). These results suggest that oxidative stress and mitochondrial dysfunction are features of α-Syn toxicity.
α-Syn Drosophila Models as Platforms to Identify Potential Therapeutic Compounds
Based on the functions and mechanisms revealed by α-Syn Drosophila models, several pharmacological interventions have been developed in Drosophila to ameliorate α-Syn toxicity. Geldamamycin, an Hsp90 inhibitor and chaperone inductor, was able to protect α-Syn-expressing neurons in Drosophila (104). Nicotinamide, the principal form of niacin (vitamin B3), has been demonstrated to improve the motor dysfunction in α-Syn transgenic flies through improvement of oxidative mitochondrial dysfunction (105). The dopamine agonists pergolide, bromocriptine, and 2,3,4,5-tetrahydro-7,8-dihydroxy-1-phenyl-1H-3-benzazepine (SK&F 38393), D-519, and D-520 were substantially effective on improvement of locomotor function of α-Syn flies (106, 107). Atropine, the prototypical muscarinic cholinergic receptor antagonist, was effective (106). A potent dopamine D2/D3 receptor agonist D-607 exhibited significant neuroprotection in a Drosophila model of synucleinopathy (108). In addition, HDAC inhibitors such as sodium butyrate or SAHA, and SIRT2 inhibitors have been identified to protect against α-Syn-induced neurotoxicity in flies (109, 110). Taken together, these studies suggest that protein quality control systems, oxidative stress, mitochondrial function, and DA biosynthesis pathways are potential targets for developing therapeutic agents for α-Syn toxicity.
Modeling GBA-Associated PD in Drosophila
Heterozygous mutations in glucocerebrosidase (GCase, encoded by GBA1 gene) are recently emerging to be the most common known genetic risk factor for PD (111). GCase is a lysosomal protein and homozygous mutations cause Gaucher’s disease, a lysosomal storage disorder (112). As a lysosomal enzyme, GCase is synthesized in the endoplasmic reticulum (ER). At ER, it undergoes N-linked glycosylation on four asparagines. After correctly folded, it processes to the Golgi for further modifications on its N-linked glycans, and finally it traffics to the lysosomes (113). GCase cleaves the β-glucosyl linkage of glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph). Mutations in GBA cause accumulation of lipid substrates of GCase such as GlcCer and GlcSph (114). Recent reports demonstrated that mutations in GBA not only contribute to the occurrence of PD but also lead to more significant and rapid cognitive decline in PD (115). Most disease causing mutations of GBA are thought to be dominant-negative mutations that lead to the GBA deficiency, compromised GlcCer metabolism and the subsequent failure of lysosomal mediated degradation of GBA substrates and α-Syn. A severe heterozygous mutation L444P and a mild heterozygous mutation N370S are the most common mutations of GBA in PD. Mutations L444P and N370S cause ER stress, decreased lysosomal GCase, and accumulation of α-Syn aggregates (116, 117). Drosophila as one of the major model systems so far for studying GBA-associated PD has revealed significant insights (Table 3).
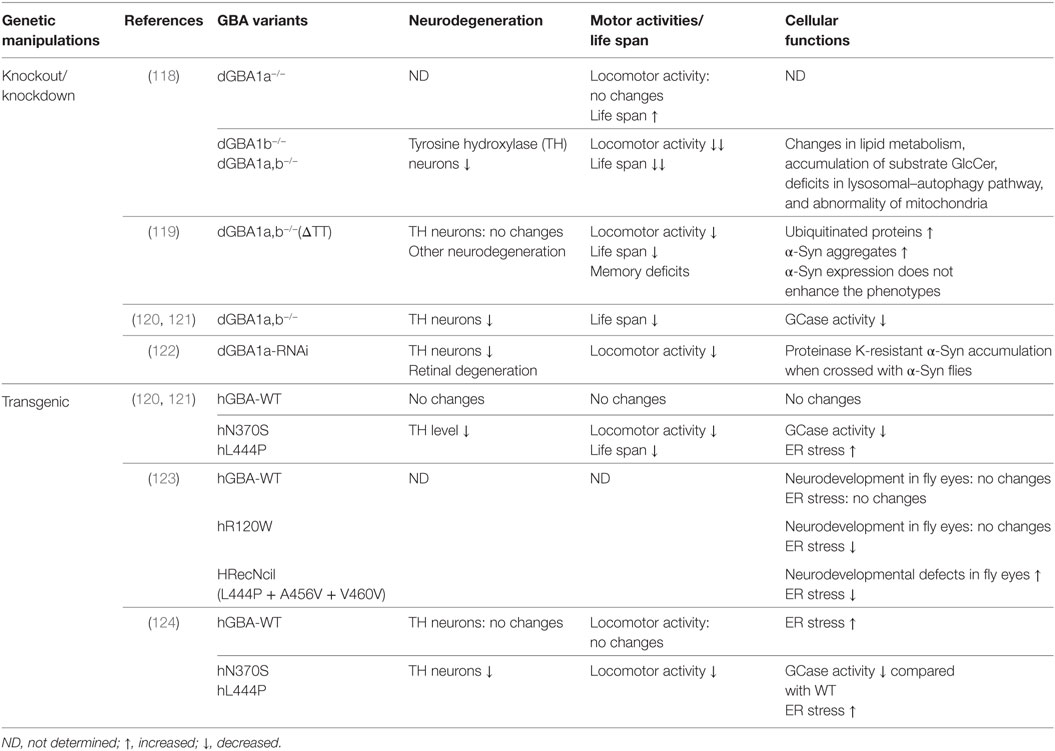
Table 3. Drosophila models of glucocerebrosidase (GBA)-associated Parkinson’s disease.
GBA Drosophila Models
GBA Knockout/Knockdown Drosophila Models
Drosophila has two homologs of the human GBA1 gene, CG31148 and CG31414, which are referred to dGBA1a and dGBA1b, respectively, and shares 32% amino acid identity. These two genes are found on the same chromosome with the CG31413 gene in between and show differential tissue expression. dGBA1b is expressed in the adult brain at low levels as well as in the adult fat body, whereas dGBA1a is predominantly expressed in the adult fly gut but not in the adult brain (FlyAtlas) (125). As GBA mutations in PD show a dominant-negative function, the loss-of-function of GBA was investigated in Drosophila by either knockdown or knockout dGBA1.
