 Nádia Pereira Gonçalves
Nádia Pereira Gonçalves Christian Bjerggaard Vægter
Christian Bjerggaard Vægter Lone Tjener Pallesen
Lone Tjener Pallesen- 1Department of Biomedicine, Nordic-EMBL Partnership for Molecular Medicine, Danish Research Institute of Translational Neuroscience (DANDRITE), Aarhus University, Aarhus, Denmark
- 2The International Diabetic Neuropathy Consortium (IDNC), Aarhus University, Aarhus, Denmark
The global prevalence of diabetes is rapidly increasing, affecting more than half a billion individuals within the next few years. As diabetes negatively affects several physiological systems, this dramatic increase represents not only impaired quality of life on the individual level but also a huge socioeconomic challenge. One of the physiological consequences affecting up to half of diabetic patients is the progressive deterioration of the peripheral nervous system, resulting in spontaneous pain and eventually loss of sensory function, motor weakness, and organ dysfunctions. Despite intense research on the consequences of hyperglycemia on nerve functions, the biological mechanisms underlying diabetic neuropathy are still largely unknown, and treatment options lacking. Research has mainly focused directly on the neuronal component, presumably from the perspective that this is the functional signal-transmitting unit of the nerve. However, it is noteworthy that each single peripheral sensory neuron is intimately associated with numerous glial cells; the neuronal soma is completely enclosed by satellite glial cells and the length of the longest axons covered by at least 1,000 Schwann cells. The glial cells are vital for the neuron, but very little is still known about these cells in general and especially how they respond to diabetes in terms of altered neuronal support. We will discuss current knowledge of peripheral glial cells and argue that increased research in these cells is imperative for a better understanding of the mechanisms underlying diabetic neuropathy.
Introduction
Peripheral neuropathy is a disorder affecting the peripheral sensory and autonomic nerves as a consequence of trauma or disease. The most common cause of peripheral neuropathy in the United States and Europe is type 2 diabetes, affecting 30–50% of diabetic patients (1–3). The global prevalence of diabetic neuropathy (DN) can be appreciated by the fact that an estimated 415 million people worldwide aged 20–79 had diabetes in 2015, and this number is expected to grow to 640 million by 2040, predominantly as a result of the increasing prevalence of type 2 diabetes. Furthermore, 318 million adults are estimated to have impaired glucose tolerance, or pre-diabetes, giving them a high risk of developing the disease (4).
In the peripheral nervous system (PNS), diabetes may induce several kinds of neuropathies. The most common type is known as the “stocking and glove” distribution of neuropathy, i.e., a bilateral and symmetric damage to nerves of the feet and hands (5). Patients commonly experience sensory impairment, which involves either loss of sensation or spontaneous feelings of touch, vibration, pricking, and hot and cold pain. The condition may also be accompanied by hypersensitivity (6, 7). Later, in the course of the disease, motor weakness and multiple organ dysfunction may be observed, arising from affected motor and autonomic nerves, respectively (8, 9). Unfortunately, no effective treatments for DN have been developed, and the main clinical strategy is still glucose control. However, a 2012 Cochrane review of all available clinical studies revealed that even rigorous glucose control is only able to decrease the incidence of DN in type 1 diabetes, whereas little to no effect was observed in type 2 diabetes (10), emphasizing the continuous need for more research.
The development of targeted therapies for DN has been hindered by a lack of understanding of the complex and different etiologies of this disorder. A number of molecular mechanisms have been proposed, ranging from metabolic to vascular hypotheses, with components of inflammation. These approaches have certainly contributed to our understanding of the various pathophysiological processes occurring in peripheral neurons under diabetic conditions. However, they have offered little help in preventing or reversing the symptoms of DN. It is noteworthy that the vast majority of clinical and basic research in DN has focused on the neuronal component of the nerves, presumably from the perspective that the neurons are the functional signal-transmitting cells. However, substantial amounts of data from research in the development and regeneration of the PNS categorically define the glial cells as indispensable components when it comes to maintaining neuronal structure and function as well as nourishing the axons and promoting survival and growth upon injury. Obviously, there is a huge gap in our understanding of how PNS glial cells respond to the diabetic conditions, and how this affects their important roles of nourishing and promoting neuronal survival and health. In this review, we address the evolving concepts relating PNS glial cells, namely satellite glial cells (SGCs) and Schwann cells, to diabetic neuropathogenesis based on insights from in vitro studies, animal models, and patients.
Satellite Glial Cells in Diabetic Neuropathy (DN)
Satellite Glial Cells—An Introduction
Satellite glial cells are localized in the sensory and autonomic ganglia of the PNS, forming a thin and tight sheath around each individual neuronal soma (Figure 1). The number of SCGs enclosing a neuron is proportional to the size of the neuronal soma and is in the range of 4–10 SGCs per neuron in mice and 8–12 SGCs per neuron in rats (11), while the numbers in humans are still to be determined. The SGCs surrounding a single soma are connected and communicate via gap junctions, forming a distinct functional unit (12). An extremely narrow gap of 20 nm between the neuronal soma and the SGCs enables tight control of the neuronal extracellular space by both the neuron and the SGCs. Evidence for bidirectional communication between neurons and SGCs is found in the sensory ganglia (13–15), largely mediated by purinergic P2 receptors, but our understanding of this communication is still limited.
