 Amol Mehta
Amol Mehta Adam M. Sonabend
Adam M. Sonabend- Department of Neurological Surgery, Northwestern University Feinberg School of Medicine, Chicago, IL, United States
Despite advances in surgery, radiotherapy, and chemotherapy, glioblastoma (GBM) remains a malignancy with poor prognosis. The molecular profile of GBM is diverse across patients, and individual responses to therapy are highly variable. Yet, patients diagnosed with GBM are treated with a rather uniform paradigm. Exploiting these molecular differences and inter-individual responses to therapy may present an opportunity to improve the otherwise bleak prognosis of patients with GBM. This review aims to examine one group of chemotherapeutics: Topoisomerase 2 (TOP2) poisons, a class of drugs that enables TOP2 to induce DNA damage, but interferes with its ability to repair it. These potent chemotherapeutic agents are currently used for a number of malignancies and have shown promise in the treatment of GBM. Despite their robust efficacy in vitro, some of these agents have fallen short of achieving similar results in clinical trials for this tumor. In this review, we explore reasons for this discrepancy, focusing on drug delivery and individual susceptibility differences as challenges for effective TOP2-targeting for GBM. We critically review the evidence implicating genes in susceptibility to TOP2 poisons and categorize this evidence as experimental, correlative or both. This is important as mere experimental evidence does not necessarily lead to identification of genes that serve as good biomarkers of susceptibility for personalizing the use of these drugs.
Introduction
Glioblastoma (GBM) is the most common primary type of brain malignancy, representing 28% of all Central Nervous System (CNS) tumors and 80% of the malignant subset (1). These tumors carry a dismal prognosis; the average survival for GBM patients is 13 months, with a 2-year survival of 27%, and a 5-year survival of 5.1% (2–4). All patients diagnosed with GBM undergo the same non-curative treatment, consisting of surgery, chemotherapy and radiation. This homogeneous treatment paradigm stands in stark contrast to the heterogeneous molecular profile found in GBM which has led to the classification of these tumors into subtypes based on their patterns of gene expression, genetic alterations, and DNA methylation (5, 6). These expression patterns and their underlying mechanisms could provide a unique tumoral vulnerability. These tumors are also rather unpredictable with regards to their response to therapies, and a growing body of literature is focused on the prediction of inter-individual response to treatment with the goal of personalizing therapies for GBM. This review aims to critically examine one such class of chemotherapeutics: Topoisomerase-2 (TOP2) poisons, one of the most powerful and common groups of chemotherapeutic agents used for cancer. We discuss these drugs given recent evidence suggesting they are highly effective for a subset of GBM, eliciting the idea that a refined assessment of their efficacy in select patient populations may show their value for treating this disease and provide a strategy for their use with a personalized or precision-medicine approach. In this context, we provide an overview of the literature that evaluates cancer susceptibility to TOP2-targeting drugs.
TOP2 Targeting Drugs Background
Topoisomerase Biology
Topoisomerases are ubiquitously expressed enzymes that execute many crucial cellular functions. They are necessary, as the double helical nature of DNA leads to torsional forces that need to be relaxed for processes like DNA replication, repair, transcription, and chromosomal segregation (7). Also, by creating single or double stranded DNA breaks (DSB), topoisomerases remove supercoils generated by the continuous unwinding and rewinding of double stranded DNA (7, 8). These enzymes also perform decatenating functions, which are necessary to remove interlinked DNA products (catenanes) formed during replication (Figure 1) (7, 8).
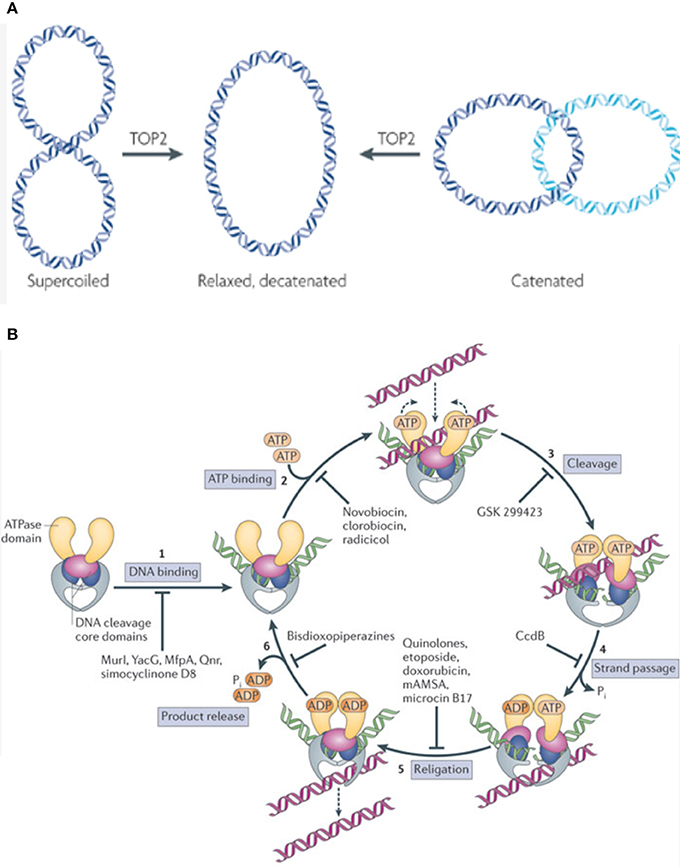
Figure 1. (A) TOP2 functions by relieving supercoils generated as a function of the double stranded nature of DNA. Additionally, TOP2 removes catenanes (interlinked DNA products) formed during replication. This allows the cell to carry out vital functions like replication, repair, and transcription. Figure modified and reproduced with permission from Nature Publishing Group. (B) TOP2 inhibitors function by interfering with various steps in the enzyme's catalytic cycle. Some Topoisomerase inhibitors work by inhibiting DNA binding, thus preventing the formation of the DNA-enzyme complex (1), while others act at the next step, preventing ATP from binding to the DNA-enzyme complex which in turn does not allow for the formation of a closed clamp structure (2). Some agents prevent the enzymatic generation of a double stranded DNA break (DSB) (3). Other downstream agents function by preventing the passage of the intact strand through the already generated ds-DNA break (4). Etoposide, doxorubicin, and analogous agents function by inhibiting DNA religation, thus stabilizing the DSB-enzyme complex and triggering apoptosis (5). Some inhibitors work at the last step of the catalytic cycle, and preventing the release of product from the enzyme (6). Figure reproduced with permission from Nature Publishing Group.