Kinghorn et al. generated dGBA1a, dGBA1b single knockouts (dGBA1a−/−, dGBA1b−/−) or dGBA1a/b double knockouts (dGBA1a,b−/−) using ends-out homologous recombination (118). dGBA1b−/− and dGBA1a,b−/− showed a significantly decreased survival and age-dependent locomotor deficits compared with control flies while dGBA1a−/− showed opposite phenotype with increased survival and without significant effect on climbing ability over time. As dGBA1b is expressed in the adult brain, the study was focusing on dGBA1b−/− and dGBA1a,b−/−, both of which showed similar phenotypes (118). Knockout of dGBA resulted in changes in lipid metabolism, accumulation of substrate GlcCer, deficits in lysosomal–autophagy pathway, neurodegeneration, and abnormality of mitochondria. Importantly, mTOR inhibitor rapamycin partially ameliorated the lifespan, locomotor, and starvation phenotypes in dGBA deficient flies (118). Another group used publicly available transposon insertions in dGBA1a and dGBA1b to create deletion of dGBA1 (GBA1ΔTT) (119). Using this approach, they removed the majority (first 433aa) of dGBA1b, small potion (33 codons) of c-terminal of dGBA1a, and the whole CG31413 gene in between. Consistent with the study by Kinghorn et al. GBA1ΔTT homozygotes exhibit shortened lifespan, behavioral phenotypes, memory deficits and neurodegeneration but no DA neuronal loss (119). GBA1ΔTT homozygotes increased accumulation of ubiquitinated proteins and α-Syn aggregates. However, α-Syn expression does not enhance GBA1ΔTT fly phenotypes (119). In addition to these two knockout dGBA1 fly lines, Maor et al. took advantage of two other fly lines, each of which has a minos insertion in dGBA1a and dGBA1b, respectively, to cause premature termination of dGBA1a and dGBA1b. By crossing these two lines, double heterozygous flies have been generated. This fly model exhibited about 30% decrease in GCase activity and has decreased TH immunoreactivity, shortened lifespan, and an age-dependent DA neurodegeneration (120, 121). Besides the knockouts of dGBA1, Suzuki et al. used transgenic RNAi flies to knock down dGBA1a and dGBA1b (122). dGBA1a-RNAi flies exhibited a bout 80–90% decrease in GCase activity while dGBA1b-RNAi flies only showed about 20% decrease. Thus, the study focused on dGBA1a-RNAi flies, which exacerbated the locomotor dysfunction, loss of DA neurons, retinal degeneration, and accumulation of proteinase K-resistant α-Syn in α-Syn-expressing flies (122).
Both knockdown and knockout of dGBA1 in fly have been consistently shown shortened lifespan, behavioral phenotypes and accumulation of α-Syn aggregates, despite of different phenotypes in DA neurodegeneration. These GBA fly models provide useful platforms for further study of GBA function in PD.
GBA Transgenic Drosophila Models
Heterozygous mutations L444P and N370S are the most common and thought to be dominant-negative mutations of GBA in PD. To investigate GBA functions in PD, transgenic Drosophila expressing human WT, N370S and L444P were generated (120, 121, 124). N370S and L444P transgenic flies exhibited significant decreased GCase activity by 82 and 75%, respectively, compared with GBA WT transgenic flies despite equivalent expression levels of GBA protein (124). N370S and L444P transgenic flies consistently showed shortened life span, a progressive climbing defect, increased level of ER stress and DA neurodegeneration (121, 123, 124). This suggests that those transgenic flies can recapitulate some PD signs.
GBA Drosophila Models Reveal GBA Functions in PD
Two major functions of GBA have been implicated in GBA Drosophila models. One is the function in ER stress and unfolded protein response (UPR) in the ER. Mutant GCase are recognized as misfolded proteins and undergo various degrees of ER associated degradation. The accumulation of midfolded molecules in the ER activate signaling events known as UPR (120). Immunostaining in GBA transgenic flies revealed that a significant amount of GCase colocalized with ER and N370S and L444P caused abnormal aggregates and swelling within the ER (124). To measure UPR activation, an ER stress reporter transgene Xbp1 was used. N370S and L444P GBA mutations induced significant higher Xbp1 level compared to WT GBA flies, suggesting of an increased level of ER stress (120, 124). Another function revealed by GBA fly models is in lysosomal-autophagic pathway. Using LysoTracker and LC3 as markers to monitor lysosomal and autophagic pathology, Kinghorn et al. demonstrated that enlarged and abnormal lysosomes and accumulated Atg8, the fly LC3 homolog, were present in dGBA knockout fly brains (118). The probable downstream effects of lysosomal-autophagic dysfunction could be the accumulation of p62, a marker for lysosomal-autophagic degradation, and polyubiquitinated proteins (118). While these studies provide significant phenotyping investigation on GBA functions, the detailed molecular mechanisms are still largely unknown. Using these GBA Drosophila models, further dissection of the molecules involved in these pathways is warranted.
GBA Drosophila Models as Platforms to Identify Potential Therapeutic Compounds
The clear evidence showed that mutant GCase causes increased ER stress and activated UPR in fly, therefore removal of mutant misfolded GCase by pharmacological chaperones from the ER should at least partially rescue the phenotype. Two chaperones, ambroxol and isofagomine, were previously used to increase amount and lysosomal activity of mutant GCase (126–129). Indeed, ambroxol and isofagomine reduced ER stress, and reversed locomotor deficits in GBA mutant flies in vivo (121, 124). This suggests that removal of mutant misfolded GCase from the ER may alleviate PD symptoms. Small chaperones can cross the blood–brain barrier, bind to GCase and stabilize proper folding and ensure delivery to lysosomes. Thus, small chaperones may be applicable for GBA-associated PD.
Modeling VPS35-Associated PD in Drosophila
Mutations in the VPS35 gene encoding a core subunit of a heteropentameric complex referred to the retromer have recently emerged as a new cause of late-onset, autosomal dominant familial PD (7, 8). The mutation, D620N, has so far been unambiguously identified to cause PD. The VPS35 protein functions as a core component of the retromer, a protein complex that associates with the endosome to facilitate recycling of transmembrane protein cargoes from both endosome-to-Golgi and endosome-to-plasma membrane transport (130). The retromer is a highly conserved multi-protein complex, the core of which consists of the subunits VPS35, VPS29, and VPS26. The only identifiable VPS35 homolog in Drosophila is encoded by CG5625.
