 Meng-Han Tsai1*
Meng-Han Tsai1* Chung-Kin Chan2Ying-Chao Chang3
Chung-Kin Chan2Ying-Chao Chang3 Chih-Hsiang Lin1Chia-Wei Liou1Wen-Neng Chang1Ching-Ching Ng2
Chih-Hsiang Lin1Chia-Wei Liou1Wen-Neng Chang1Ching-Ching Ng2 Kheng-Seang Lim4
Kheng-Seang Lim4 Daw-Yang Hwang5*
Daw-Yang Hwang5*- 1Department of Neurology, Kaohsiung Chang Gung Memorial Hospital, College of Medicine, Chang Gung University, Kaohsiung, Taiwan
- 2Genetics and Molecular Biology, Faculty of Science, Institute of Biological Sciences, University of Malaya, Kuala Lumpur, Malaysia
- 3Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
- 4Division of Neurology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
- 5Division of Nephrology, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
Objective: Focal epilepsy is the most common subtype of epilepsies in which the influence of underlying genetic factors is emerging but remains largely uncharacterized. The purpose of this study is to determine the contribution of currently known disease-causing genes in a large cohort (n = 593) of common focal non-lesional epilepsy patients.
Methods: The customized focal epilepsy gene panel (21 genes) was based on multiplex polymerase chain reaction (PCR) and sequenced by Illumina MiSeq platform.
Results: Eleven variants (1.85%) were considered as pathogenic or likely pathogenic, including seven novel mutations. There were three SCN1A (p.Leu890Pro, p.Arg1636Ter, and p.Met1714Val), three PRRT2 (two p.Arg217Profs*8 and p.Leu298Pro), two CHRNA4 (p.Ser284Leu, p.Ile321Asn), one DEPDC5 (p.Val516Ter), one PCDH19 (p.Asp233Asn), and one SLC2A1 (p.Ser414Ter) variants. Additionally, 16 other rare variants were classified as unknown significance due to inconsistent phenotype or lack of segregation data.
Conclusion: Currently known focal epilepsy genes only explained a very small subset of focal epilepsy patients. This indicates that the underlying genetic architecture of focal epilepsies is very heterogeneous and more novel genes are likely to be discovered. Our study highlights the usefulness, challenges and limitations of using the multi-gene panel as a diagnostic test in routine clinical practice in patients with focal epilepsy.
Introduction
Focal epilepsy constitutes for about 60% of all epilepsies, which is the commonest phenotypic group of epilepsies (1). The etiology of more than half of the focal epilepsies remains uncertain despite high-quality neuroimaging studies (2–4). Some of these unsolved focal epilepsy patients may have a genetic etiology. Recently, patients with focal structural epilepsies were also found to have a genetic cause, such as mTOR pathway genes mutations in focal cortical dysplasia (5, 6). Several disease-causing genes were identified in patients presented with focal seizures as part of their phenotypic spectrums through studies of large families (7–12). For examples, LGI1 in familial lateral temporal epilepsies (13), DEPDC5 in familial focal epilepsy with various foci (9, 10), SCN1A in genetic epilepsy with febrile seizure plus (GEFS+) (14, 15) and CHRNA2, CHRNAB2, CHRNA4, KCNT1 in sleep related hypermotor epilepsies (16–19). A better understanding of the contribution of these genes in common focal epilepsies patients can be helpful in guiding appropriate tests and treatments in routine clinical care.
Recent advances in genomic medicine have significantly unveiled the influence of genetic factors in epilepsy. The targeted gene panel approach has been successfully used in specific syndromes and severe epilepsies, such as epileptic encephalopathies and familial epilepsies.(20–27) Hitherto, only two studies have addressed focal epilepsy specifically using targeted gene panel or whole exome sequencing (WES) with targeted gene analysis (28, 29). Here, we developed a more comprehensive focal epilepsy gene panel, with 21 genes, using multiplex polymerase chain reaction (PCR) based technique followed by massively parallel sequencing to study a large cohort of patients with focal epilepsies. We aim to better understand the contribution of currently known disease-causing genes to focal epilepsy and the utility of multi-gene panel in real-world clinical setting.