There are two main categories of Topoisomerases, TOP1 and TOP2, each with their own subtypes (A/B), distinct functions, and mechanisms of action. TOP2 decoils DNA by creating transient DSB and passing a separate DNA duplex through the breaks generated, enhancing chromatin accessibility (9, 10). TOP2A/B differ in their catalytic sites; the etoposide binding site of hTOP2A contains a methionine (M762) residue while hTOP2B contains a Glutamine (Q778). This difference makes the DNA-TOP2A complex more stable than the DNA-TOP2B complex, and this stability has been shown to increase cellular susceptibility to TOP2 poisions (11, 12). TOP2A is typically expressed by cycling cells, whereas TOP2B is often expressed by post-mitotic cells (7). Both enzymes are capable of re-ligating the break they generate. The period between the cleaved and ligated states is when these enzymes are susceptible to TOP2 poisons (13).
TOP2 Targeting Drugs' Mechanism of Action
TOP2 targeting drugs can be split into two broad categories; catalytic inhibitors and TOP2 poisons. Catalytic inhibitors impede an important catalytic step in the enzyme's reaction cycle (9, 8). Examples include Aclarubicin, which functions by preventing TOP2 from initially binding to DNA, and Merbarone, which prevents TOP2 from cleaving the DNA once bound (9).
Other drugs are called TOP2 poisons, as they kill cells by stabilizing transient intermediates in which the enzyme is linked to DNA and thus trigger DSB, essentially converting the topoisomerase enzyme into an agent that damages DNA, leading to apoptosis (14). Epipodophyllotoxins like etoposide perform the above functions by covalently linking to DNA while Anthracyclines do so by intercalating into DNA. Doxorubicin, an Anthracycline, is a TOP2 poison that intercalates into DNA and also dislodges histones from their chromatin, disrupting the normal DNA damage response, interfering with epigenetic regulation at damaged sites, and generating reactive oxygen species (15, 16).
Some agents, like dexrazoxane and other bisdiozopiperazines, inhibit TOP2 after it passes the duplex through its DNA break, but before it hydrolyzes ATP. This effectively prevents the closed-clamp structure of TOP2 from re-opening, and the enzyme from turning over (17).
TOP2 Poisons in Brain Tumors
Pre-clinical and Early Clinical Data
There is some evidence to suggest that the use of TOP2-targeting drugs may be efficacious in the treatment of GBM. Numerous in vitro and animal studies have demonstrated the anti-tumor effects of doxorubicin against GBM cell lines (18–21). A human ex vivo study investigating the response to TOP2 poisons in short-term cultures derived from malignant gliomas demonstrated that both etoposide and doxorubicin are toxic to these tumor cells (18), while another study in rat models designed to study combination TOP1 + TOP2 therapies showed doxorubicin toxicity toward GBM cell lines (19).
Phase II studies tested the use of systemic etoposide in recurrent gliomas and showed that a subset of recurrent GBM patients partially responded to an etoposide-containing regimen (22, 23). Other studies, however, demonstrated a lack of efficacy, which may be partially explained by variable expression of TOP2A within GBM. It is important to keep in mind some of these trials used metronomic doses of etoposide (35 mg/m2). This dose is sub-optimal given that the majority of trials that have demonstrated etoposide's efficacy against GBM have used doses of 50 mg/m2 and 100 mg/m2 (24). Additionally, many of these trials used etoposide in combination with a number of other agents (25). Additionally, a meta-analysis found that treatment with etoposide is associated with overall increased survival (24).
To investigate the relative susceptibility of gliomas to etoposide compared to other cancers, we conducted an analysis and compared the susceptibility of 667 human cancer cell lines to etoposide using publicly available data from https://www.cancerrxgene.org (Figure 2) (27). Our analysis demonstrates that testicular cancer is the most responsive to etoposide, and gliomas' response is comparable to that of lymphoma, osteosarcoma, and neuroblastoma. We found gliomas had a similar response to etoposide as small cell lung cancer (SCLC) and myeloma, two cancers that have traditionally been treated with etoposide.
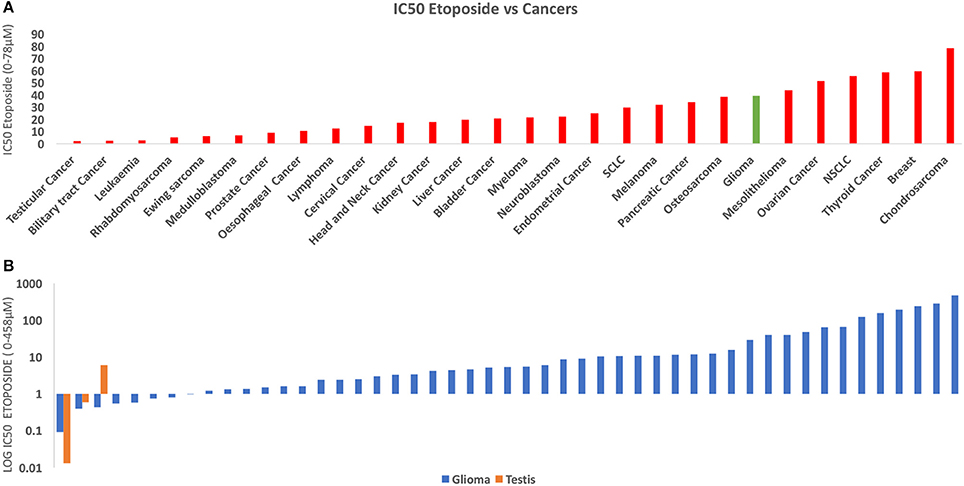
Figure 2. (A) This figure depicts the IC50 of Etoposide against human cancers derived from 900 cell lines. The data was derived from Cancerxgene. The IC50 for each cancer group was averaged and the standard deviation was then determined. Testicular cancer demonstrated the highest susceptibility to etoposide. The response of Glioma (red) was similar to many of these cell lines including SCLC and Osteosarcoma, both of which are traditionally treated with Etoposide (26). (B) Chart derived from the same data comparing IC50 Etoposide for Glioma (Orange) and Testicular Cancer (Blue). Some glioma cell lines demonstrate a similar response to etoposide as do testicular cancer cell lines.
Current Challenges in TOP2-Targeting Therapy for Brain Tumors
While early data for TOP2 poisons is promising, their pharmacokinetic profile and poor blood-brain barrier (BBB) penetrance have limited their efficacy in the treatment of GBM. The underperformance of etoposide can be attributed to low levels and wide ranges of intra-tumoral drug concentrations. Concentrations have been shown to range between 12 and 36% of blood concentration, with intratumoral concentrations ranging between 2–6 μM (28–30). Conventional systemic delivery beyond this dosing is limited by toxicity. Similarly, the primary explanation for doxorubicin's disappointing efficacy in vivo has been its lack of ability to penetrate the BBB, due to its high molecular weight and low lipophilicity (31).