VPS35 Drosophila Models
VPS35 Knockout/Knockdown Drosophila Models
Two lines of null mutation in VPS35 were generated by either imprecise excision of a P-element inserted at the 5′ end of CG5625 (P[EPgy2]CG5625EY14200), or by a deletion of nearly 2 kb, which removes the first three exons including the translation start site (VPS35MH20) (Table 4). Both mutants die at late larval or pupal stages, indicating the essential function of VPS35. VPS35-null mutants and RNAi lines (the Vienna Drosophila RNAi Center) were consistently demonstrated to reduce Wingless secretion but not Hedgehog signaling by reducing the recycle of Wntless from endosomes to the trans-Golgi network (131–133). Loss of VPS35 inhibits scavenger receptor ligand endocytosis, causes signaling defects at the NMJ, and leads to over proliferation of blood cells in larvae, which suggests VPS35 has tumor suppressor properties (134). Mechanistically, the endocytic and signaling defects of VPS35 mutants maybe due to VPS35 negatively regulation of actin polymerization (134). As these studies were at early stages before mutations of VPS35 has been identified to associate with PD, the PD pathological phenotypes in VPS35 knockout or knockdown mutants were not investigated. In recent studies, knockdown of VPS35 in Drosophila induced the accumulation of the detergent-insoluble α-Syn in the brain and exacerbated locomotor deficits, compound eye disorganization, and interommatidial bristle loss in α-Syn transgenic flies (135). These findings indicate that the retromer may play a crucial role in α-Syn degradation. The loss of Drosophila VPS35 (dVPS35) affects synaptic vesicle recycling, DA synaptic release and sleep behavior (46). The manipulation of Drosophila LRRK2 dLRRK together with Rab5 and Rab11 improves the VPS35 synaptic phenotypes (46). Taken together, VPS35 knockout/knockdown Drosophila models mimic some pathogenesis of PD, indicating that these fly models could be useful platforms to study VPS35-associated PD.
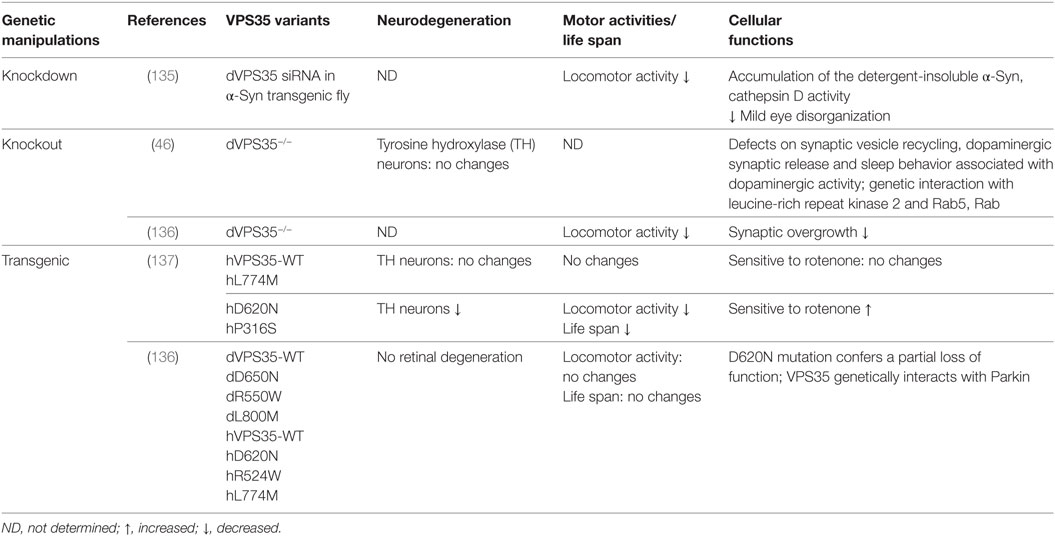
Table 4. Drosophila models of vacuolar protein sorting 35 (VPS35)-associated Parkinson’s disease.
VPS35 Transgenic Drosophila Models
Vacuolar protein sorting 35-linked PD is inherited as a dominant trait, which may imply that the mutation of VPS35 has a gain-of-function toxicity (Table 4). One study demonstrated that VPS35 D620N transgenic flies led to late-onset loss of DA neurons, locomotor deficits, shortened lifespans, and increased sensitivity to a PD-linked environmental toxin, rotenone (137). However, Malik and colleagues did not find evidence of dominant toxicity from any variants including the pathogenic D620N mutation, even with aging. By a definitive test to determine whether transgene expression can rescue endogenous VPS35 mutant phenotypes, they concluded that the D620N mutation confers a partial loss of function (136). This notion is further supported by other studies in fly or mouse systems that VPS35 DN mutation acts as a dominant-negative function (47, 48, 138, 139).
VPS35 Drosophila Models Reveal VPS35 Functions in PD
To date, VPS35 Drosophila models have revealed three major functions of VPS35 in trafficking pathways in neuronal system. First, VPS35 regulates synaptic vesicle endocytosis through the endosomal pathway. Loss of VPS35 increases the number of synaptic boutons of the NMJ in larval motor neurons (46, 134). It has been demonstrated that VPS35 cooperates with LRRK2 to regulate synaptogenesis, synaptic dynamics and endocytosis, and synaptic vesicles regeneration through the Rab-mediated endocytic pathway (46). Importantly, it in turn regulates DA activity and survival, a key element of PD etiology (46). Second, VPS35 mediates endolysosomal and Golgi apparatus sorting. Wild-type VPS35, but not a familial PD-associated mutant form, can rescue LRRK2 led to endolysosomal and Golgi apparatus sorting defects (48). In addition, it has been reported by several groups that VPS35 functions in endosome-to-Golgi retrieval are required for Wingless secretion (131–133). However, whether this function is related to DA neurodegeneration is unknown. Third, VPS35 functions in lysosomal degradation pathway. VPS35 dysfunction impairs the maturation of a lysosome protease cathepsin D in regulating the proteolytic pathway that is important for α-Syn metabolism, and in turn exacerbates neurotoxicity and causes eye degeneration and motor disability (135). These findings indicate that VPS35 may play a crucial role in α-Syn degradation and might thereby contribute to the pathogenesis of the disease. While it remains unclear if these functions are causally for DA neurodegeneration caused by VPS35 PD mutant, these studies have provided important insights into cellular pathways that are perturbed by VPS35 mutations in neurons.