Methods
Patients and Phenotyping
Patients with focal epilepsies were recruited for the Department of Neurology, Kaohsiung Chang Gung Memorial Hospital, Taiwan and the Neurology clinic, University of Malaya Medical Center, Malaysia. The clinical information, electroencephalography (EEG) and neuroimaging results were obtained from a direct interview or review of medical records. Most (506/593, 85.3%) of them underwent 3T or 1.5T brain MRI, the remaining 87 had brain CT scans. Patients who had focal structural epilepsy due to stroke, trauma, brain tumor, or focal cortical dysplasia were excluded. Patients with isolated generalized epilepsies were also excluded, but those who have both generalized and focal seizures were still included because some of the genes included in the panel were known to have both presentations. Positive family history was defined as the presence of epilepsy or seizures in the first or second-degree relatives. All of them were recruited regardless of family history and none had received prior genetic testing. All available family members were included for segregation analysis. This study was approved by the local human research ethics committees and written consents were obtained from all subjects. In minors and those with intellectual disabilities, consents were obtained from their legal guardian.
Focal Epilepsy Gene Panel
Venous blood was obtained and genomic DNA was extracted from peripheral blood leukocytes using QIAGEN DNA extraction kits (Qiagen, Germany), according to the manufacturer instructions (30). A customized focal epilepsy gene panel was used, including 21 genes: SCN1A, SCN1B, SCN2A, SCN9A, DEPDC5, GRIN2A, GRIN2B, PRRT2, SLC2A1, PCDH19, KCNT1, KCNQ2, KCNQ3, KCNA2, CHRNA4, CHRNB2, CHRNA2, LGI1, GABRG2, HCN1, CHD2. All coding exons and at least 10 base pair (bp) flanking sequences of the intron/exon boundaries were amplified using targeted specific primers, with a total 69,787 bp region. The amplicon sizes ranged from 204 to 432 bp with an average of 315 bp. Universal primer sequences, 5′-ACACTGACGACATGGTTCTACA-3′ and 5′-TACGGTAGCAGAGACTTGGTCT-3′ were added to the 5' end of all target-specific forward and reverse primers, respectively. Primers were pooled to generate six-plex primer pools per PCR with a final concentration of 1 uM. Libraries were prepared by using the Fluidigm Access 48.48 Array platform (Fluidigm, South San Francisco, California). Harvested amplicon pools underwent another PCR step to barcode the products according to the manufacturer's protocol. Barcoded PCR products were pooled and submitted to an Illumina MiSeq using 2 x 300 bp paired-end runs.
Bioinformatics Analysis
Raw read data was processed with FastQC, FastQ groomer, Trimmomatics to remove primer sequences, and then mapped to human reference genome (version GRCh37) with Burrows-Wheeler Aligner (BWA-MEM, version 0.7.15, http://bio-bwa.sourceforge.net/) (31, 32). The aligned BAM file was processed with SAM tool (http://www.htslib.org/) and Picard (http://picard.sourceforge.net/) to remove low quality mapped reads as well as duplicate reads. Indel realignment was performed using GATK tool as recommended by the Broad Institute GATK Best Practice (33, 34). Single nucleotide variants and small indels were called using FreeBayes (35). The read depth and coverage of each BAM files were calculated using BEDtools (36). Variants that did not adhere to the following criteria were excluded from further analysis: mapping quality <30, base quality <20, coverage <20, variants with strand bias and clustered variants. The variant calling was performed using the Galaxy platform (http://usegalaxy.org). Variants were annotated with wANNOVAR (http://wannovar.wglab.org). Only nonsense, nonsynonymous, splice-site and frameshift variants were further evaluated. Variants presented in the Thousand Genome Project (TGP, http://www.internationalgenome.org/), the Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/), more than 1 hit in the Board Institute Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org), and more than five hits in the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org) were excluded (37). Four prediction programs, including SIFT (v1.03) (38), PolyPhen-2 (v2.2.2 build r394) (39), MutationTaster 2 (40), and Combined Annotation Dependent Depletion (CADD v1.2)(41) were used to prioritize variants. The cutoff value of CADD was set at 20. Only variants predicted probably damaging by more than three in silico programs were further validated by Sanger sequencing.