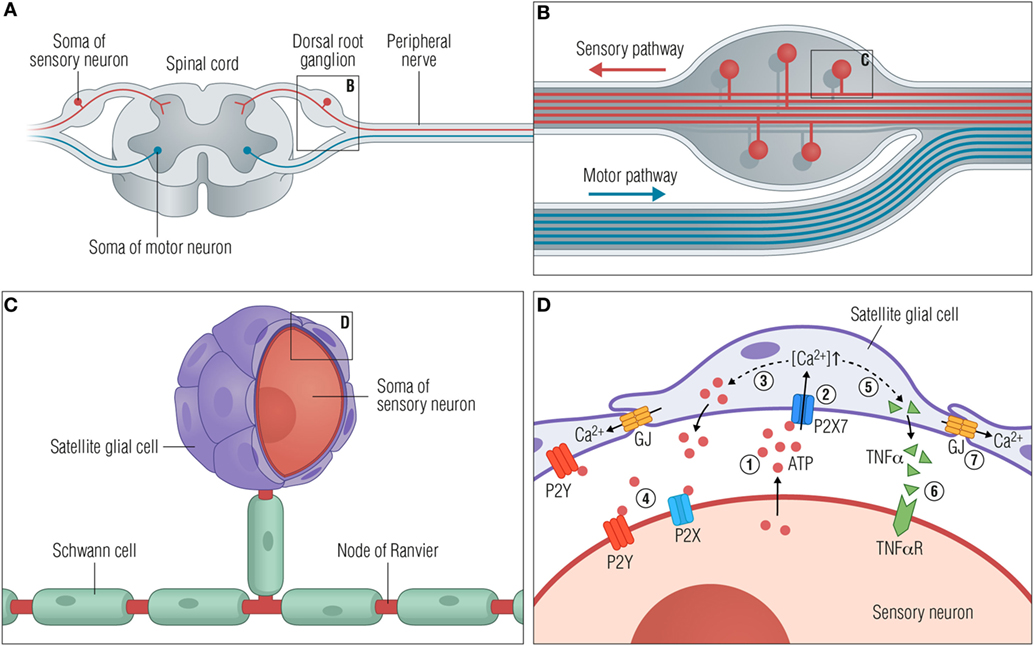
Figure 1. Satellite glial cells respond to neuronal stress. (A) A peripheral ganglion exemplified by the dorsal root ganglion (DRG). (B) Organization of the DRG. The pseudounipolar DRG neurons branch off with a peripheral process that connects with target tissues in the periphery and a central process in the spinal cord dorsal horn. (C) Close-up view of a single neuronal soma with myelinating Schwann cells along the axons and satellite glial cells (SGCs) closely enwrapping the soma. (D) Schematic diagram showing the communication between SGCs and the soma of the sensory neurons following neuronal stress. The consequences of injury or stress are various changes, some of which are outlined here. (1) ATP is released from the soma to the extracellular space. (2) Secreted ATP activates purinergic receptors such as P2X7 on the SGCs, causing an increase in [Ca2+]in. (3) The higher [Ca2+]in leads to greater release of ATP from SGCs (dashed line), further increasing the extracellular ATP concentration and subsequently (4) activation of P2X and P2Y receptors on the neurons. (5) Activation of P2X7 receptors furthermore facilitates the release of cytokines such as TNFα (dashed line) from the SGC, which then (6) binds and activates neuronal TNFα receptors. (7) Activated SGCs are characterized by an upregulation of connexin channels and thereby an increase in gap junctions and intercellular coupling of SGCs by Ca2+ waves. Abbreviations: GJ, gap junctions; TNFα, tumor necrosis factor α; TNFαR, TNFα receptor.
The SGCs are flattened cells, and the intimate spatial relationship between SGCs and neurons complicates their isolation and analysis. Consequently, the biology of SGCs is inadequately characterized under normal as well as pathological conditions. However, in recent decades, an increasing amount of research utilizing rodent models of nerve injury and dorsal root ganglion (DRG) explant assays have established SGCs as homeostatic cells providing support and modulation of sensory neurons; it is now clear that SGCs express various neurotransmitter receptors, transporters, and ion channels, allowing them to monitor neuronal activity and homeostasis. Furthermore, they release neurotransmitters and neuroactive substances such as ATP and cytokines as part of a glia-to-neuron signaling (12, 14, 16–20).
SGCs Respond to Neuronal Stress
It has become evident that a wide variety of neuronal stress situations trigger a state of activation in the SGCs. This was demonstrated in animal models of traumatic nerve injury (21–24), but also in animal models of diabetes (25–27), inflammation (28, 29), chemotherapy (30), and herpes simplex infection (31). Altogether, these findings strongly suggest that SGC activation is a normal physiological response to neuronal stress.
Activated SGCs are characterized by profound changes [for extensive reviews, see Ref. (17, 32, 33)] that generally appear as cell proliferation (22), upregulation of glial fibrillary acidic protein (GFAP) (34, 35), and upregulation of connexin channels (17, 36, 37). Injured or stressed neurons have been shown to release ATP, targeting SGC purinergic P2 receptors (13, 38) and thereby initiating gap junction-dependent Ca2+ waves between the SGCs (17, 39). The principal purinergic receptors expressed in SGCs are the metabotropic P2Y receptor subtypes 1, 2, 4, 6, 12, and 13 (20, 40), and the ionotropic P2X7 receptor in both rodents (41, 42) and human DRGs (43).
Interestingly, SGC-to-neuron signaling is mediated by the activation of SGC P2X7 receptors, resulting in the secretion of both tumor necrosis factor α (TNFα) and ATP from SGC (44) and contributing to hyperexcitability of the enwrapped neurons (41, 44–46). Furthermore, recent publications demonstrate how SGCs surrounding neighboring sensory neurons respond to nerve injury by inducing neuronal activity coupling, arguing that neuronal stress may spread within a ganglion (23, 45, 47). In accordance with these observations, it has also been shown how the blocking of gap junctions attenuates hyperexcitability of DRG neurons and mechanical hyperalgesia (48).
These and other findings establish SGCs as important components for neuronal homeostasis and activity and show that SGC activation is a normal consequence of neuronal stress and injury. It is, therefore, a realistic perspective that targeting SGCs is a feasible strategy for modulating neuronal activity and obtain improved pain management and neuronal regeneration in patients suffering from various peripheral neuropathies.
A Perspective on SGC Involvement in DN
Characteristics of DN are nerve dysfunction and degeneration of nerve endings in the extremities, likely due to a complex combination of factors such as metabolic imbalances, inflammation, hyperglycemia, oxidative stress, and lower oxygen tension (9, 49). Analyses of DRGs indeed show that the DRG neurons are susceptible to injury by metabolic and hypoxic stressors in models of diabetes, as the neurons show altered gene expression of, e.g., voltage-gated calcium channels and sodium channels, increased neuronal excitability as well as mitochondrial dysfunction leading to excess formation of reactive oxygen species, deregulation of Ca2+ homeostasis, and Ca2+ signaling (9, 50–54).
As outlined above, SGCs respond to a wide range of neuronal stress situations, but only very few reports on SGCs in a diabetic context have been published. Hanani et al. described how SGCs are activated in a streptozotocin-induced type 1 diabetes model in rodents, revealed by a fourfold increase in the number of neurons surrounded by GFAP-positive SGCs in the mouse DRG and a fivefold increase in rats (25). In line with this, it was recently found that preventing P2X7 receptor upregulation in a rat diabetic model inhibited the activation of SGCs as evaluated by GFAP expression in the DRG (26). Moreover, inhibiting upregulation of P2X7 receptors reduced the release of TNFα, thereby inhibiting the excitability of DRG neurons and reducing mechanical and thermal hyperalgesia in diabetic rats.
Further support for SGC responsiveness came from the observation that streptozotocin-induced diabetes in mice substantially enhanced DRG neuron expression and activity of pyruvate dehydrogenase kinases (PDK2 and PDK4), key regulatory enzymes in glucose metabolism (27). This upregulation seemed necessary to induce SGC reactivity, as PDK2/4 deficiency significantly attenuates SGC GFAP immunoreactivity following streptozotocin-treatment. The authors indicated that PDK2/4 may be expressed by activated SGCs; however, the presented data are not sufficiently convincing.