Convergent Mechanisms
Dissecting genetic interaction among PD genes will be crucial to establish convergent functional pathways of these genes or risk factors. Drosophila as a classic genetic model provides powerful tools to study genetic interactions between different genes. Genetic dissection revealed that LRRK2 interacts with other PD genes or risk factors such as Parkin, DJ-1, PINK1, VPS35, and RAB7L (31, 46–48) and implicated several potential functions. Genetic interaction between LRRK2 and VPS35 or Rab7L indicates LRRK2 function in retromer and lysosomal pathways (46–48). Genetic interaction between LRRK2 and Parkin or PINK1 indicates LRRK2 function in mitochondria dysfunction and also suggests that dominant PD genes may act via common pathways with the recessive PD genes (31). Furthermore, VPS35 genetically interacts with Parkin but interestingly not with PINK1(136). Notably, α-Syn, LRRK2, and PINK1 have recently been linked to Rab proteins (48, 92–98), and the manipulation of Drosophila LRRK2 dLRRK together with Rab5 and Rab11 improves the VPS35 synaptic phenotypes (46). All the studies are convergent to implicate an important emerging role for defects in trafficking pathways. The accumulation of altered proteins including α-Syn and damaged mitochondria ultimately might overwhelm the disposal mechanisms, in turn cause DA neurodegeneration.
Concluding Remarks
While the rodent models generally attack more attention and efforts on studying human disorders because of their high conservation of basal ganglia circuit with human, modeling PD in rodents using genetics has been viewed as difficult (23, 140). The rodent models of PD could not fulfill all the key features of PD (140). The reason that the rodents are “imperfect” for modeling PD might be compensatory mechanisms in the rodents, and/or incomplete penetrance of some PD gene mutations such as LRRK2 disease causing mutations in human, and/or the combination effects of non-cell-autonomous and cell-autonomous processes (23, 140).
To this caveat, Drosophila models have provided significant contributions to our understanding of the mechanisms of PD pathogenesis in a comparatively short time frame and cost effective mode. Overexpression of PD dominant traits (LRRK2 and α-Syn) or knockout of dominant-negative genes (GBA and VPS35) in fly has been consistently demonstrated to mimic the essential PD signs such as DA neurodegeneration and behavioral deficits. Based on these fly models, genetic modifiers and small molecular compounds have been rapidly identified. Moreover, the combination roles of the genetic and environmental factors such as oxidative stress have been explored in PD. The important functions of LRRK2 in trafficking and protein translation, the critical contribution of α-Syn aggregation and phosphorylation, were initially discovered in fly. The Drosophila models so far are one of the major model systems to study GBA function in PD. Thus, the use of Drosophila models opened tremendous opportunities to explore the basic function of disease causing genes and to model the disease pathogenesis.
However, Drosophila is a relatively simple model organism, far less complex brain circuit than humans. For example, Drosophila does not have α-Syn homolog and a true human LRRK2 homolog. α-Syn neuropathology in the form of LBs is the hallmark of PD pathogenesis. Whether α-Syn is required for developing PD models has been raised. In addition, Drosophila has limited cell death effectors. Some aspects of human diseases may not be evident in fly. Thus, validation of findings from Drosophila to mammalian systems, including rodent models, human postmortem tissue, and human DA neuronal cultures, is warranted. Therefore, the strategy would be to identify the basic aspects of the underlying disease mechanisms using simple organism model systems such as Drosophila and further characterize and validate the findings in mammalian conditions.
Author Contributions
YX and JY analyzed the literatures and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from NIH/NIA K01-AG046366, the William N. & Bernice E. Bumpus Foundation Innovation Awards, the Kansas INBRE: P20 GM103418, American Parkinson Disease Association (APDA) Research Grant, Johnson Cancer Research Center Innovative Research Awards and Equipment Awards at Kansas State University, start-up fund and SUCCESS-FYI Intramural Grant from Kansas State University College of Veterinary Medicine.
Abbreviations
PD, Parkinson’s disease; LRRK2, leucine-rich repeat kinase 2; PINK1, phosphatase and tensin homolog deleted on chromosome 10-induced putative kinase 1; VPS35, vacuolar protein sorting 35; GBA, glucocerebrosidase; DA, dopaminergic; SNpc, substantia nigra pars compacta; EMS, ethyl methanesulfonate; SJ1, synaptojanin 1; EndoA, endophilin A; GSK3β, glycogen synthase kinase 3β; GWASs, genome-wide association studies; LBs, Lewy bodies; α-Syn, α-synuclein; UPS, ubiquitin-protease system; HDAC6, histone deacetylase 6; TRAP1, tumor necrosis factor receptor associated protein-1; ER, endoplasmic reticulum; GlcSph, glucosylceramide (GlcCer) and glucosylsphingosine; GD, Gaucher’s disease; UPR, unfolded protein response.