Criteria for Pathogenicity of Filtered Variants
The confirmed rare variants were classified into pathogenic, likely pathogenic, and variants of unknown significance (VUS) modified from previous guidelines (42, 43). Variants presented in the disease databases (HGMD, http://www.hgmd.org/; ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/; LOVD, http://www.lovd.nl/) were classified as being known pathogenic. Null variants (including frameshift mutations, nonsense mutations, obligatory splicing sites mutations, and mutations affecting the initial codon) identified in known epilepsy genes, where loss of function is a known disease mechanism, were also considered to be pathogenic.
Ultra-rare missense variants (not present in TGP, EVS, ≤1 in ExAc and ≤5 in gnomAD) predicted to be deleterious or damaging by more than three of the four prediction programs were classified as likely pathogenic if their phenotypes correlate with the reported literature. If available, functional data and segregation analysis were taken into consideration. Variants that passed in silico prediction but the patient's phenotype was not previously associated with the gene were classified as VUS.
Statistical Analysis
Fisher exact test was used for comparison of categorical data. The statistical analysis was performed with R software, version 3.2.1 (44).
Results
Patient Characteristics
Five hundred and ninety-three patients, including 298 (50.3%) Taiwanese and 295 (49.7%) Malaysian patients, were recruited and underwent customized focal epilepsy gene panel screening. Among them, 315 (53.1%) had temporal lobe epilepsies, 153 (25.8%) frontal lobe epilepsies, 26 (4.4%) occipital lobe epilepsies, 11 parietal lobe epilepsies (1.8%), 13 (2.2%) benign childhood epilepsy with centrotemporal spikes, and 20 (3.4%) had other syndromes with focal seizures, including 12 Dravet syndrome, 5 Lennox-Gastaut syndrome, 2 epilepsy aphasia spectrum disorders and one genetic epilepsy with febrile seizure plus (GEFS+). The localization was undefined in 55 (9.3%) patients. There were 99 (16.7%) patients had a positive family history and the remaining 494 (83.3%) were sporadic cases.
Customized Focal Gene Panel Study
Total 593 patients were screened with the focal epilepsy gene panel with a mean read depth of 142.4x, and 83.8% coverage of the target region for at least 20 reads.
A total of 27 variants were confirmed by Sanger sequencing in 25 individuals (4.2%), where two individuals had two different variants. Eleven variants (1.85%) were considered as pathogenic or likely pathogenic, including 4 reported and 7 novel mutations (Table 1); the remaining 16 variants were classified as VUS (Supplemental Table 1). Pathogenic and likely pathogenic variants were found in SCN1A (3 patients), PRRT2 (3 patients), CHRNA4 (2 patients), followed by one patient each in DEPDC5, PCDH19, and SLC2A1 (Table 1). The pedigrees, clinical phenotypes, and characteristics of patients with pathogenic or likely pathogenic variants were summarized in Figure 1, Table 2 and detailed in below. The clinical phenotypes, and characteristics of patients with variants of unknown significance were summarized in Supplemental Table 2. Pathogenic or likely pathogenic variants were found in 4 out of 99 focal epilepsy patients with a positive family history (4%) compared to 7 out of 494 sporadic focal epilepsy patients (1.4%, p = 0.094). We further divided our cohort into patients with specific syndromes and focal epilepsies with intellectual disabilities vs. “non-syndromic” focal epilepsies, the diagnostic rate was higher 12.8% (5/39) in syndromic/ID group than in “non-syndromic” group, which was 1.26% (7/554).