Together, these data establish that SGCs indeed respond to the diabetes-induced neuronal stress and, therefore, may participate in the generation and maintenance of DN. It may, however, also be that increased glucose acts upon the SGCs directly, initiating SGC reactivity without prior neuronal stress. Such a view is supported by the observation that aldose reductase is expressed by SGCs but not neurons in the DRG of both normal and diabetic rats (55). As described in further detail in the Section “Schwann Cells in DN” below, increased flux through the polyol pathway converts glucose to sorbitol by aldose reductase, increasing cellular stress by increasing osmolarity and depleting cellular stores of nicotinamide adenine dinucleotide phosphate (NADPH). However, in contrast to Schwann cells, the role of the polyol pathway in SGCs in relation to DN is completely unexplored.
Unfortunately, it has proven very difficult to isolate and study the SGCs due to their extremely close spatial relationship with the neurons. Attempts to study SGCs are made using primary cultures of isolated SGC for in vitro studies (56, 57). However, it has been reported how mouse trigeminal SGCs rapidly change morphology and gene expression in culture when they lose their tight association with the neuronal soma (58). This is likely a general feature also applying to DRG and autonomic SGCs, complicating both in vitro identification of SGCs and a meaningful interpretation of results.
To overcome these obstacles, we have recently developed a protocol that allows complete dissociation and isolation of highly pure SGCs by fluorescence-activated cell sorting (FACS) using SGC-specific antibodies. The method further allows purification of high-quality RNA from the fixed and permeabilized cells (59), and proteomic data can also be obtained. We believe that this strategy will initiate a deeper investigation of SGCs at transcriptional and translational levels in vivo, increasing our knowledge on SGC biology and their role in the complex pathobiology underlying DN.
Schwann Cells in DN
Pathological Hallmarks
Schwann cells are the most abundant glial cells of the PNS, ensheathing all axons of peripheral nerves as either myelinating or non-myelinating cells (Figure 2A). Schwann cells are not merely passive insulators of axons but dynamic partners with the potential to modulate neuronal biology by providing metabolic support (60), enrichment of sodium channels (61), as well as modulating responses to tissue injury (62, 63). In disease states such as diabetes, Schwann cell function might be disturbed, compromising glial–axon communication, and nerve homeostasis, ultimately leading to fiber loss, neurodegeneration, and pain. Signs of Schwann cell stress are well demonstrated by unspecific morphological changes found in nerve preparations from humans as well as cat and rodent models of disease (64–67), indicating compromised Schwann cell functionality. As pathology proceeds, mild segmental axonal demyelination and remyelination begin to occur in the presence of a normal axon (68, 69), suggesting that Schwannopathy may very well underlie the primary damage to nerve fibers and be the first step in the pathogenesis of DN.
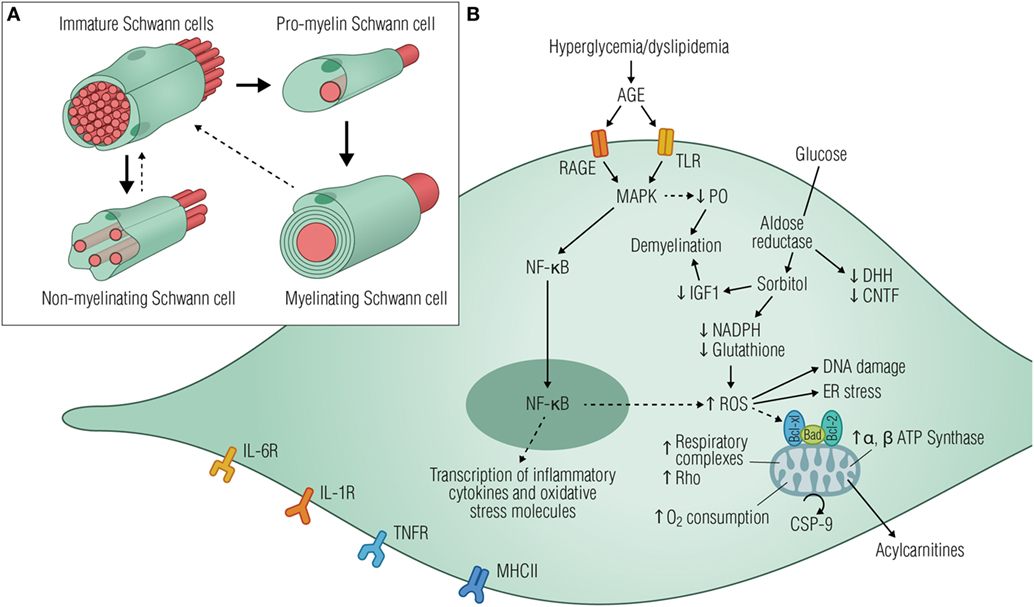
Figure 2. Diabetic stressors lead to Schwann cell dysfunction and neurodegeneration. (A) Schematic illustration of the main developmental transitions of Schwann cells. Dashed arrows denote the reversibility of the final stage, where mature myelinating and non-myelinating cells might dedifferentiate as a result of injury or disease. (B) Elevated glucose levels are shunted into the polyol pathway by aldose reductase, depleting cytosolic NADH, and ultimately reducing regeneration of glutathione, an important cellular antioxidant. Consequently, glucose metabolism generates local oxidative damage, reduces the production of neurotrophic factors, activates the ER stress, and causes DNA damage, all of which drives the cells to an immature phenotype. Signs of mitochondrial stress triggered by hyperglycemia are also evident in Schwann cells by the increased expression of ATP synthase subunits and respiratory chain complexes I, III, IV, and V, thus increasing oxygen consumption and apoptotic signaling events. As a result, acylcarnitines are released from stressed Schwann cells, leading to axonal degeneration and fiber loss. AGE activation of RAGE and/or TLR induces downstream signaling events mediated, at least in part, by the activation of MAPK and NF-κB with consequent transcription of inflammatory cytokines and oxidative stress molecules. Schwann cells have been recognized as immune-competent cells, expressing MHC class II molecules and several cytokine receptors such as IL-6R, IL-1R, and TNFR. Since MHC II antigen reactivity was detected in Schwann cells from patients suffering from diabetic neuropathy, it is plausible that these cells might be functioning as antigen processing and presenting cells contributing to immune responses within the peripheral nerves. Abbreviations: AGE, advanced glycation end products; RAGE, receptor for advanced glycation end products; TLRS, toll-like receptors; MAPK, mitogen-activated protein kinase; P0, protein 0; NF-κB, nuclear factor κB; NADPH, nicotinamide adenine dinucleotide phosphate-oxidase; IGF1, insulin-like growth factor 1; ROS, reactive oxygen species; ER, endoplasmic reticulum; CSP-9, caspase-9; IL-6R, interleukin-6 receptor; IL-1R, IL-1 receptor; TNFR, tumor necrosis factor receptor; MHCII, major histocompatibility complex II.