References
1. Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet (2009) 373:2055–66. doi:10.1016/S0140-6736(09)60492-X
2. Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest (2006) 116:1744–54. doi:10.1172/JCI29178
3. Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet (2011) 12:301–25. doi:10.1146/annurev-genom-082410-101440
4. Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, Lincoln SJ, Lepretre F, Hulihan MM, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet (2011) 89:398–406. doi:10.1016/j.ajhg.2011.08.009
5. Scholz SW, Jeon BS. GBA mutations and Parkinson disease: when genotype meets phenotype. Neurology (2015) 84:866–7. doi:10.1212/WNL.0000000000001321
6. Vilarino-Guell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet (2014) 23:1794–801. doi:10.1093/hmg/ddt570
7. Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet (2011) 89:162–7. doi:10.1016/j.ajhg.2011.06.001
8. Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet (2011) 89:168–75. doi:10.1016/j.ajhg.2011.06.008
9. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron (2004) 44:595–600. doi:10.1016/j.neuron.2004.10.023
10. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron (2004) 44:601–7. doi:10.1016/j.neuron.2004.11.005
11. Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci (2010) 11:791–7. doi:10.1038/nrn2935
12. Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci (2006) 29:286–93. doi:10.1016/j.tins.2006.03.006
13. Xiong Y, Dawson VL, Dawson TM. LRRK2 GTPase dysfunction in the pathogenesis of Parkinson’s disease. Biochem Soc Trans (2012) 40:1074–9. doi:10.1042/BST20120093
14. Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol (2008) 7:583–90. doi:10.1016/S1474-4422(08)70117-0
15. Benamer HT, de Silva R. LRRK2 G2019S in the north African population: a review. Eur Neurol (2010) 63:321–5. doi:10.1159/000279653
16. Lesage S, Patin E, Condroyer C, Leutenegger AL, Lohmann E, Giladi N, et al. Parkinson’s disease-related LRRK2 G2019S mutation results from independent mutational events in humans. Hum Mol Genet (2010) 19:1998–2004. doi:10.1093/hmg/ddq081
17. Berwick DC, Harvey K. LRRK2 signaling pathways: the key to unlocking neurodegeneration? Trends Cell Biol (2011) 21(5):257–65. doi:10.1016/j.tcb.2011.01.001
18. Islam MS, Moore DJ. Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation. Biochem Soc Trans (2017) 45:163–72. doi:10.1042/BST20160264
19. Cookson MR. LRRK2 pathways leading to neurodegeneration. Curr Neurol Neurosci Rep (2015) 15:42. doi:10.1007/s11910-015-0564-y
20. Martin I, Kim JW, Dawson VL, Dawson TM. LRRK2 pathobiology in Parkinson’s disease. J Neurochem (2014) 131:554–65. doi:10.1111/jnc.12949
21. Nikonova EV, Xiong Y, Tanis KQ, Dawson VL, Vogel RL, Finney EM, et al. Transcriptional responses to loss or gain of function of the leucine-rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Hum Mol Genet (2012) 21:163–74. doi:10.1093/hmg/ddr451
22. Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, et al. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet (2010) 6:e1000902. doi:10.1371/journal.pgen.1000902
23. Xiong Y, Dawson TM, Dawson VL. Models of LRRK2-associated Parkinson’s disease. Adv Neurobiol (2017) 14:163–91. doi:10.1007/978-3-319-49969-7_9
24. Xiong Y, Neifert S, Karuppagounder SS, Liu Q, Stankowski JN, Lee BD, et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc Natl Acad Sci U S A (2018) 115(7):1635–40. doi:10.1073/pnas.1712648115
25. Xiong Y, Neifert S, Karuppagounder SS, Stankowski JN, Lee BD, Grima JC, et al. Overexpression of Parkinson’s disease-associated mutation LRRK2 G2019S in mouse forebrain induces behavioral deficits and alpha-synuclein pathology. eNeuro (2017) 4(2). doi:10.1523/ENEURO.0004-17.2017
26. Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun (2007) 358:534–9. doi:10.1016/j.bbrc.2007.04.156
27. Wang D, Tang B, Zhao G, Pan Q, Xia K, Bodmer R, et al. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol Neurodegener (2008) 3:3. doi:10.1186/1750-1326-3-3
28. Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, et al. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J (2008) 27:2432–43. doi:10.1038/emboj.2008.163
29. Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci (2009) 12:1129–35. doi:10.1038/nn.2372
30. Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, et al. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A (2008) 105:2693–8. doi:10.1073/pnas.0708452105
31. Venderova K, Kabbach G, Abdel-Messih E, Zhang Y, Parks RJ, Imai Y, et al. Leucine-rich repeat kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum Mol Genet (2009) 18:4390–404. doi:10.1093/hmg/ddp394
32. Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, et al. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci (2009) 29:11257–62. doi:10.1523/JNEUROSCI.2375-09.2009
33. Lin CH, Tsai PI, Wu RM, Chien CT. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J Neurosci (2010) 30:13138–49. doi:10.1523/JNEUROSCI.1737-10.2010
34. Hindle S, Afsari F, Stark M, Middleton CA, Evans GJ, Sweeney ST, et al. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum Mol Genet (2013) 22:2129–40. doi:10.1093/hmg/ddt061
35. Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, et al. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun (2014) 5:5245. doi:10.1038/ncomms6245
36. Cording AC, Shiaelis N, Petridi S, Middleton CA, Wilson LG, Elliott CJH. Targeted kinase inhibition relieves slowness and tremor in a Drosophila model of LRRK2 Parkinson’s disease. NPJ Parkinsons Dis (2017) 3:34. doi:10.1038/s41531-017-0036-y
37. Islam MS, Nolte H, Jacob W, Ziegler AB, Putz S, Grosjean Y, et al. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet (2016) 25:5365–82. doi:10.1093/hmg/ddw352
38. Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature (2016) 539:207–16. doi:10.1038/nature20414
39. Hewitt VL, Whitworth AJ. Mechanisms of Parkinson’s disease: lessons from Drosophila. Curr Top Dev Biol (2017) 121:173–200. doi:10.1016/bs.ctdb.2016.07.005
40. Dodson MW, Zhang T, Jiang C, Chen S, Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet (2012) 21:1350–63. doi:10.1093/hmg/ddr573