In order to circumvent these challenges, there have been a number of attempts to optimize chemotherapeutic delivery to the CNS. Attempts of using alternative delivery methods like Convection Enhanced Delivery (CED) have yielded promising results. A recent study by our group demonstrated direct intratumoral delivery of high concentrations of etoposide and increased anti-tumor effects against the proneural subtype of GBM (25). In this study, we found that intratumoral delivery of etoposide at a 4uM concentration, which is similar to what is achieved following intravenous delivery, only led to transient decrease in tumor growth with no effect on survival. Yet, a concentration of 80 μM of etoposide delivered intra-tumorally led to a robust survival benefit for transgenic mouse models of proneural gliomas, a subtype of glioma that has been shown to highly express 5, TOP2A and TOP2B (25). Direct intra-tumoral delivery of 680 μM led to cure of most treated mice, and remained well tolerated (Figure 3) (25). It is important to recognize that this study does not establish a causal link between the proneural gene signature with etoposide susceptibility. There is, in fact, causal evidence linking other genes with etoposide susceptibility, and ultimately, there may be better biomarkers to predict etoposide response than the proneural gene signature.
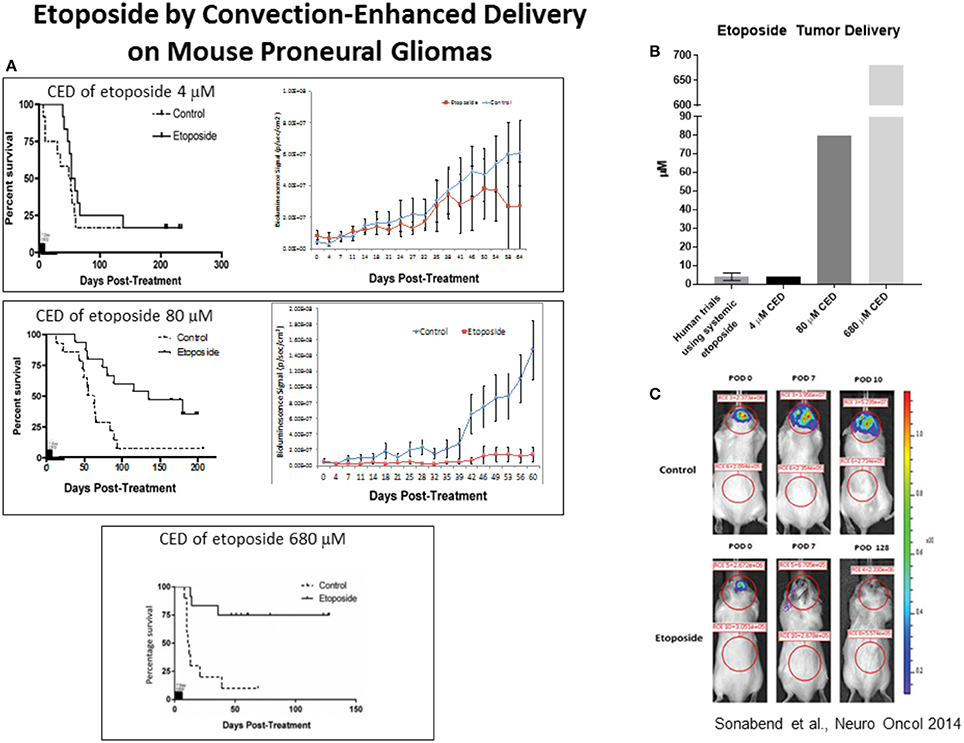
Figure 3. (A) In a recent study, we found that mice treated with 80 μM etoposide via Convection Enhanced Delivery (CED) demonstrated prolonged survival when compared control mice treated with 4 μM (the mean concentration achieved in previous studies using etoposide). Additionally, mice treated with 680 μM had a significant survival benefit when compared to control mice, with a 75% cure rate. (B) Graphic illustrating different doses of etoposide delivered in this study; 4 μM, 80 μM, 680 μM. (C) Mice treated with 680 μM of etoposide using CED also demonstrated lower bioluminescence signal, measured by using bioluminescence imaging to monitor luciferase signaling. Figures reproduced with permission from Oxford University Press.
Other attempts to optimize doxorubicin delivery include the use of liposomes (32), nanoparticles (33, 34), focused ultrasound (35), minicells (36), and direct injection (37). In a rat model, MRI-guided focused ultrasound was demonstrated to achieve intratumoral doxorubicin concentrations of 886 ± 327 ng/g tissue which is within the therapeutic range of 819 ± 482 ng/g tumor compared with the control intratumoral concentration of 215 ± 119 ng/g tissue regardless of the dose administered (35).
Combining TOP2 poisons with other agents is another attempt to increase the narrow therapeutic window of these drugs as certain combinations can enhance cytotoxicity as well as increase selectivity (32, 38, 39). Nanoliposomal topotecan in combination with pegylated liposomal doxorubicin administered through CED was found to be associated with a significantly increased median survival, and an additive effect was observed between these two agents in a rodent model (40). Additionally, in certain instances combination therapy may allow for intermittent rather than continuous dosing regiments, allowing for better tolerability while maintaining a similar efficacy. For example, poly (ADP-ribose) polymerase (PARP) inhibition is typically necessary throughout DNA damage and repair processes. However, when combined with TOP2 poisons, which are DNA damaging agents, continuous PARP inhibition may not be necessary as long as a critical inhibitory level is met during only DNA repair (41). This combination of TOP2 poison and PARP inhibitors has been studied in ovarian cancer; a phase 1 dose escalation study with pegylated liposomal doxorubicin in combination with olaparib, a PARP inhibitor, demonstrated that this combination was generally well tolerated, with only 3 out of 44 patients demonstrating dose-limiting toxicities, while 33% of the patients responded to therapy (41).
Ongoing Trials Testing TOP2 Drugs for GBM
There are currently four clinical trials investigating doxorubicin's role in the treatment of GBM. Some are investigating doxorubicin in the setting of novel delivery mechanisms such as Laser Interstitial Thermal Therapy (LITT) mediated disruption of the BBB (ClinicalTrials.gov-NCT01851733), and nanoparticle delivery targeting cells using bispecific antibodies (ClinicalTrials.gov-NCT02766699). Another is studying the safety and efficacy of aldoxorubicin in subjects with unresectable GBM whose tumors have progressed following prior treatment with surgery, radiation, and temozolomide (ClinicalTrials.gov-NCT02014844). Another trial is investigating the safety and efficacy of prolonged administration of doxorubicin in combination with radiotherapy, temozolomide, and histone deacetylase inhibitor valproic acid in pediatric and adult patients with newly diagnosed GBM and diffuse intrinsic pontine glioma (ClinicalTrials.gov-NCT02758366).