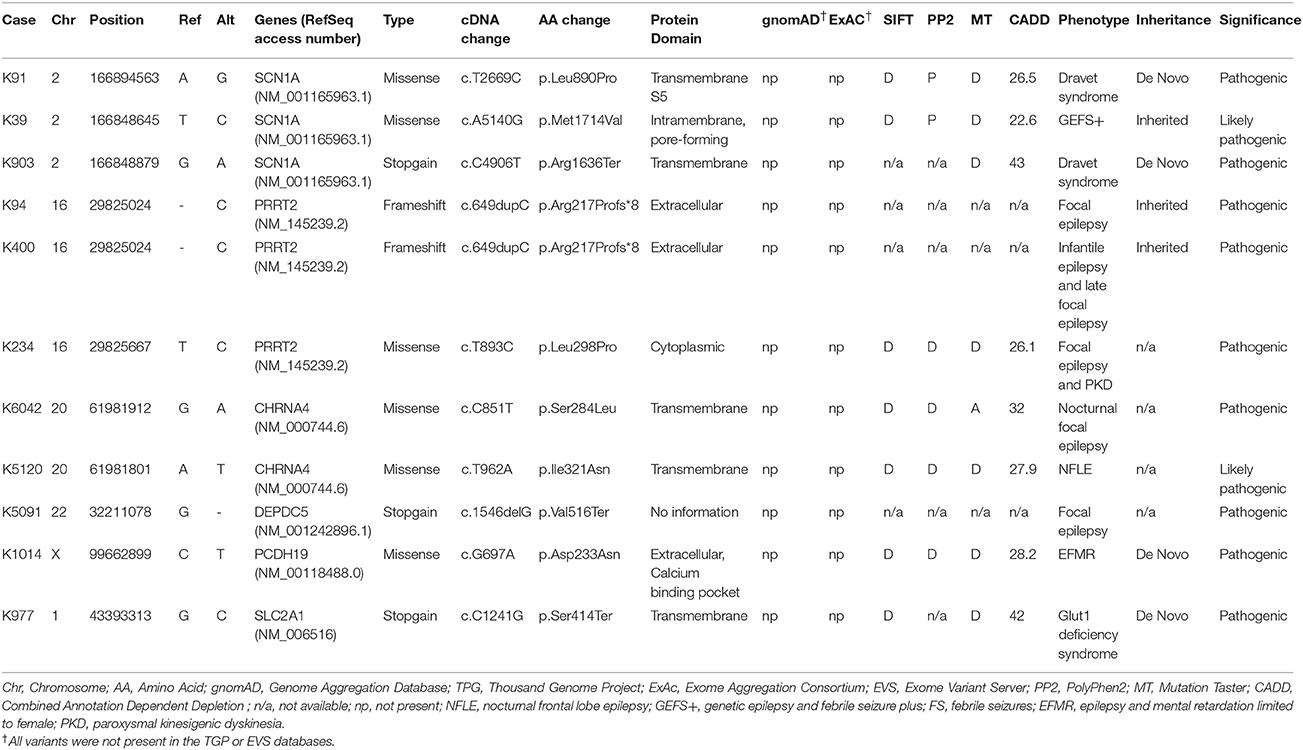
Table 1. The pathogenic or likely pathogenic variants identified by customized focal epilepsy gene panel.