41. Dodson MW, Leung LK, Lone M, Lizzio MA, Guo M. Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis Model Mech (2014) 7:1351–63. doi:10.1242/dmm.017020
42. Schreij AM, Chaineau M, Ruan W, Lin S, Barker PA, Fon EA, et al. LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep (2015) 16:79–86. doi:10.15252/embr.201438714
43. Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron (2012) 75:1008–21. doi:10.1016/j.neuron.2012.08.022
44. Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, et al. A LRRK2-dependent endophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron (2016) 92:829–44. doi:10.1016/j.neuron.2016.09.037
45. Soukup SF, Verstreken P. EndoA/Endophilin-A creates docking stations for autophagic proteins at synapses. Autophagy (2017) 13:971–2. doi:10.1080/15548627.2017.1286440
46. Inoshita T, Arano T, Hosaka Y, Meng H, Umezaki Y, Kosugi S, et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum Mol Genet (2017) 26:2933–48. doi:10.1093/hmg/ddx179
47. Linhart R, Wong SA, Cao J, Tran M, Huynh A, Ardrey C, et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of leucine-rich repeat kinase 2 (LRRK2). Mol Neurodegener (2014) 9:23. doi:10.1186/1750-1326-9-23
48. MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron (2013) 77:425–39. doi:10.1016/j.neuron.2012.11.033
49. Xiong Y, Yuan C, Chen R, Dawson TM, Dawson VL. ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. J Neurosci (2012) 32:3877–86. doi:10.1523/JNEUROSCI.4566-11.2012
50. Lin CH, Li H, Lee YN, Cheng YJ, Wu RM, Chien CT. Lrrk regulates the dynamic profile of dendritic Golgi outposts through the golgin Lava lamp. J Cell Biol (2015) 210(3):471–83. doi:10.1083/jcb.201411033
51. Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature (2010) 466:637–41. doi:10.1038/nature09191
52. Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell (2014) 157:472–85. doi:10.1016/j.cell.2014.01.064
53. Penney J, Tsurudome K, Liao EH, Kauwe G, Gray L, Yanagiya A, et al. LRRK2 regulates retrograde synaptic compensation at the Drosophila neuromuscular junction. Nat Commun (2016) 7:12188. doi:10.1038/ncomms12188
54. Lee S, Liu HP, Lin WY, Guo H, Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci (2010) 30:16959–69. doi:10.1523/JNEUROSCI.1807-10.2010
55. Liu Z, Hamamichi S, Lee BD, Yang D, Ray A, Caldwell GA, et al. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum Mol Genet (2011) 20:3933–42. doi:10.1093/hmg/ddr312
56. Yang D, Li T, Liu Z, Arbez N, Yan J, Moran TH, et al. LRRK2 kinase activity mediates toxic interactions between genetic mutation and oxidative stress in a Drosophila model: suppression by curcumin. Neurobiol Dis (2012) 47:385–92. doi:10.1016/j.nbd.2012.05.020
57. Sun X, Ran D, Zhao X, Huang Y, Long S, Liang F, et al. Melatonin attenuates hLRRK2-induced sleep disturbances and synaptic dysfunction in a Drosophila model of Parkinson’s disease. Mol Med Rep (2016) 13:3936–44. doi:10.3892/mmr.2016.4991
58. Lin CH, Lin HI, Chen ML, Lai TT, Cao LP, Farrer MJ, et al. Lovastatin protects neurite degeneration in LRRK2-G2019S parkinsonism through activating the Akt/Nrf pathway and inhibiting GSK3beta activity. Hum Mol Genet (2016) 25(10):1965–78. doi:10.1093/hmg/ddw068
59. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science (1997) 276:2045–7. doi:10.1126/science.276.5321.2045
60. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature (1997) 388:839–40. doi:10.1038/42166
61. Koprich JB, Kalia LV, Brotchie JM. Animal models of alpha-synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci (2017) 18:515–29. doi:10.1038/nrn.2017.75
62. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet (2014) 46:989–93. doi:10.1038/ng.3043
63. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet (2009) 41:1308–12. doi:10.1038/ng.487
64. Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron (2013) 79:1044–66. doi:10.1016/j.neuron.2013.09.004
65. Wong YC, Krainc D. alpha-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med (2017) 23:1–13. doi:10.1038/nm.4269
66. Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J. Alpha-synuclein oligomers: a new hope. Acta Neuropathol (2017) 134:819–38. doi:10.1007/s00401-017-1755-1
67. Brundin P, Melki R. Prying into the prion hypothesis for Parkinson’s disease. J Neurosci (2017) 37:9808–18. doi:10.1523/JNEUROSCI.1788-16.2017
68. Surmeier DJ, Obeso JA, Halliday GM. Parkinson’s disease is not simply a prion disorder. J Neurosci (2017) 37:9799–807. doi:10.1523/JNEUROSCI.1787-16.2017
69. Mizuno H, Fujikake N, Wada K, Nagai Y. alpha-Synuclein transgenic Drosophila as a model of Parkinson’s disease and related synucleinopathies. Parkinsons Dis (2010) 2011:212706. doi:10.4061/2011/212706
70. Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature (2000) 404:394–8. doi:10.1038/35006074
71. Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science (2002) 295:865–8. doi:10.1126/science.1067389
72. Takahashi M, Kanuka H, Fujiwara H, Koyama A, Hasegawa M, Miura M, et al. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci Lett (2003) 336:155–8. doi:10.1016/S0304-3940(02)01258-2
73. Pesah Y, Burgess H, Middlebrooks B, Ronningen K, Prosser J, Tirunagaru V, et al. Whole-mount analysis reveals normal numbers of dopaminergic neurons following misexpression of alpha-synuclein in Drosophila. Genesis (2005) 41:154–9. doi:10.1002/gene.20106
74. Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci (2005) 8:657–63. doi:10.1038/nn1443
75. Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci (2007) 27:3338–46. doi:10.1523/JNEUROSCI.0285-07.2007
76. Karpinar DP, Balija MB, Kugler S, Opazo F, Rezaei-Ghaleh N, Wender N, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J (2009) 28:3256–68. doi:10.1038/emboj.2009.257
77. Chen L, Periquet M, Wang X, Negro A, McLean PJ, Hyman BT, et al. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J Clin Invest (2009) 119:3257–65. doi:10.1172/JCI39088
78. Gajula Balija MB, Griesinger C, Herzig A, Zweckstetter M, Jackle H. Pre-fibrillar alpha-synuclein mutants cause Parkinson’s disease-like non-motor symptoms in Drosophila. PLoS One (2011) 6:e24701. doi:10.1371/journal.pone.0024701