A number of clinical trials studying the etoposide's utility are also ongoing. One trial is investigating combination therapy with sodium thiosulfate in the treatment of gliomas in order to determine if the addition of sodium thiosulfate can protect against chemotherapy related thrombocytopenia (ClinicalTrials.gov-NCT00075387). Another clinical trial is studying the side effects and optimal method of delivering vorinostat with isotretinoin and chemotherapy including IV etoposide phosphate and other agents like carboplatin, cisplatin, cyclophosphamide, thiotepa, vincristine sulfate, and vorinostat against embryonal CNS tumors like medulloblastoma and pineoblastoma. There is a trial investigating the use of LITT to disrupt peritumoral BBB to enhance delivery and efficacy of therapeutic agents including etoposide in the treatment of pediatric brain tumors (ClinicalTrials.gov-NCT02372409). Another clinical trial is investigating overall survival after administration of various chemotherapeutic agents (including etoposide) followed by autologous peripheral stem cell transplantation in the treatment of numerous solid and CNS tumors (ClinicalTrials.gov-NCT01505569).
Topoisomerase Poison Susceptibility: Biomarkers to Personalize Therapy
For many of the 1 million patients treated annually with a TOP2 poison, therapy is often ineffective and can lead to a number of side effects (16). Many tumors are either initially resistant to these therapies, or become resistant at time of relapse (42). Recognizing underlying mechanisms of resistance/susceptibility (Figure 4) and their tumor molecular signature presents an opportunity to personalize our treatments, and expand our currently narrow therapeutic window by increasing the benefit of these drugs while decreasing unnecessary side effects. The studies that have implicated these underlying mechanisms in susceptibility can be split into two broad categories. The first category, called “experimental” evidence, includes studies in which the gene was experimentally disrupted, leading to cancer cell resistance/susceptibility. The second category, “correlative” evidence, includes genes that are implicated as their natural disruption or variations of expression in some tumors correlates with susceptibility (Table 1).
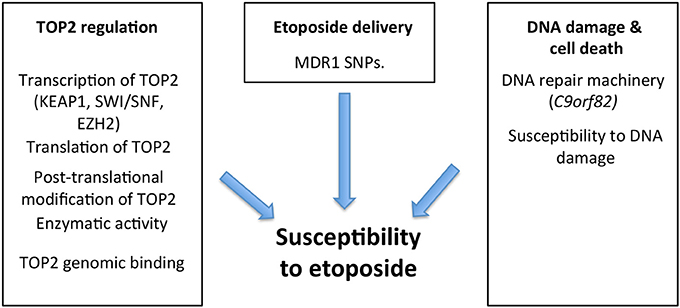
Figure 4. There are several processes that might modulate cancer susceptibility to etoposide, including levels of TOP2 expression (determined by transcription and translation), genomic binding of TOP2 as well as its enzymatic activity and post-translational modifications. Mutations in KEAP1, SWI/SNF complex, and EZH2 influence TOP2 at a transcriptional level, consequently influencing TOP2 resistance/susceptibility. Additionally, etoposide delivery is influenced by resistance proteins like the MDR1 efflux pump, which dictate intracellular concentration and therefore cytotoxicity. The cytotoxic effect of these agents largely stems from their ability to stabilize double-stranded breaks (DSB) in DNA, triggering cellular apoptosis. Therefore, the cell's existing DNA repair machinery, and its overall susceptibility to DNA damage plays a role in determining the success of TOP2 poisons such as etoposide and mutations such as those in C9orf82 have been found to influence this (43, 44).
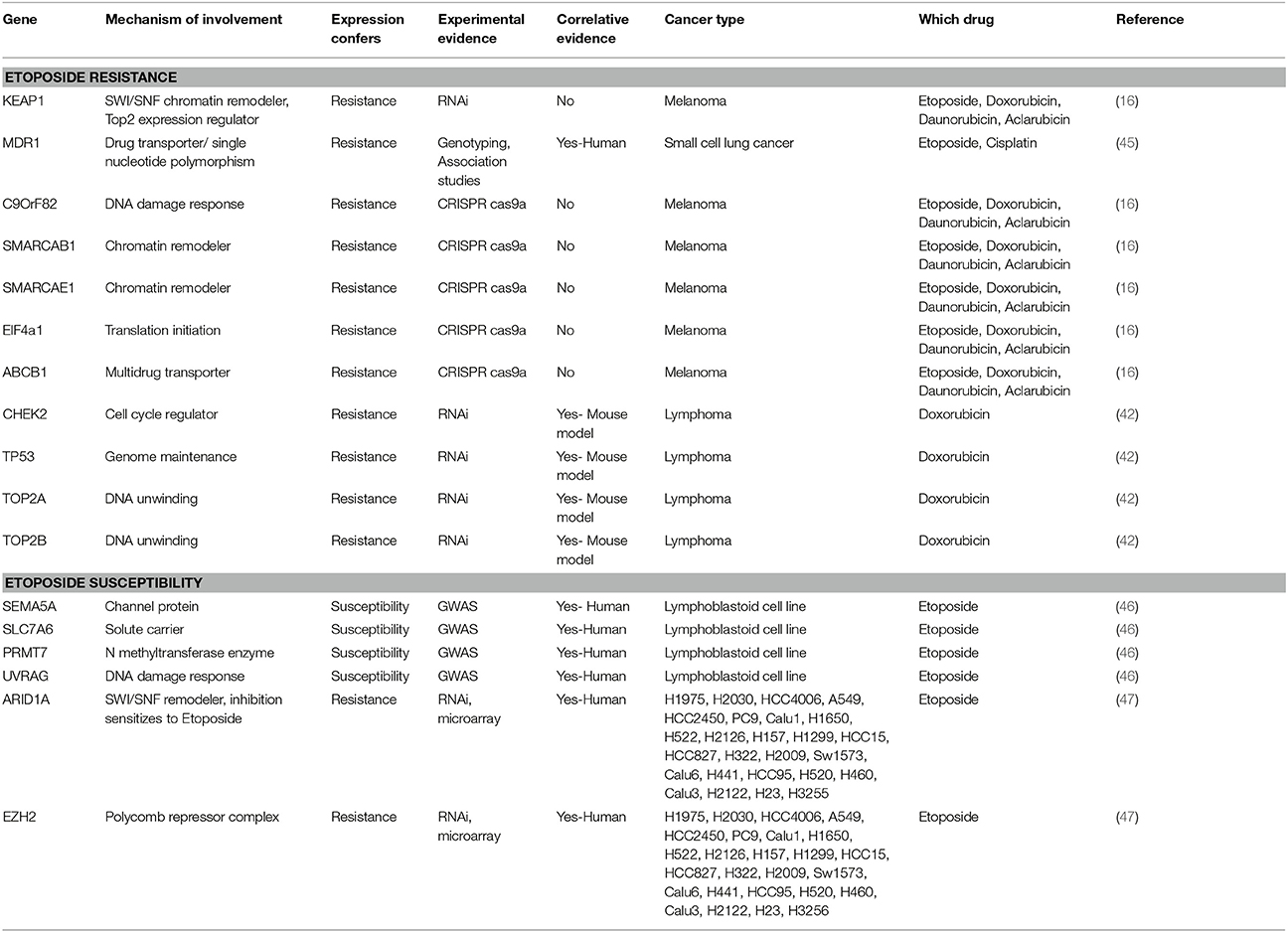
Table 1. This table shows curation of genes identified in (A) etoposide /doxorubicin resistance or (B) susceptibility, their mechanism of action, means of experimental validation of the genes, correlation to survival, cancers in which they were identified, and the associated reference.