We will now review metabolic pathways known to be activated in Schwann cells during DN (summarized in Figure 2B), with an emphasis on the concept that Schwannopathy might be a central mechanism that leads to PNS damage in diabetes.
Revisiting the Polyol Pathway
Hyperglycemia-driven increased flux through the polyol pathway is the most studied pathogenic mechanism of DN. Excess glucose is converted to sorbitol by aldose reductase, increasing cellular osmolarity and depleting stores of NADPH, an important component for the generation of nitric oxide and restoration of the crucial antioxidant glutathione. As a result, oxidative stress will contribute to cellular damage and dysfunction, impairing nerve structure and homeostasis (9) (Figure 2B). Strikingly, in the endoneurium, aldose reductase is essentially expressed by myelinating Schwann cells (55), implying that increased flux through aldose reductase is an essential factor in hyperglycemic toxicity to these cells. Although diabetic rodents only show demyelination after long-term neuropathy or when in the presence of hypertension (70), there is nevertheless evidence of Schwann cell alterations in these models such as reduced production of myelin proteins (71) and neurotrophic factors like CNTF (72), NGF, and NT-3 (73, 74). Reduced desert hedgehog (DHH) expression by the Schwann cells further contributes to nerve fiber loss and conduction velocity impairment, which together with induced endothelial dysfunction contribute to the development of DN (75, 76).
Perturbations in Schwann cell metabolism will affect glial–axon communication and promote direct deleterious effects on neuronal function, resulting from myelin disruption, impaired regeneration, and slower nerve conduction velocity (49). One hypothesis, based on cell culture studies, is that hyperglycemia leads to sorbitol accumulation and reduces the expression of insulin-like growth factor 1, promoting Schwann cell dedifferentiation into an immature phenotype (77). Other studies highlight the reduced expression of glycoprotein P0, myelin basic protein, and myelin-associated glycoprotein by the Schwann cells as main culprits for myelin loss (78, 79). Reduced P0, in particular, was shown to be dependent on Schwann cell upregulation of the mitogen-activated protein kinase (MAPK) signaling, which in turn contributes to keeping the cell in a dedifferentiated state (79), creating a pathogenic feedback loop (Figure 2B).
Preclinical tests with aldose reductase inhibitors became the hot topic in the 1990s. In spite of promising results in the animal models (80), all human clinical trials failed, possibly due to poor trial design, incorrect dose, limited drug potency, or the inability of the compounds to cross the blood–nerve barrier (81), pointing toward the importance of new lines of research.
Diabetic Stressors Increase Schwann Cell Dysfunction and Death
When cultured in an hyperglycemic environment, Schwann cells change their spindle-shaped morphology and look thin and short, with less extended cell bodies and failed process outgrowth (79, 82), consistent with glucose-triggered apoptotic signaling events such as cleavage of caspase-3 and DNA fragmentation (83).
High glucose-induced death in Schwann cells in vitro was associated with altered expression of caspase-9, BAX, and Bcl-2 (84), thus indicating mitochondrial internal stress (Figure 2B). In fact, mitochondrial dysfunction has been associated with the pathogenesis of DN in multiple ways: data from primary sensory neurons associated mitochondrial dysfunction with perturbed calcium homeostasis (85, 86). More recently, studies have demonstrated how mitochondrial deficits in Schwann cells seem to activate a maladaptive stress response, causing accumulation of acylcarnitines, which ultimately induced axonal degeneration upon release (87, 88).
Hyperglycemia has also been associated with remodeling of the Schwann cell mitochondrial proteome, with increased expression of α and β subunits of ATP synthase. Other consequences of high glucose are suboptimal respiratory capacity triggered by the increased overall rate of oxygen consumption, thus suggesting deficient oxidative phosphorylation in Schwann cells (89) (Figure 2B). Proteomic and metabolomic data from the peripheral nerve of a rodent model of type 1 diabetes showed upregulation of mitochondrial respiratory complexes and Rho GTPase 1. The fact that no similar differences were found in sensory or trigeminal ganglia assign the described changes to a disrupted Schwann cell mitochondrial function (90) and is believed to engage the glial demyelination program (91). Indeed, ultrastructural abnormalities in Schwann cell mitochondria are present in peripheral nerves from both human diabetic patients and animal models of disease (92, 93). Hence, targeting Schwann cell mitotoxicity might offer new insights into the therapeutics of DN by preventing demyelination and increasing axonal survival.
Activation of an Immune-Like Phenotype in Schwann Cells
The non-enzymatic glycation of proteins leading to their dysfunction is a long-established feature of diabetes that has been linked to neuropathy. In recent years, this concept has been expanded to include pathological signaling of advanced glycation end products through its receptor (RAGE), which is located on axons and Schwann cells of the peripheral nerve (94). In fact, RAGE overexpression was evident in Schwann cells of the sural nerve from diabetic patients, possibly linking oxidative modification and inflammation with preclinical features of disease (95). The induced downstream events resulting from RAGE signaling involve activation of MAPKs and nuclear factor κB, with consequent transcription of inflammatory intermediates, production of ROS, and vasoconstriction (96) (Figure 2B). This scenario is similar to that of traumatic nerve injury where Schwann cells dedifferentiate and adapt a repair-mediating phenotype essential for myelin degradation and nerve regeneration (97).
However, with continuous exposure to deleterious signals such as hyperglycemia and modified lipoproteins, a feed-forward loop of injury is created due to enhanced immune responses, increased cellular oxidative, endoplasmic, and nitrosative stress (98–100). Schwann cells have also been recognized as immune-competent cells, as they express major histocompatibility complex II molecules and several toll-like and inflammatory receptors (Figure 2B) and produce several cytokines known to be involved in the pathogenesis of DN (101–105). As inflammatory cytokines can sensitize Aδ and C-fibers, these observations highlight the potential involvement of Schwann cells in the development of painful DN. In addition, inflammatory mediators produced by Schwann cells may contribute to the recruitment of immune cells like macrophages and T cells, which in turn can induce Schwann cell damage by triggering apoptosis (106–108). Overall, a deeper investigation of the Schwann cell inflammatory response might offer new insights into the stressful cellular mechanisms contributing to glial cell dysfunction, disruption of neuronal metabolism, axonal transport, or repair capabilities in DN.