79. Chen AY, Wilburn P, Hao X, Tully T. Walking deficits and centrophobism in an alpha-synuclein fly model of Parkinson’s disease. Genes Brain Behav (2014) 13:812–20. doi:10.1111/gbb.12172
80. Chen AY, Xia S, Wilburn P, Tully T. Olfactory deficits in an alpha-synuclein fly model of Parkinson’s disease. PLoS One (2014) 9:e97758. doi:10.1371/journal.pone.0097758
81. Ordonez DG, Lee MK, Feany MB. alpha-Synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron (2018) 97(1):108–124.e6. doi:10.1016/j.neuron.2017.11.036
82. Whitworth AJ, Wes PD, Pallanck LJ. Drosophila models pioneer a new approach to drug discovery for Parkinson’s disease. Drug Discov Today (2006) 11:119–26. doi:10.1016/S1359-6446(05)03693-7
83. Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol (2007) 170:1725–38. doi:10.2353/ajpath.2007.061232
84. Du G, Liu X, Chen X, Song M, Yan Y, Jiao R, et al. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol Biol Cell (2010) 21:2128–37. doi:10.1091/mbc.E10-03-0200
85. Butler EK, Voigt A, Lutz AK, Toegel JP, Gerhardt E, Karsten P, et al. The mitochondrial chaperone protein TRAP1 mitigates alpha-synuclein toxicity. PLoS Genet (2012) 8:e1002488. doi:10.1371/journal.pgen.1002488
86. Lee FK, Wong AK, Lee YW, Wan OW, Chan HY, Chung KK. The role of ubiquitin linkages on alpha-synuclein induced-toxicity in a Drosophila model of Parkinson’s disease. J Neurochem (2009) 110:208–19. doi:10.1111/j.1471-4159.2009.06124.x
87. Davies SE, Hallett PJ, Moens T, Smith G, Mangano E, Kim HT, et al. Enhanced ubiquitin-dependent degradation by Nedd4 protects against alpha-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol Dis (2014) 64:79–87. doi:10.1016/j.nbd.2013.12.011
88. Alexopoulou Z, Lang J, Perrett RM, Elschami M, Hurry ME, Kim HT, et al. Deubiquitinase Usp8 regulates alpha-synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci U S A (2016) 113:E4688–97. doi:10.1073/pnas.1523597113
89. Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain (2009) 2:5. doi:10.1186/1756-6606-2-5
90. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol (2002) 4:160–4. doi:10.1038/ncb748
91. Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol (2003) 62:644–54. doi:10.1093/jnen/62.6.644
92. Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science (2006) 313:324–8. doi:10.1126/science.1129462
93. Breda C, Nugent ML, Estranero JG, Kyriacou CP, Outeiro TF, Steinert JR, et al. Rab11 modulates alpha-synuclein-mediated defects in synaptic transmission and behaviour. Hum Mol Genet (2015) 24:1077–91. doi:10.1093/hmg/ddu521
94. Dinter E, Saridaki T, Nippold M, Plum S, Diederichs L, Komnig D, et al. Rab7 induces clearance of alpha-synuclein aggregates. J Neurochem (2016) 138:758–74. doi:10.1111/jnc.13712
95. Yin G, Lopes da Fonseca T, Eisbach SE, Anduaga AM, Breda C, Orcellet ML, et al. alpha-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol Dis (2014) 70:149–61. doi:10.1016/j.nbd.2014.06.018
96. Lai YC, Kondapalli C, Lehneck R, Procter JB, Dill BD, Woodroof HI, et al. Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J (2015) 34:2840–61. doi:10.15252/embj.201591593
97. Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, et al. Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J (2018) 37(1):1–18. doi:10.15252/embj.201798099
98. Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife (2016) 5:e12813. doi:10.7554/eLife.12813
99. Botella JA, Bayersdorfer F, Schneuwly S. Superoxide dismutase overexpression protects dopaminergic neurons in a Drosophila model of Parkinson’s disease. Neurobiol Dis (2008) 30:65–73. doi:10.1016/j.nbd.2007.11.013
100. Bayersdorfer F, Voigt A, Schneuwly S, Botella JA. Dopamine-dependent neurodegeneration in Drosophila models of familial and sporadic Parkinson’s disease. Neurobiol Dis (2010) 40:113–9. doi:10.1016/j.nbd.2010.02.012
101. Trinh K, Moore K, Wes PD, Muchowski PJ, Dey J, Andrews L, et al. Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson’s disease. J Neurosci (2008) 28:465–72. doi:10.1523/JNEUROSCI.4778-07.2008
102. Jahromi SR, Haddadi M, Shivanandappa T, Ramesh SR. Attenuation of neuromotor deficits by natural antioxidants of Decalepis hamiltonii in transgenic Drosophila model of Parkinson’s disease. Neuroscience (2015) 293:136–50. doi:10.1016/j.neuroscience.2015.02.048
103. Knight AL, Yan X, Hamamichi S, Ajjuri RR, Mazzulli JR, Zhang MW, et al. The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson’s models. Cell Metab (2014) 20:145–57. doi:10.1016/j.cmet.2014.04.017
104. Auluck PK, Meulener MC, Bonini NM. Mechanisms of suppression of {alpha}-synuclein neurotoxicity by geldanamycin in Drosophila. J Biol Chem (2005) 280:2873–8. doi:10.1074/jbc.M412106200
105. Jia H, Li X, Gao H, Feng Z, Li X, Zhao L, et al. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J Neurosci Res (2008) 86:2083–90. doi:10.1002/jnr.21650
106. Pendleton RG, Parvez F, Sayed M, Hillman R. Effects of pharmacological agents upon a transgenic model of Parkinson’s disease in Drosophila melanogaster. J Pharmacol Exp Ther (2002) 300:91–6. doi:10.1124/jpet.300.1.91
107. Yedlapudi D, Joshi GS, Luo D, Todi SV, Dutta AK. Inhibition of alpha-synuclein aggregation by multifunctional dopamine agonists assessed by a novel in vitro assay and an in vivo Drosophila synucleinopathy model. Sci Rep (2016) 6:38510. doi:10.1038/srep38510
108. Das B, Rajagopalan S, Joshi GS, Xu L, Luo D, Andersen JK, et al. A novel iron (II) preferring dopamine agonist chelator D-607 significantly suppresses alpha-syn- and MPTP-induced toxicities in vivo. Neuropharmacology (2017) 123:88–99. doi:10.1016/j.neuropharm.2017.05.019
109. Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet (2006) 15:3012–23. doi:10.1093/hmg/ddl243
110. Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science (2007) 317:516–9. doi:10.1126/science.1143780
111. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med (2009) 361:1651–61. doi:10.1056/NEJMoa0901281
112. Aflaki E, Westbroek W, Sidransky E. The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron (2017) 93:737–46. doi:10.1016/j.neuron.2017.01.018
113. Erickson AH, Ginns EI, Barranger JA. Biosynthesis of the lysosomal enzyme glucocerebrosidase. J Biol Chem (1985) 260:14319–24.
114. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet (2008) 372:1263–71. doi:10.1016/S0140-6736(08)61522-6
115. Barkhuizen M, Anderson DG, Grobler AF. Advances in GBA-associated Parkinson’s disease – pathology, presentation and therapies. Neurochem Int (2016) 93:6–25. doi:10.1016/j.neuint.2015.12.004
116. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell (2011) 146:37–52. doi:10.1016/j.cell.2011.06.001
117. Siebert M, Sidransky E, Westbroek W. Glucocerebrosidase is shaking up the synucleinopathies. Brain (2014) 137:1304–22. doi:10.1093/brain/awu002
118. Kinghorn KJ, Gronke S, Castillo-Quan JI, Woodling NS, Li L, Sirka E, et al. A Drosophila model of neuronopathic Gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin. J Neurosci (2016) 36:11654–70. doi:10.1523/JNEUROSCI.4527-15.2016
119. Davis MY, Trinh K, Thomas RE, Yu S, Germanos AA, Whitley BN, et al. Glucocerebrosidase deficiency in Drosophila results in alpha-synuclein-independent protein aggregation and neurodegeneration. PLoS Genet (2016) 12:e1005944. doi:10.1371/journal.pgen.1005944
120. Maor G, Rencus-Lazar S, Filocamo M, Steller H, Segal D, Horowitz M. Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J Rare Dis (2013) 8:140. doi:10.1186/1750-1172-8-140
121. Maor G, Cabasso O, Krivoruk O, Rodriguez J, Steller H, Segal D, et al. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum Mol Genet (2016) 25:2712–27. doi:10.1093/hmg/ddw129
122. Suzuki M, Fujikake N, Takeuchi T, Kohyama-Koganeya A, Nakajima K, Hirabayashi Y, et al. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant alpha-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet (2015) 24:6675–86. doi:10.1093/hmg/ddv372
123. Suzuki T, Shimoda M, Ito K, Hanai S, Aizawa H, Kato T, et al. Expression of human Gaucher disease gene GBA generates neurodevelopmental defects and ER stress in Drosophila eye. PLoS One (2013) 8:e69147. doi:10.1371/journal.pone.0069147
124. Sanchez-Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep (2016) 6:31380. doi:10.1038/srep31380
125. Robinson SW, Herzyk P, Dow JA, Leader DP. FlyAtlas: database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res (2013) 41:D744–50. doi:10.1093/nar/gks1141
126. Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis (2015) 82:235–42. doi:10.1016/j.nbd.2015.06.008
127. Bendikov-Bar I, Maor G, Filocamo M, Horowitz M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol Dis (2013) 50:141–5. doi:10.1016/j.bcmd.2012.10.007
128. Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol (2016) 80:766–75. doi:10.1002/ana.24790
129. Narita A, Shirai K, Itamura S, Matsuda A, Ishihara A, Matsushita K, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann Clin Transl Neurol (2016) 3:200–15. doi:10.1002/acn3.292
130. Williams ET, Chen X, Moore DJ. VPS35, the retromer complex and Parkinson’s disease. J Parkinsons Dis (2017) 7:219–33. doi:10.3233/JPD-161020
131. Belenkaya TY, Wu Y, Tang X, Zhou B, Cheng L, Sharma YV, et al. The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev Cell (2008) 14:120–31. doi:10.1016/j.devcel.2007.12.003
132. Franch-Marro X, Wendler F, Guidato S, Griffith J, Baena-Lopez A, Itasaki N, et al. Wingless secretion requires endosome-to-Golgi retrieval of Wntless/Evi/Sprinter by the retromer complex. Nat Cell Biol (2008) 10:170–7. doi:10.1038/ncb1678
133. Port F, Kuster M, Herr P, Furger E, Banziger C, Hausmann G, et al. Wingless secretion promotes and requires retromer-dependent cycling of Wntless. Nat Cell Biol (2008) 10:178–85. doi:10.1038/ncb1687
134. Korolchuk VI, Schutz MM, Gomez-Llorente C, Rocha J, Lansu NR, Collins SM, et al. Drosophila Vps35 function is necessary for normal endocytic trafficking and actin cytoskeleton organisation. J Cell Sci (2007) 120:4367–76. doi:10.1242/jcs.012336
135. Miura E, Hasegawa T, Konno M, Suzuki M, Sugeno N, Fujikake N, et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol Dis (2014) 71:1–13. doi:10.1016/j.nbd.2014.07.014
136. Malik BR, Godena VK, Whitworth AJ. VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum Mol Genet (2015) 24:6106–17. doi:10.1093/hmg/ddv322
137. Wang HS, Toh J, Ho P, Tio M, Zhao Y, Tan EK. In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol Brain (2014) 7:73. doi:10.1186/s13041-014-0073-y
138. Ishizu N, Yui D, Hebisawa A, Aizawa H, Cui W, Fujita Y, et al. Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum Mol Genet (2016) 25:4507–17. doi:10.1093/hmg/ddw279
139. Munsie LN, Milnerwood AJ, Seibler P, Beccano-Kelly DA, Tatarnikov I, Khinda J, et al. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum Mol Genet (2015) 24:1691–703. doi:10.1093/hmg/ddu582
Keywords: Parkinson’s disease, Drosophila, modeling, leucine-rich repeat kinase 2, α-synuclein, glucocerebrosidase, vacuolar protein sorting 35
Citation: Xiong Y and Yu J (2018) Modeling Parkinson’s Disease in Drosophila: What Have We Learned for Dominant Traits? Front. Neurol. 9:228. doi: 10.3389/fneur.2018.00228
Received: 09 January 2018; Accepted: 23 March 2018;
Published: 09 April 2018
Edited by:
Graziella Madeo, National Institutes of Health (NIH), United StatesReviewed by:
Matteo Bologna, Sapienza Università di Roma, ItalyPaolo Calabresi, University of Perugia, Italy
Copyright: © 2018 Xiong and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yulan Xiong, eXVsYW54aW9uZ0Brc3UuZWR1;
Jianzhong Yu, amlhbnpob25neXVAa3N1LmVkdQ==