Levels of Expression of TOP2 Protein
A number of studies have thus far shown an association between levels of topoisomerase expression and cellular susceptibility to TOP2 poisons (48, 49). TOP2A suppression is thought to result in resistance by decreasing the amount of enzyme-DNA complex, in turn decreasing the amount of DNA damage (42). Our group found a correlation between the transcript levels of TOP2B and etoposide susceptibility across 139 human cancer cell lines (25). We demonstrated that TOP2A levels were significantly elevated in platelet-derived growth factor (PDGF)+ and phosphatase tensin homolog (PTEN)−/− mouse proneural tumors which were susceptible to intratumorally delivered etoposide by CED (correlative evidence) (Figure 3) (25). Several additional studies have demonstrated that TOP2 poison susceptibility is related to levels of TOP2 expression. One particular study decreased levels of TOP2A expression using gene suppressor elements (GSE's) and demonstrated a resistance to etoposide in different mammalian cell lines (experimental evidence) (50). Another study explored the genetic basis for response heterogeneity using a pool-based RNAi screening approach, identifying TOP2A expression levels as major determinants of doxorubicin response in a mouse model of lymphoma. In addition, by decreasing TOP2A expression in vivo using retrovirally encoded shRNAs (experimental evidence), this study was able to demonstrate a relationship between TOP2A levels and doxorubicin susceptibility (42). This same study noted that tumors which relapsed after doxorubicin therapy displayed highly reduced TOP2A levels (42).
Merely suppressing the expression of TOP2A and demonstrating resistance does not prove that TOP2A expression levels can be used as a biomarker to personalize therapy. Rather, this only succeeds in demonstrating etoposide and doxorubicin's mechanisms of action. The more important question is whether or not naturally occurring variations in TOP2A expression correlate with etoposide/doxorubicin susceptibility, which does not (25). This implies that TOP2A expression is necessary but perhaps not sufficient for efficacious tumor cell killing by these agents. It is possible that there are other molecular factors limiting the effectiveness of these agents, and that even a small amount of TOP2A expression is sufficient to elicit DNA damage if other conditions are met. Importantly, many studies have demonstrated that increased TOP2A expression is correlated with improved prognosis in GBM, measured as survival at 2 years (51), 5-year progression free survival and overall survival (52, 53) (correlative evidence).
Other Genes Associated With Susceptibility to TOP2 Poisons
KEAP1
Mutations in the KEAP1 (Kelch-like ECH-associated Protein-1) gene are common abnormalities in non-small cell lung cancer (NSCLC), gallbladder, liver (54), ovarian (55), endometrial (56), and lung papillary cancers (57). Keap1 is an E3 ubiquitin ligase involved in degrading Nrf2, which regulates transcription of genes that mediate the response to oxidative stress (58). Studies have suggested that decreased Keap1 expression and increased Nrf2 expression enhances tumor cell growth. A systematic analysis of the KEAP1 genomic locus in NSCLC cell lines demonstrated bi-allelic inactivation in KEAP1 was associated with constitutive activation of Nrf2-mediated gene expression (59). Another study demonstrated similar findings in biliary tract and gallbladder cancers. Amplifying alterations in KEAP1 are found in approximately 0.8% of GBM tumors (43, 44). Mutations in the Keap1-Nrf2 pathway have been implicated in TOP2 poison resistance (60). For instance, Wang, et. al demonstrated that stable overexpression of Nrf2 resulted in increased resistance to agents such as etoposide, cisplatin, and doxorubicin. Conversely, they demonstrated down-regulation of the Nrf2 response was associated with increased susceptibility to the above therapies (experimental evidence) (61). Also, a recent study used a genome wide knock down approach and demonstrated that KEAP1 mutations also confer resistance to TOP2 poisons by decreasing TOP2A expression levels (experimental evidence) (16). Therefore, assessing the mutational status of KEAP1 prior to initiating therapy may provide a more informed and personalized approach to treatment.
ABCB1
Drug efflux is a well-researched mechanism of resistance to chemotherapy, where increased activity of a molecular pump decreases the intracellular accumulation of a therapeutic agent (62). In particular, three such transporters—multidrug resistance protein 1 (MDR1), multidrug resistance associated protein 1 (MRP1), and breast cancer resistance protein (BCRP) have been implicated in cancer resistance to a number of chemotherapeutic agents (62). MDR1 encodes P-glycoprotein, a protein that functions as an active efflux pump, removing lipophilic compounds from the cell, thus decreasing the intracellular accumulation of chemotherapeutics. Many studies have demonstrated a negative correlation between P-glycoprotein and chemotherapeutic efficacy (correlative evidence) (63–65). This protein is especially important for the treatment of neurological malignancies as it removes drugs at the BBB, preventing access of agents to the CNS (16). Animal studies have demonstrated that specifically inhibiting P-glycoprotein can increase the level of drug in the brain while also improving the drug's efficacy against implanted human tumors (66, 67). However, later clinical attempts to incorporate P-glycoprotein inhibitors into chemotherapy protocols have shown contradictory and disappointing results (68–70). This discrepancy may be explained by suboptimal dosing of both the inhibitor and chemotherapeutic, with possible contribution by other pumps on the luminal side of brain capillary endothelial cells (71). The presence of P-glycoprotein may be an important molecular biomarker whose presence should be assessed before starting therapy with a TOP2 poison, however, studies are yet to determine a conclusive clinical benefit to targeting these resistance mechanisms.
C9Orf82
The activity of C9orf82 been recently implicated in resistance to TOP2 poisons. There is currently no consensus as to its precise function in the cell, with some data suggesting it negatively regulates apoptosis mediated by caspases (72), and other data suggesting it is involved with the repair of DNA after DSB induced by TOP2 proteins (experimental evidence) (16). Alterations in C9orf82, predominantly deletions, are found in approximately between 6 and11% of GBM tumors (43, 44). A mutation in the C9orf82 gene was shown to confer resistance to TOP2 poisons (experimental evidence) (16). The role of C9orf82 needs to be examined further before its clinical use as a reliable biomarker.