Conclusion
Despite the increasing knowledge into the importance of glial cells for nerve tissue function and homeostasis, most research on DN is focused on the peripheral sensory neurons. Significantly less is known about the peripheral glial cells, and particularly, the SGCs have hardly been touched in a diabetic context. Important insights into the fundamental biology of PNS glia have been obtained in models of nerve injury and inflammation, and we believe that many aspects of glia responsiveness can be extrapolated into other disease states such as diabetes mellitus. Future work should focus on obtaining a comprehensive understanding of the genetic and molecular architecture of SGCs and Schwann cells, unraveling how they support neurons and engage in bidirectional communication to modulate neuronal activity. Knowledge on the pharmacological properties of these glia will help unravel novel mechanisms and targets for peripheral neuropathies such as DN.
Author Contributions
NG, CV, and LP equally contributed to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Lundbeck Foundation, the Independent Research Fund Denmark, the Dagmar Marshall Fund and the Riisfort Foundation.
References
1. Abbott CA, Malik RA, van Ross ERE, Kulkarni J, Boulton AJM. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care (2011) 34:2220–4. doi:10.2337/dc11-1108
2. Van Acker K, Bouhassira D, De Bacquer D, Weiss S, Matthys K, Raemen H, et al. Prevalence and impact on quality of life of peripheral neuropathy with or without neuropathic pain in type 1 and type 2 diabetic patients attending hospital outpatients clinics. Diabetes Metab (2009) 35:206–13. doi:10.1016/j.diabet.2008.11.004
3. Ziegler D, Rathmann W, Dickhaus T, Meisinger C, Mielck A, KORA Study Group. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: the MONICA/KORA Augsburg Surveys S2 and S3. Diabetes Care (2008) 31:464–9. doi:10.2337/dc07-1796
4. International Diabetes Federation. IDF Diabetes Atlas. 8th ed. Brussels (2015). Available from: http://www.diabetesatlas.org (Accessed: April 13, 2018).
5. Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol (2012) 11:521–34. doi:10.1016/S1474-4422(12)70065-0
6. Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol (2014) 13:924–35. doi:10.1016/S1474-4422(14)70102-4
7. Truini A, Biasiotta A, Di Stefano G, La Cesa S, Leone C, Cartoni C, et al. Peripheral nociceptor sensitization mediates allodynia in patients with distal symmetric polyneuropathy. J Neurol (2012) 260:761–6. doi:10.1007/s00415-012-6698-9
8. Verrotti A, Prezioso G, Scattoni R, Chiarelli F. Autonomic neuropathy in diabetes mellitus. Front Endocrinol (2014) 5:579. doi:10.3389/fendo.2014.00205
9. Feldman EL, Nave K-A, Jensen TS, Bennett DLH. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron (2017) 93:1296–313. doi:10.1016/j.neuron.2017.02.005
10. Callaghan BC, Little AA, Feldman EL, Hughes RAC. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst Rev (2012) (6):CD007543. doi:10.1002/14651858.CD007543.pub2
11. Ledda M, De Palo S, Pannese E. Ratios between number of neuroglial cells and number and volume of nerve cells in the spinal ganglia of two species of reptiles and three species of mammals. Tissue Cell (2004) 36:55–62. doi:10.1016/j.tice.2003.09.001
12. Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev (2005) 48:457–76. doi:10.1016/j.brainresrev.2004.09.001
13. Feldman-Goriachnik R, Hanani M. Functional study of endothelin B receptors in satellite glial cells in trigeminal ganglia. Neuroreport (2011) 22:465–9. doi:10.1097/WNR.0b013e3283472487
14. Suadicani SO, Cherkas PS, Zuckerman J, Smith DN, Spray DC, Hanani M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol (2010) 6:43–51. doi:10.1017/S1740925X09990408
15. Huang L-YM, Gu Y, Chen Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia (2013) 61:1571–81. doi:10.1002/glia.22541
16. Gu Y, Chen Y, Zhang X, Li G-W, Wang C, Huang L-YM. Neuronal soma-satellite glial cell interactions in sensory ganglia and the participation of purinergic receptors. Neuron Glia Biol (2010) 6:53–62. doi:10.1017/S1740925X10000116
17. Hanani M. Intercellular communication in sensory ganglia by purinergic receptors and gap junctions: implications for chronic pain. Brain Res Rev (2012) 1487:183–91. doi:10.1016/j.brainres.2012.03.070
18. Jasmin L, Vit J-P, Bhargava A, Ohara PT. Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol (2010) 6:63–71. doi:10.1017/S1740925X10000098
19. Takeda M, Takahashi M, Matsumoto S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci Biobehav Rev (2009) 33:784–92. doi:10.1016/j.neubiorev.2008.12.005
20. Villa G, Fumagalli M, Verderio C, Abbracchio MP, Ceruti S. Expression and contribution of satellite glial cells purinoceptors to pain transmission in sensory ganglia: an update. Neuron Glia Biol (2010) 6:31–42. doi:10.1017/S1740925X10000086
21. Humbertson A, Zimmermann E, Leedy M. A chronological study of mitotic activity in satellite cell hyperplasia associated with chromatolytic neurons. Z Zellforsch Mikrosk Anat (1969) 100:507–15. doi:10.1007/BF00344371
22. Donegan M, Kernisant M, Cua C, Jasmin L, Ohara PT. Satellite glial cell proliferation in the trigeminal ganglia after chronic constriction injury of the infraorbital nerve. Glia (2013) 61:2000–8. doi:10.1002/glia.22571
23. Hanani M, Huang TY, Cherkas PS, Ledda M, Pannese E. Glial cell plasticity in sensory ganglia induced by nerve damage. Neuroscience (2002) 114:279–83. doi:10.1016/S0306-4522(02)00279-8
24. Vit J-P, Jasmin L, Bhargava A, Ohara PT. Satellite glial cells in the trigeminal ganglion as a determinant of orofacial neuropathic pain. Neuron Glia Biol (2007) 2:247–57. doi:10.1017/S1740925X07000427
25. Hanani M, Blum E, Liu S, Peng L, Liang S. Satellite glial cells in dorsal root ganglia are activated in streptozotocin-treated rodents. J Cell Mol Med (2014) 18:2367–71. doi:10.1111/jcmm.12406
26. Liu S, Zou L, Xie J, Xie W, Wen S, Xie Q, et al. LncRNA NONRATT021972 siRNA regulates neuropathic pain behaviors in type 2 diabetic rats through the P2X7 receptor in dorsal root ganglia. Mol Brain (2016) 9:348. doi:10.1186/s13041-016-0226-2