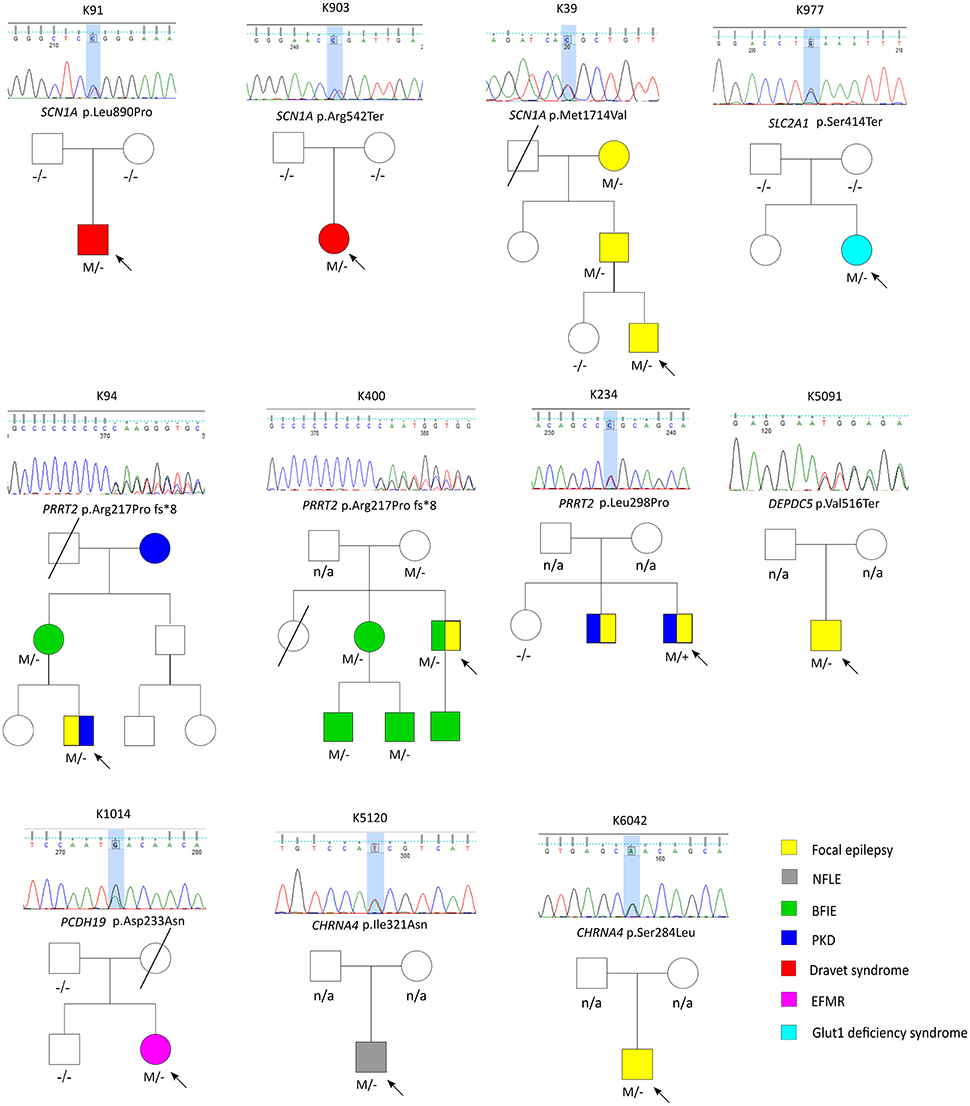
Figure 1. The pedigrees of pathogenic and likely pathogenic variants identified by targeted multigene panel.
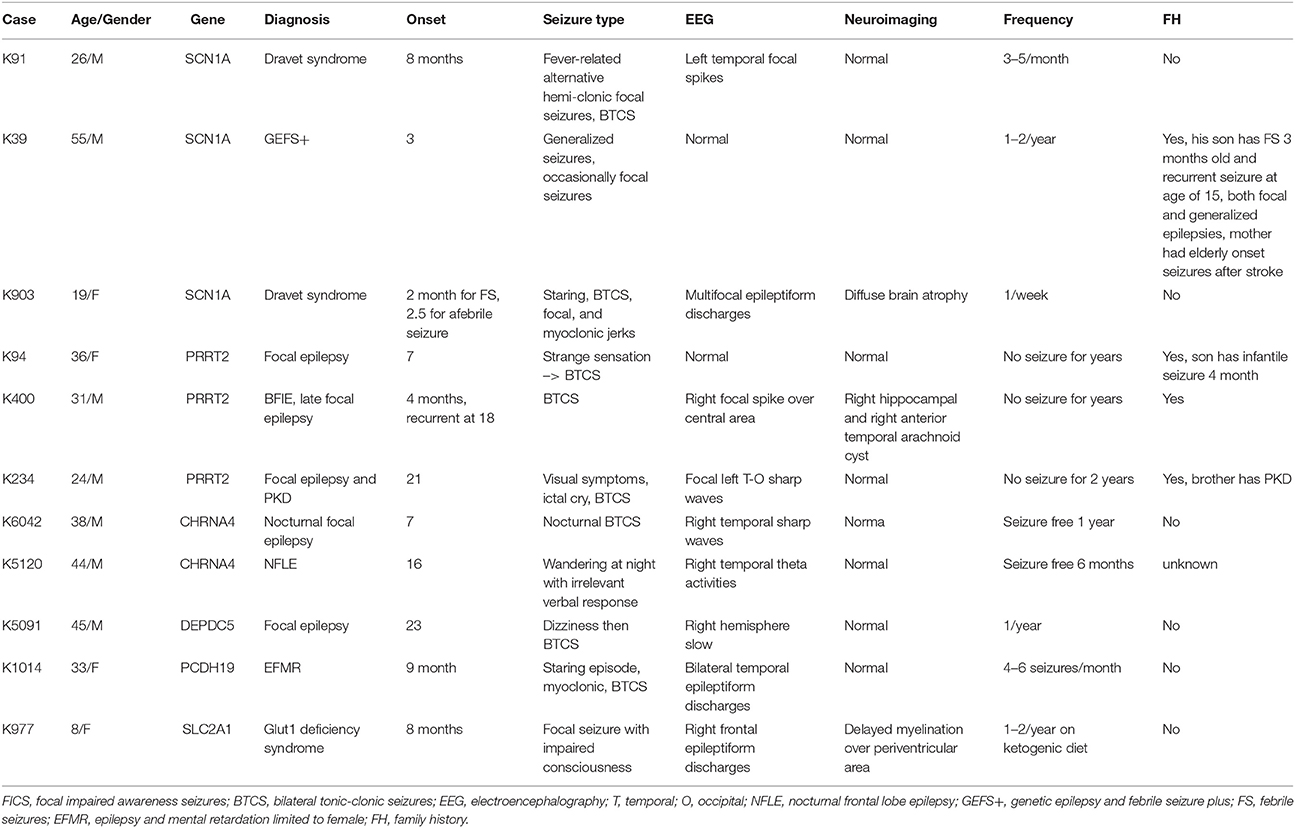
Table 2. The clinical phenotypes and characteristics of patients presented with pathogenic or likely pathogenic variants.
SCN1A
Three variant were found in SCN1A, including two patients with Dravet syndromes (p.Leu890Pro, p.Arg1636Ter) and one family with genetic epilepsy with febrile seizure plus (GEFS+) (p.Met1714Val, Figure 1). The missense mutation p.Leu890Pro is de novo and located in the pore-forming transmembrane S5 domain, while the inherited missense mutation in GEFS+ family (p.Met1714Val) is located in the pore-forming loop between S5 and S6 domain. Both are novel mutations and located in the hot-spot for disease-related missense mutations (45). The p.Met1714Val missense variant was also found in the affected son and proband's mother who had focal seizures in old age (Figure 1).
The patient with de novo p.Leu890Pro mutation had more than 10 seizures a month on Carbamazepine, Vigabatrin and Levetiracetam before the genetic diagnosis. His medication was changed to Topiramate, Levetiracetam and Clobazam in the following months after receiving the results and his seizure frequency drastically reduced to only 1-2 seizure a month.
PRRT2
Three variants were found in PRRT2 gene, including two hotspot p.Arg217Profs*8 frameshift mutations that were inherited in the families with benign infantile epilepsies (Figure 1). The third missense variant p.Leu298Pro is novel and found in a patient with both focal epilepsy and paroxysmal kinesigenic dyskinesia. In silico programs predicted this missense mutation to be deleterious/damaging/disease causing. The available unaffected sister did not have the mutation, the affected brother had paroxysmal kinesigenic dyskinesia and epilepsy but was not available for testing. Functional study of this variant showed lack of membrane localization of the mutant protein, similar to the hotspot truncating mutation p.Arg217Profs*8 (Tsai et al., under review). Therefore, the variant is classified as pathogenic based on consistent phenotype and functional data.
CHRNA4
Two missense variants (p.Ser284Leu, p.Ile321Asn) were found in CHRNA4, both located in the transmembrane domain; the novel missense variant p.Ile321Asn is predicted to be disease-causing by all in silico programs. Both patients had nocturnal frontal lobe epilepsy.