SWI/SNF Complex
The SWI/SNF complex is involved in regulating gene expression via chromatin remodeling. This complex controls DNA accessibility to transcription factors by altering histone-DNA interactions in an ATP-dependent manner (73, 74). This complex is composed of a number of subunits, including SNF5/SMARCB1, SMARCE1, BRG1, PBRM1/BAF180 (75). The SWI/SNF complex has been implicated in malignant rhabdoid tumors (76), and specific subunits of this complex (SNF5/SMARCB1, SMARCE1, BRG1, PBRM1/BAF180) have been identified in approximately 20% of human cancers including renal carcinoma (77), and NSCLC (78). Alterations in individual components of the SWI/SNF complex are also seen in GBM, with mutation, fusion, deletion, or amplification occurring in between 0.25 and 3.5% of tumors depending on the subunit and the underlying gene (43, 44). This complex can influence susceptibility to TOP2 inhibitor therapy as units like SMARCB1 are involved in loading TOP2A onto DNA, and thus can determine how many DNA breaks a cancer cell will have when exposed to a TOP2 poison (16). This is consistent with in vitro findings (experimental evidence) (16) and clinical observations, as malignant rhabdoid tumors are unresponsive to doxorubicin (79).
EZH2
EZH2 (Enhancer of Zeste Homolog-2) is another chromatin remodeler implicated in cancer resistance to chemotherapy (80). This molecule is a subunit of a larger complex called the Polycomb Repressive Complex 2 which is involved in the tri-methylation of histone 3 at lysine 27 to negatively regulate transcription (81). Evidence implicating EZH2 in oncogenesis exists in a variety of cancer types including prostate, breast cancer, melanoma, and bladder cancer (82, 83). EZH2 has been implicated in the sensitivity/resistance to TOP2 poisons; NSCLC cell lines have demonstrated enhanced sensitivity to EZH2 inhibition with etoposide therapy both in vitro and in vivo (47).
Conclusion
As the prognosis of patients diagnosed with GBM remains poor, further efforts should be dedicated to improving the efficiency of our medical therapies. The genetic heterogeneity of GBM cell lines presents a unique opportunity to investigate possible biomarkers to personalize therapy. A precision medicine approach to chemotherapy for brain tumors can potentially enhance efficacy of these treatments, and avoid unnecessary exposure to toxic agents that are not helpful for some cases. The use of TOP2 poisons is an apt illustration of this opportunity, with a growing body of research identifying biomarkers and unique tumoral characteristics that influence susceptibility. Additionally, the use of novel delivery techniques may allow us to achieve therapeutic intratumoral concentrations of TOP2 poisons without having to administer potentially toxic systemic doses, which up to this point has limited their effectiveness. Together, these offer a glimpse into precision therapy and personalized medicine, allowing us to enhance the efficacy of existing therapies in efforts to make progress in a disease that has proved extremely difficult to treat.
Author Contributions
AM contributed to the writing and reviewing of the manuscript, reviewing the literature, editing the manuscript, and submitting the manuscript. CA contributed to the editing and writing of the manuscript, contributing scientific expertise, conducting original analysis based on existing data. AS contributed to the overall vision behind the manuscript, writing the manuscript, editing the manuscript, supervising all other aspects of its creation, and contributing scientific expertise.
Funding
We thank the Robert H. Lurie Comprehensive Cancer Center of Northwestern University in Chicago, IL for providing developmental funds. The Lurie Cancer Center is supported in part by a NCI Cancer Center Support Grant #P30CA060553 (AS). We also would like to acknowledge that this work was supported by the NIH grant T35 AF044303 (AM), NIH Office of the Director 1DP5OD021356 (AS), and SPORE for Translational Approaches to Brain Cancer P50CA221747 (AS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Robert H. Lurie Comprehensive Cancer Center of Northwestern University in Chicago, IL for providing developmental funds. The Lurie Cancer Center is supported in part by a NCI Cancer Center Support Grant #P30CA060553.
References
1. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol. (2013) 15 (Suppl. 2):ii1–56. doi: 10.1093/neuonc/not151
2. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
3. Kanu OO, Mehta A, Di C, Lin N, Bortoff K, Bigner DD, et al. Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets (2009) 13:701–18. doi: 10.1517/14728220902942348
4. Stupp R, Hegi ME, Mason WP, Van Den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
5. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
6. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell (2012) 22:425–37. doi: 10.1016/j.ccr.2012.08.024
7. Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer (2009) 9:327–37. doi: 10.1038/nrc2608
8. Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. (2013) 8:82–95. doi: 10.1021/cb300648v
9. Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer (2009) 9:338–50. doi: 10.1038/nrc2607
10. Schmidt BH, Osheroff N, Berger JM. Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat Struct Mol Biol. (2012) 19:1147–54. doi: 10.1038/nsmb.2388
11. Yeo CQ, Alexander I, Lin Z, Lim S, Aning OA, Kumar R, et al. p53 Maintains genomic stability by preventing interference between transcription and replication. Cell Rep. (2016) 15:132–46. doi: 10.1016/j.celrep.2016.03.011
12. Zdraljevic S, Strand C, Seidel HS, Cook DE, Doench JG, Andersen EC. Natural variation in a single amino acid substitution underlies physiological responses to topoisomerase II poisons. PLoS Genet. (2017) 13:e1006891. doi: 10.1371/journal.pgen.1006891
13. Seol Y, Zhang H, Pommier Y, Neuman KC. A kinetic clutch governs religation by type IB topoisomerases and determines camptothecin sensitivity. Proc Natl Acad Sci USA. (2012) 109:16125–30. doi: 10.1073/pnas.1206480109
14. Rubin EH, Hait WN. How Drugs “Poison” Topoisomerases. In: Kufe DW, Pollock RE, Weichselbaum RR, Bast Jr. RC, Gansler TS, Holland JF, Frei III E, editors. Holland-Frei Cancer Medicine. 6th Edn, Hamilton, ON: BC Decker (2003).
15. Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun. (2013) 4:1908. doi: 10.1038/ncomms2921
16. Wijdeven RH, Pang B, Van Der Zanden SY, Qiao X, Blomen V, Hoogstraat M, et al. Genome-wide identification and characterization of novel factors conferring resistance to topoisomerase II poisons in cancer. Cancer Res. (2015) 75:4176–87. doi: 10.1158/0008-5472.CAN-15-0380
17. Roca J, Ishida R, Berger JM, Andoh T, Wang JC. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc Natl Acad Sci USA. (1994) 91:1781–5. doi: 10.1073/pnas.91.5.1781
18. Darling JL, Thomas DG. Response of short-term cultures derived from human malignant glioma to aziridinylbenzoquinone, etoposide and doxorubicin: an in vitro phase II trial. Anticancer Drugs (2001) 12:753–60. doi: 10.1097/00001813-200110000-00007
19. Pavillard V, Kherfellah D, Richard S, Robert J, Montaudon D. Effects of the combination of camptothecin and doxorubicin or etoposide on rat glioma cells and camptothecin-resistant variants. Br J Cancer (2001) 85:1077–83. doi: 10.1054/bjoc.2001.2027
20. Saul JM, Annapragada A, Natarajan JV, Bellamkonda RV. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J Control Release (2003) 92:49–67. doi: 10.1016/S0168-3659(03)00295-5
21. Lin R, Shi Ng L, Wang CH. In vitro study of anticancer drug doxorubicin in PLGA-based microparticles. Biomaterials (2005) 26:4476–85. doi: 10.1016/j.biomaterials.2004.11.014
22. Kesari S, Schiff D, Doherty L, Gigas DC, Batchelor TT, Muzikansky A, et al. Phase II study of metronomic chemotherapy for recurrent malignant gliomas in adults. Neuro Oncol. (2007) 9:354–63. doi: 10.1215/15228517-2007-006
23. Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Sampson JH, Sathornsumetee S, et al. Metronomic chemotherapy with daily, oral etoposide plus bevacizumab for recurrent malignant glioma: a phase II study. Br J Cancer (2009) 101:1986–94. doi: 10.1038/sj.bjc.6605412
24. Leonard A, Wolff JE. Etoposide improves survival in high-grade glioma: a meta-analysis. Anticancer Res. (2013) 33:3307–3315.