27. Rahman MH, Jha MK, Kim J-H, Nam Y, Lee MG, Go Y, et al. Pyruvate dehydrogenase kinase-mediated glycolytic metabolic shift in the dorsal root ganglion drives painful diabetic neuropathy. J Biol Chem (2016) 291:6011–25. doi:10.1074/jbc.M115.699215
28. Nascimento DSM, Castro-Lopes JM, Neto FLM. Satellite glial cells surrounding primary afferent neurons are activated and proliferate during monoarthritis in rats: is there a role for ATF3? PLoS One (2014) 9:e108152. doi:10.1371/journal.pone.0108152
29. Blum E, Procacci P, Conte V, Sartori P, Hanani M. Long term effects of lipopolysaccharide on satellite glial cells in mouse dorsal root ganglia. Exp Cell Res (2016) 350(1):236–41. doi:10.1016/j.yexcr.2016.11.026
30. Warwick RA, Hanani M. The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur J Pain (2013) 17:571–80. doi:10.1002/j.1532-2149.2012.00219.x
31. Warwick RA, Hanani M. Involvement of aberrant calcium signalling in herpetic neuralgia. Exp Neurol (2016) 277:10–8. doi:10.1016/j.expneurol.2015.12.002
32. Hanani M. Satellite glial cells in sympathetic and parasympathetic ganglia: in search of function. Brain Res Rev (2010) 64:304–27. doi:10.1016/j.brainresrev.2010.04.009
33. Hanani M. Role of satellite glial cells in gastrointestinal pain. Front Cell Neurosci (2015) 9:4733. doi:10.3389/fncel.2015.00412
34. Elfvin LG, Björklund H, Dahl D, Seiger A. Neurofilament-like and glial fibrillary acidic protein-like immunoreactivities in rat and guinea-pig sympathetic ganglia in situ and after perturbation. Cell Tissue Res (1987) 250:79–86. doi:10.1007/BF00214657
35. Woodham P, Anderson PN, Nadim W, Turmaine M. Satellite cells surrounding axotomised rat dorsal root ganglion cells increase expression of a GFAP-like protein. Neurosci Lett (1989) 98:8–12. doi:10.1016/0304-3940(89)90364-9
36. Ohara PT, Vit J-P, Bhargava A, Romero M, Sundberg C, Charles AC, et al. Gliopathic pain: when satellite glial cells go bad. Neuroscientist (2009) 15:450–63. doi:10.1177/1073858409336094
37. Pannese E, Ledda M, Cherkas PS, Huang TY, Hanani M. Satellite cell reactions to axon injury of sensory ganglion neurons: increase in number of gap junctions and formation of bridges connecting previously separate perineuronal sheaths. Anat Embryol (2003) 206:337–47. doi:10.1007/s00429-002-0301-6
38. Feldman-Goriachnik R, Hanani M. The effects of endothelin-1 on satellite glial cells in peripheral ganglia. Neuropeptides (2017) 63:37–42. doi:10.1016/j.npep.2017.03.002
39. Verkhratsky A, Fernyhough P. Calcium signalling in sensory neurones and peripheral glia in the context of diabetic neuropathies. Cell Calcium (2014) 56:362–71. doi:10.1016/j.ceca.2014.07.005
40. Ceruti S, Fumagalli M, Villa G, Verderio C, Abbracchio MP. Purinoceptor-mediated calcium signaling in primary neuron-glia trigeminal cultures. Cell Calcium (2008) 43:576–90. doi:10.1016/j.ceca.2007.10.003
41. Zhang X, Chen Y, Wang C, Huang LYM. Neuronal somatic ATP release triggers neuron-satellite glial cell communication in dorsal root ganglia. Proc Natl Acad Sci U S A (2007) 104:9864–9. doi:10.1073/pnas.0611048104
42. Kushnir R, Cherkas PS, Hanani M. Peripheral inflammation upregulates P2X receptor expression in satellite glial cells of mouse trigeminal ganglia: a calcium imaging study. Neuropharmacology (2011) 61:739–46. doi:10.1016/j.neuropharm.2011.05.019
43. Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain (2005) 114:386–96. doi:10.1016/j.pain.2005.01.002
44. Song J, Ying Y, Wang W, Liu X, Xu X, Wei X, et al. The role of P2X7R/ERK signaling in dorsal root ganglia satellite glial cells in the development of chronic postsurgical pain induced by skin/muscle incision and retraction (SMIR). Brain Behav Immun (2017) 69:180–9. doi:10.1016/j.bbi.2017.11.011
45. Spray DC, Hanani M. Gap junctions, pannexins and pain. Neurosci Lett (2017). doi:10.1016/j.neulet.2017.06.035
46. Dubový P, Jančálek R, Klusáková I, Svíženská I, Pejchalová K. Intra- and extraneuronal changes of immunofluorescence staining for TNF- and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell Mol Neurobiol (2006) 26:1203–15. doi:10.1007/s10571-006-9006-3
47. Kim YS, Anderson M, Park K, Zheng Q, Agarwal A, Gong C, et al. Coupled activation of primary sensory neurons contributes to chronic pain. Neuron (2016) 91:1085–96. doi:10.1016/j.neuron.2016.07.044
48. Huang T-Y, Belzer V, Hanani M. Gap junctions in dorsal root ganglia: possible contribution to visceral pain. Eur J Pain (2010) 14:.e1–11. doi:10.1016/j.ejpain.2009.02.005
49. Gonçalves NP, Vaegter CB, Andersen H, Østergaard L, Calcutt NA, Jensen TS. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat Rev Neurol (2017) 13:135–47. doi:10.1038/nrneurol.2016.201
50. Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem (2004) 279:29341–50. doi:10.1074/jbc.M404167200
51. Latham JR, Pathirathna S, Jagodic MM, Joo Choe W, Levin ME, Nelson MT, et al. Selective T-type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes (2009) 58:2656–65. doi:10.2337/db08-1763
52. Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, et al. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci (2007) 27:3305–16. doi:10.1523/JNEUROSCI.4866-06.2007
53. Duzhyy DE, Viatchenko-Karpinski VY, Khomula EV, Voitenko NV, Belan PV. Upregulation of T-type Ca2+ channels in long-term diabetes determines increased excitability of a specific type of capsaicin-insensitive DRG neurons. Mol Pain (2015) 11:29. doi:10.1186/s12990-015-0028-z
54. Huang Y, Zang Y, Zhou L, Gui W, Liu X, Zhong Y. The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem Int (2014) 75:112–9. doi:10.1016/j.neuint.2014.05.012
55. Jiang Y, Calcutt NA, Ramos KM, Mizisin AP. Novel sites of aldose reductase immunolocalization in normal and streptozotocin-diabetic rats. J Peripher Nerv Syst (2006) 11:274–85. doi:10.1111/j.1529-8027.2006.00099.x
56. da Silva LB, Poulsen JN, Arendt-Nielsen L, Gazerani P. Botulinum neurotoxin type A modulates vesicular release of glutamate from satellite glial cells. J Cell Mol Med (2015) 19:1900–9. doi:10.1111/jcmm.12562
57. Poulsen JN, Larsen F, Duroux M, Gazerani P. Primary culture of trigeminal satellite glial cells: a cell-based platform to study morphology and function of peripheral glia. Int J Physiol Pathophysiol Pharmacol (2014) 6:1–12.
58. Belzer V, Shraer N, Hanani M. Phenotypic changes in satellite glial cells in cultured trigeminal ganglia. Neuron Glia Biol (2010) 6:237–43. doi:10.1017/S1740925X1100007X
59. Jager SB, Pallesen LT, Vaegter CB. Isolation of satellite glial cells for high-quality RNA purification. J Neurosci Methods (2018) 297:1–8. doi:10.1016/j.jneumeth.2018.01.001
60. Brown AM, Evans RD, Black J, Ransom BR. Schwann cell glycogen selectively supports myelinated axon function. Ann Neurol (2012) 72:406–18. doi:10.1002/ana.23607
61. Voas MG, Glenn TD, Raphael AR, Talbot WS. Schwann cells inhibit ectopic clustering of axonal sodium channels. J Neurosci (2009) 29:14408–14. doi:10.1523/JNEUROSCI.0841-09.2009
62. Scheib JL, Höke A. An attenuated immune response by Schwann cells and macrophages inhibits nerve regeneration in aged rats. Neurobiol Aging (2016) 45:1–9. doi:10.1016/j.neurobiolaging.2016.05.004
63. Scheib J, Höke A. Advances in peripheral nerve regeneration. Nat Rev Neurol (2013) 9:668–76. doi:10.1038/nrneurol.2013.227
64. Mizisin AP. Mechanisms of diabetic neuropathy: Schwann cells. Handb Clin Neurol (2014) 126:401–28. doi:10.1016/B978-0-444-53480-4.00029-1
65. Lennertz RC, Medler KA, Bain JL, Wright DE, Stucky CL. Impaired sensory nerve function and axon morphology in mice with diabetic neuropathy. J Neurophysiol (2011) 106:905–14. doi:10.1152/jn.01123.2010
66. Vincent AM, Brownlee M, Russell JW. Oxidative stress and programmed cell death in diabetic neuropathy. Ann N Y Acad Sci (2002) 959:368–83. doi:10.1111/j.1749-6632.2002.tb02108.x
67. Eckersley L. Role of the Schwann cell in diabetic neuropathy. Int Rev Neurobiol (2002) 50:293–321. doi:10.1016/S0074-7742(02)50081-7
68. Malik RA, Tesfaye S, Newrick PG, Walker D, Rajbhandari SM, Siddique I, et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia (2005) 48:578–85. doi:10.1007/s00125-004-1663-5
69. Mizisin AP, Shelton GD, Wagner S, Rusbridge C, Powell HC. Myelin splitting, Schwann cell injury and demyelination in feline diabetic neuropathy. Acta Neuropathol (1998) 95:171–4. doi:10.1007/s004010050783
70. Gregory JA, Jolivalt CG, Goor J, Mizisin AP, Calcutt NA. Hypertension-induced peripheral neuropathy and the combined effects of hypertension and diabetes on nerve structure and function in rats. Acta Neuropathol (2012) 124:561–73. doi:10.1007/s00401-012-1012-6
71. Kawashima R, Kojima H, Nakamura K, Arahata A, Fujita Y, Tokuyama Y, et al. Alterations in mRNA expression of myelin proteins in the sciatic nerves and brains of streptozotocin-induced diabetic rats. Neurochem Res (2007) 32:1002–10. doi:10.1007/s11064-006-9260-2
72. Calcutt NA, Muir D, Powell HC, Mizisin AP. Reduced ciliary neuronotrophic factor-like activity in nerves from diabetic or galactose-fed rats. Brain Res Rev (1992) 575:320–4. doi:10.1016/0006-8993(92)90097-S
73. Suzuki T, Sekido H, Kato N, Nakayama Y, Yabe-Nishimura C. Neurotrophin-3-induced production of nerve growth factor is suppressed in Schwann cells exposed to high glucose: involvement of the polyol pathway. J Neurochem (2004) 91:1430–8. doi:10.1111/j.1471-4159.2004.02824.x
74. Dey I, Midha N, Singh G, Forsyth A, Walsh SK, Singh B, et al. Diabetic Schwann cells suffer from nerve growth factor and neurotrophin-3 underproduction and poor associability with axons. Glia (2013) 61:1990–9. doi:10.1002/glia.22570
75. Calcutt NA, Allendoerfer KL, Mizisin AP, Middlemas A, Freshwater JD, Burgers M, et al. Therapeutic efficacy of sonic hedgehog protein in experimental diabetic neuropathy. J Clin Invest (2003) 111:507–14. doi:10.1172/JCI200315792
76. Chapouly C, Yao Q, Vandierdonck S, Larrieu-Lahargue F, Mariani JN, Gadeau A-P, et al. Impaired hedgehog signalling-induced endothelial dysfunction is sufficient to induce neuropathy: implication in diabetes. Cardiovasc Res (2016) 109:217–27. doi:10.1093/cvr/cvv263
77. Hao W, Tashiro S, Hasegawa T, Sato Y, Kobayashi T, Tando T, et al. Hyperglycemia promotes Schwann cell de-differentiation and de-myelination via sorbitol accumulation and Igf1 protein down-regulation. J Biol Chem (2015) 290:17106–15. doi:10.1074/jbc.M114.631291
78. Rachana KS, Manu MS, Advirao GM. Insulin influenced expression of myelin proteins in diabetic peripheral neuropathy. Neurosci Lett (2016) 629:110–5. doi:10.1016/j.neulet.2016.06.067
79. Liu D, Liang X, Zhang H. Effects of high glucose on cell viability and differentiation in primary cultured Schwann cells: potential role of ERK signaling pathway. Neurochem Res (2016) 41(6):1281–90. doi:10.1007/s11064-015-1824-6
80. Stavniichuk R, Shevalye H, Hirooka H, Nadler JL, Obrosova IG. Interplay of sorbitol pathway of glucose metabolism, 12/15-lipoxygenase, and mitogen-activated protein kinases in the pathogenesis of diabetic peripheral neuropathy. Biochem Pharmacol (2012) 83:932–40. doi:10.1016/j.bcp.2012.01.015
81. Grewal AS, Bhardwaj S, Pandita D, Lather V, Sekhon BS. Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Rev Med Chem (2016) 16:120–62. doi:10.2174/1389557515666150909143737
82. Gadau SD. Nitrosative stress induces proliferation and viability changes in high glucose-exposed rat Schwannoma cells. Neuro Endocrinol Lett (2012) 33:279–84.