25. Sonabend AM, Carminucci AS, Amendolara B, Bansal M, Leung R, Lei L, et al. Convection-enhanced delivery of etoposide is effective against murine proneural glioblastoma. Neuro Oncol. (2014) 16:1210–9. doi: 10.1093/neuonc/nou026
26. Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature (2016) 533:333–7. doi: 10.1038/nature17987
27. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. (2017) 45:D777–83. doi: 10.1093/nar/gkw1121
28. Stewart DJ, Richard MT, Hugenholtz H, Dennery JM, Belanger R, Gerin-Lajoie J, et al. Penetration of VP-16 (etoposide) into human intracerebral and extracerebral tumors. J Neurooncol. (1984) 2:133–9. doi: 10.1007/BF00177899
29. Zucchetti M, Rossi C, Knerich R, Donelli MG, Butti G, Silvani V, et al. Concentrations of VP16 and VM26 in human brain tumors. Ann Oncol. (1991) 2:63–6. doi: 10.1093/oxfordjournals.annonc.a057826
30. Kiya K, Uozumi T, Ogasawara H, Sugiyama K, Hotta T, Mikami T, et al. Penetration of etoposide into human malignant brain tumors after intravenous and oral administration. Cancer Chemother Pharmacol. (1992) 29:339–42. doi: 10.1007/BF00686001
31. Neuwelt EA, Pagel M, Barnett P, Glassberg M, Frenkel EP. Pharmacology and toxicity of intracarotid adriamycin administration following osmotic blood-brain barrier modification. Cancer Res. (1981) 41:4466–70.
32. Chen X, Yuan M, Zhang Q, Ting Yang Y, Gao H, He Q. Synergistic combination of doxorubicin and paclitaxel delivered by blood brain barrier and glioma cells dual targeting liposomes for chemotherapy of brain glioma. Curr Pharm Biotechnol. (2016) 17:636–50. doi: 10.2174/1389201017666160401144440
33. Juillerat-Jeanneret L. The targeted delivery of cancer drugs across the blood-brain barrier: chemical modifications of drugs or drug-nanoparticles? Drug Discov Today (2008) 13:1099–106. doi: 10.1016/j.drudis.2008.09.005
34. Cui Y, Xu Q, Chow PK, Wang D, Wang CH. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials (2013) 34:8511–20. doi: 10.1016/j.biomaterials.2013.07.075
35. Treat LH, Mcdannold N, Zhang Y, Vykhodtseva N, Hynynen K. Improved anti-tumor effect of liposomal doxorubicin after targeted blood-brain barrier disruption by MRI-guided focused ultrasound in rat glioma. Ultrasound Med Biol. (2012) 38:1716–25. doi: 10.1016/j.ultrasmedbio.2012.04.015
36. Macdiarmid JA, Langova V, Bailey D, Pattison ST, Pattison SL, Christensen N, et al. Targeted doxorubicin delivery to brain tumors via minicells: proof of principle using dogs with spontaneously occurring tumors as a model. PLoS ONE (2016) 11:e0151832. doi: 10.1371/journal.pone.0151832
37. Voulgaris S, Partheni M, Karamouzis M, Dimopoulos P, Papadakis N, Kalofonos HP. Intratumoral doxorubicin in patients with malignant brain gliomas. Am J Clin Oncol. (2002) 25:60–4. doi: 10.1097/00000421-200202000-00013
38. Kim KY, Kim SH, Yu SN, Park SK, Choi HD, Yu HS, et al. Salinomycin enhances doxorubicin-induced cytotoxicity in multidrug resistant MCF-7/MDR human breast cancer cells via decreased efflux of doxorubicin. Mol Med Rep. (2015) 12:1898–904. doi: 10.3892/mmr.2015.3633
39. Da Ros M, Iorio AL, Consolante D, Cardile F, Muratori M, Fantappie O, et al. Morphine modulates doxorubicin uptake and improves efficacy of chemotherapy in an intracranial xenograft model of human glioblastoma. Am J Cancer Res. (2016) 6:639–48.
40. Yamashita Y, Krauze MT, Kawaguchi T, Noble CO, Drummond DC, Park JW, et al. Convection-enhanced delivery of a topoisomerase I inhibitor (nanoliposomal topotecan) and a topoisomerase II inhibitor (pegylated liposomal doxorubicin) in intracranial brain tumor xenografts. Neuro Oncol. (2007) 9:20–8. doi: 10.1215/15228517-2006-016
41. Del Conte G, Sessa C, Von Moos R, Vigano L, Digena T, Locatelli A, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer (2014) 111:651–9. doi: 10.1038/bjc.2014.345
42. Burgess DJ, Doles J, Zender L, Xue W, Ma B, Mccombie WR, et al. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci USA. (2008) 105:9053–8. doi: 10.1073/pnas.0803513105
43. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
44. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) 6:pl1. doi: 10.1126/scisignal.2004088
45. Sohn JW, Lee SY, Lee SJ, Kim EJ, Cha SI, Kim CH, et al. MDR1 polymorphisms predict the response to etoposide-cisplatin combination chemotherapy in small cell lung cancer. Jpn J Clin Oncol. (2006) 36:137–41. doi: 10.1093/jjco/hyi231
46. Bleibel WK, Duan S, Huang RS, Kistner EO, Shukla SJ, Wu X, et al. Identification of genomic regions contributing to etoposide-induced cytotoxicity. Hum Genet. (2009) 125:173–80. doi: 10.1007/s00439-008-0607-4
47. Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature (2015) 520:239–42. doi: 10.1038/nature14122
48. Walker JV, Nitiss JL. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Invest. (2002) 20:570–89. doi: 10.1081/CNV-120002156
49. Sevim H, Parkinson JF, Mcdonald KL. Etoposide-mediated glioblastoma cell death: dependent or independent on the expression of its target, topoisomerase II alpha? J Cancer Res Clin Oncol. (2011) 137:1705–12. doi: 10.1007/s00432-011-1046-5
50. Gudkov AV, Zelnick CR, Kazarov AR, Thimmapaya R, Suttle DP, Beck WT, et al. Isolation of genetic suppressor elements, inducing resistance to topoisomerase II-interactive cytotoxic drugs, from human topoisomerase II cDNA. Proc Natl Acad Sci USA. (1993) 90:3231–5. doi: 10.1073/pnas.90.8.3231
51. Holden JA, Townsend JJ. DNA topoisomerase II-alpha as a proliferation marker in astrocytic neoplasms of the central nervous system: correlation with MIB1 expression and patient survival. Mod Pathol. (1999) 12:1094–100.