83. Delaney CL, Russell JW, Cheng HL, Feldman EL. Insulin-like growth factor-I and over-expression of Bcl-xL prevent glucose-mediated apoptosis in Schwann cells. J Neuropathol Exp Neurol (2001) 60:147–60. doi:10.1093/jnen/60.2.147
84. Han J, Tan P, Li Z, Wu Y, Li C, Wang Y, et al. Fuzi attenuates diabetic neuropathy in rats and protects Schwann cells from apoptosis induced by high glucose. PLoS One (2014) 9:e86539. doi:10.1371/journal.pone.0086539
85. Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A, et al. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes (2003) 52:2129–36. doi:10.2337/diabetes.52.8.2129
86. Verkhratsky A, Fernyhough P. Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium (2008) 44:112–22. doi:10.1016/j.ceca.2007.11.010
87. Viader A, Golden JP, Baloh RH, Schmidt RE, Hunter DA, Milbrandt J. Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J Neurosci (2011) 31:10128–40. doi:10.1523/JNEUROSCI.0884-11.2011
88. Viader A, Sasaki Y, Kim S, Strickland A, Workman CS, Yang K, et al. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron (2013) 77:886–98. doi:10.1016/j.neuron.2013.01.012
89. Zhang L, Yu C, Vasquez FE, Galeva N, Onyango I, Swerdlow RH, et al. Hyperglycemia alters the Schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J Proteome Res (2010) 9:458–71. doi:10.1021/pr900818g
90. Freeman OJ, Unwin RD, Dowsey AW, Begley P, Ali S, Hollywood KA, et al. Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy. Diabetes (2015) 65(1):228–38. doi:10.2337/db15-0835
91. Gonzalez S, Berthelot J, Jiner J, Perrin-Tricaud C, Fernando R, Chrast R, et al. Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies. J Clin Invest (2016) 126:1023–38. doi:10.1172/JCI84505
92. Kalichman MW, Powell HC, Mizisin AP. Reactive, degenerative, and proliferative Schwann cell responses in experimental galactose and human diabetic neuropathy. Acta Neuropathol (1998) 95:47–56. doi:10.1007/s004010050764
93. Mizisin AP, Nelson RW, Sturges BK, Vernau KM, Lecouteur RA, Williams DC, et al. Comparable myelinated nerve pathology in feline and human diabetes mellitus. Acta Neuropathol (2007) 113:431–42. doi:10.1007/s00401-006-0163-8
94. Martini R, Willison H. Neuroinflammation in the peripheral nerve: cause, modulator, or bystander in peripheral neuropathies? Glia (2015) 64(4):475–86. doi:10.1002/glia.22899
95. Haslbeck KM, Neundörfer B, Schlötzer-Schrehardt U, Bierhaus A, Schleicher E, Pauli E, et al. Activation of the RAGE pathway: a general mechanism in the pathogenesis of polyneuropathies? Neurol Res (2013) 29:103–10. doi:10.1179/174313206X152564
96. Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, et al. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology (2007) 148:548–58. doi:10.1210/en.2006-0073
97. Gomez-Sanchez JA, Pilch KS, van der Lans M, Fazal SV, Benito C, Wagstaff LJ, et al. After nerve injury, lineage tracing shows that myelin and Remak Schwann cells elongate extensively and branch to form repair Schwann cells, which shorten radically on remyelination. J Neurosci (2017) 37:9086–99. doi:10.1523/JNEUROSCI.1453-17.2017
98. Vincent AM, Calabek B, Roberts L, Feldman EL. Biology of diabetic neuropathy. Handb Clin Neurol (2013) 115:591–606. doi:10.1016/B978-0-444-52902-2.00034-5
99. Padilla A, Descorbeth M, Almeyda AL, Payne K, De Leon M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res Rev (2011) 1370:64–79. doi:10.1016/j.brainres.2010.11.013
100. Yang X, Yao W, Liu H, Gao Y, Liu R, Xu L. Tangluoning, a traditional Chinese medicine, attenuates in vivo and in vitro diabetic peripheral neuropathy through modulation of PERK/Nrf2 pathway. Sci Rep (2017) 7:144. doi:10.1038/s41598-017-00936-9
101. Tzekova N, Heinen A, Küry P. Molecules involved in the crosstalk between immune- and peripheral nerve Schwann cells. J Clin Immunol (2014) 34:86–104. doi:10.1007/s10875-014-0015-6
102. O’Brien PD, Hur J, Hayes JM, Backus C, Sakowski SA, Feldman EL. BTBR ob/ob mice as a novel diabetic neuropathy model: neurological characterization and gene expression analyses. Neurobiol Dis (2015) 73:348–55. doi:10.1016/j.nbd.2014.10.015
103. Menichella DM, Abdelhak B, Ren D, Shum A, Frietag C, Miller RJ. CXCR4 chemokine receptor signaling mediates pain in diabetic neuropathy. Mol Pain (2014) 10:42. doi:10.1186/1744-8069-10-42
104. Chen L, Li B, Chen B, Shao Y, Luo Q, Shi X, et al. Thymoquinone alleviates the experimental diabetic peripheral neuropathy by modulation of inflammation. Sci Rep (2016) 6:137. doi:10.1038/srep31656
105. Shi X, Chen Y, Nadeem L, Xu G. Beneficial effect of TNF-α inhibition on diabetic peripheral neuropathy. J Neuroinflammation (2013) 10:69. doi:10.1186/1742-2094-10-69
106. Nukada H, McMorran PD, Baba M, Ogasawara S, Yagihashi S. Increased susceptibility to ischemia and macrophage activation in STZ-diabetic rat nerve. Brain Res Rev (2011) 1373:172–82. doi:10.1016/j.brainres.2010.11.084
107. Conti G, Scarpini E, Baron P, Livraghi S, Tiriticco M, Bianchi R, et al. Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: a process concomitant with endoneurial induction of IL-1beta and p75NTR. J Neurol Sci (2002) 195:35–40. doi:10.1016/S0022-510X(01)00684-0
Keywords: diabetic neuropathy, satellite glial cells, Schwann cells, peripheral nervous system, dorsal root ganglion, diabetes mellitus
Citation: Gonçalves NP, Vægter CB and Pallesen LT (2018) Peripheral Glial Cells in the Development of Diabetic Neuropathy. Front. Neurol. 9:268. doi: 10.3389/fneur.2018.00268
Received: 02 December 2017; Accepted: 06 April 2018;
Published: 02 May 2018
Edited by:
Lars Dahlin, Lund University, SwedenReviewed by:
Claude Knauf, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceYulia A. Sidorova, University of Helsinki, Finland
Menachem Hanani, Hadassah Medical Center, Israel
Copyright: © 2018 Gonçalves, Vægter and Pallesen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Bjerggaard Vægter, Y3ZAYmlvbWVkLmF1LmRr