52. Bredel M, Piribauer M, Marosi C, Birner P, Gatterbauer B, Fischer I, et al. High expression of DNA topoisomerase IIalpha and Ki-67 antigen is associated with prolonged survival in glioblastoma patients. Eur J Cancer (2002) 38:1343–7. doi: 10.1016/S0959-8049(02)00065-5
53. Bredel M, Pollack IF, Hamilton RL, Birner P, Hainfellner JA, and Zentner J. DNA topoisomerase IIalpha predicts progression-free and overall survival in pediatric malignant non-brainstem gliomas. Int J Cancer (2002) 99:817–20. doi: 10.1002/ijc.10421
54. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA. (2008) 105:13568–73. doi: 10.1073/pnas.0806268105
55. Konstantinopoulos PA, Fountzilas E, Pillay K, Zerbini LF, Libermann TA, Cannistra SA, et al. Carboplatin-induced gene expression changes in vitro are prognostic of survival in epithelial ovarian cancer. BMC Med Genomics (2008) 1:59. doi: 10.1186/1755-8794-1-59
56. Wong TF, Yoshinaga K, Monma Y, Ito K, Niikura H, Nagase S, et al. Association of keap1 and nrf2 genetic mutations and polymorphisms with endometrioid endometrial adenocarcinoma survival. Int J Gynecol Cancer (2011) 21:1428–35. doi: 10.1097/IGC.0b013e31822d0eb2
57. Li QK, Singh A, Biswal S, Askin F, Gabrielson E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J Hum Genet. (2011) 56:230–4. doi: 10.1038/jhg.2010.172
58. Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia (2011) 13:864–73. doi: 10.1593/neo.11750
59. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. (2006) 3:e420. doi: 10.1371/journal.pmed.0030420
60. Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology (2008) 135:1358–68.e4 doi: 10.1053/j.gastro.2008.06.082
61. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis (2008) 29:1235–43. doi: 10.1093/carcin/bgn095
62. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (2014) 6:1769–92. doi: 10.3390/cancers6031769
63. Campling BG, Young LC, Baer KA, Lam YM, Deeley RG, Cole SP, et al. Expression of the MRP and MDR1 multidrug resistance genes in small cell lung cancer. Clin Cancer Res. (1997) 3:115–22.
64. Kawasaki M, Nakanishi Y, Kuwano K, Takayama K, Kiyohara C, Hara N. Immunohistochemically detected p53 and P-glycoprotein predict the response to chemotherapy in lung cancer. Eur J Cancer (1998) 34:1352–57. doi: 10.1016/S0959-8049(98)00067-7
65. Hsia TC, Lin CC, Wang JJ, Ho ST, Kao A. Relationship between chemotherapy response of small cell lung cancer and P-glycoprotein or multidrug resistance-related protein expression. Lung (2002) 180:173–9. doi: 10.1007/s004080000091
66. Kemper EM, Cleypool C, Boogerd W, Beijnen JH, Van Tellingen O. The influence of the P-glycoprotein inhibitor zosuquidar trihydrochloride (LY335979) on the brain penetration of paclitaxel in mice. Cancer Chemother Pharmacol. (2004) 53:173–8. doi: 10.1007/s00280-003-0720-y
67. Zibell G, Unkruer B, Pekcec A, Hartz AM, Bauer B, Miller DS, et al. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology (2009) 56:849–55. doi: 10.1016/j.neuropharm.2009.01.009
68. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist (2003) 8:411–24. doi: 10.1634/theoncologist.8-5-411
69. Chen J, Balmaceda C, Bruce JN, Sisti MB, Huang M, Cheung YK, et al. Tamoxifen paradoxically decreases paclitaxel deposition into cerebrospinal fluid of brain tumor patients. J Neurooncol. (2006) 76:85–92. doi: 10.1007/s11060-005-4171-7
70. Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. (2007) 7:447–59. doi: 10.1586/14737140.7.4.447
71. Miller DS, Bauer B, Hartz AM. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. (2008) 60:196–209. doi: 10.1124/pr.107.07109
72. Zhang Y, Johansson E, Miller ML, Janicke RU, Ferguson DJ, Plas D, et al. Identification of a conserved anti-apoptotic protein that modulates the mitochondrial apoptosis pathway. PLoS ONE (2011) 6:e25284. doi: 10.1371/journal.pone.0025284
73. Becker PB, Horz W. ATP-dependent nucleosome remodeling. Annu Rev Biochem. (2002) 71:247–73. doi: 10.1146/annurev.biochem.71.110601.135400
74. Schiaffino-Ortega S, Balinas C, Cuadros M, Medina PP. SWI/SNF proteins as targets in cancer therapy. J Hematol Oncol. (2014) 7:81. doi: 10.1186/s13045-014-0081-5
75. Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. (2014) 30:356–63. doi: 10.1016/j.tig.2014.05.001
76. Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature (1998) 394:203–6. doi: 10.1038/28212
77. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature (2011) 469:539–42. doi: 10.1038/nature09639
78. Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. (2000) 60:6171–7.
79. Tomlinson GE, Breslow NE, Dome J, Guthrie KA, Norkool P, Li S, et al. Rhabdoid tumor of the kidney in the National Wilms' Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol. (2005) 23:7641–5. doi: 10.1200/JCO.2004.00.8110
81. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature (2011) 469:343–9. doi: 10.1038/nature09784
82. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature (2002) 419:624–9. doi: 10.1038/nature01075
83. Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. (2006) 24:268–73. doi: 10.1200/JCO.2005.01.5180
Keywords: topoisomerase 2 Poisons, glioblastoma multiforme, personalized therapy, drug delivery, tumor susceptibility
Citation: Mehta A, Awah CU and Sonabend AM (2018) Topoisomerase II Poisons for Glioblastoma; Existing Challenges and Opportunities to Personalize Therapy. Front. Neurol. 9:459. doi: 10.3389/fneur.2018.00459
Received: 27 March 2018; Accepted: 30 May 2018;
Published: 20 June 2018.
Edited by:
Marcos Vinicius Calfat Maldaun, Hospital Sírio-Libanês, BrazilReviewed by:
Kamalakannan Palanichamy, The Ohio State University, United StatesSunit Das, St. Michael's Hospital, Canada
Copyright © 2018 Mehta, Awah and Sonabend. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam M. Sonabend, YWRhbS5zb25hYmVuZEBubS5vcmc=