DEPDC5
The pathogenic nonsense mutation p.Val516Ter in DEPDC5 is not presented in any control databases and is predicted to cause nonsense-mediated decay. Therefore, the mutation is likely to cause haploinsufficiency of the DEPDC5 protein, consistent with the currently known molecular mechanism. Clinically, the patient had focal epilepsy, consistent with the DEPDC5 phenotypic spectrum.
PCDH19
The patient had febrile seizures at 9 months old and later developed fever sensitive seizure clusters and intellectual disability. The novel missense variant p.Asp233Asn is located in the extracellular domain; the amino acid forms part of the calcium binding pocket that is critical to the homophilic binding function of PCDH19. The variant is classified as de novo pathogenic because both unaffected parents did not carry the mutation.
SLC2A1
The patient had early onset focal seizures, intellectual disability, and low CSF glucose level, and clinically suspected GLUT1 deficiency syndrome. The novel nonsense variant p.Ser414Ter is located in the transmembrane domain and both unaffected parents did not have the mutation, thus the variant is classified as de novo pathogenic. The patient received ketogenic diet and responded partially to the therapy.
Discussion
The real-world utility and experience of the multi-gene panel in focal epilepsies, the most common form of epilepsies, is very limited (28, 29). We screened a large cohort of focal epilepsy patients and found that 1.85% (11/594) can be attributed to a pathogenic or likely pathogenic variant. Our study highlights the usefulness but also challenges and limitations of using the multi-gene panel in focal epilepsies. The determination of the significance of identified genetic variants is complicated in real-world situation, which requires correct correlation between phenotypes and genotypes. It becomes more difficult when the phenotypes are not previously associated with the genes where variants are identified. It requires more studies to explore the boundaries of the phenotypic spectrum associated with each epilepsy gene. Some of the VUS may be reclassified as pathogenic or likely pathogenic when the phenotype-genotype relationship redefined. Moreover, we noted lack of segregation data is a common obstacle due to limited availability of the family members in routine clinical setting, which makes the determination of the pathogenicity of variants more difficult.
Previous studies using multi-gene panel in epilepsy with various genes (n = 35–327) have generated a diagnostic yield ranged from 10 to 48.5% (11, 21, 23–27, 32). Those studies selected patients with epileptic encephalopathy (21, 27), epileptic syndrome with suspected genetic etiology (12, 21, 32) or enriched for positive family history (21). The higher diagnostic yield was likely due to early onset epilepsies and severe/specific phenotypes such as epileptic encephalopathies (21), which are known to have a stronger genetic underpinning.
Our results are consistent with those reported by Hildebrand et. al., suggesting that currently known focal epilepsy genes only explain a small proportion (0.8–1.85%) of all focal epilepsy patients (29). After excluding patients with clinical suspected specific epilepsy syndromes, such as Dravet syndrome, GEFS+, EFMR and patients with intellectual disability, the diagnostic rate for “garden-variety” focal epilepsies was 1.26%. The reason for the slightly higher diagnostic rate in our study could be explained by the fact that most of our patients had not previously received genetic testing and 10 more genes were included in our panel (29). Interestingly, a recent study used WES based targeted gene analysis of 64 genes on 40 consecutive patients with focal epilepsies with suspected genetic etiology. (28) They reported a much higher positive rate at 12.5% (5/40), three variants were found when limiting to the 21 genes we studied (3/40, 7.5%). The higher yield rate could be explained by the presence of a positive family history of this study. In our study, patients with a positive family history also have a higher diagnostic rate (4% vs. 1.4%) although not statistically significant. Moreover, we did not include copy number variation (CNV), in-frame indels and splice-region variants that are not on the canonical site in this study, which may underestimate the diagnostic rate.
Taken together, our study found a multi-gene panel provides genetic diagnosis of a relatively small percentage of real-world patients with focal epilepsies. Our data indicate that the underlying genetic architecture of focal epilepsies is very heterogeneous and more genes await discovery. Supporting this, a recent study using WES reported positive findings in 38% of patients with focal epilepsy, including discovery of novel genes in 7% (46). The positive rate is expected to increase in the future when more causative genes are identified in focal epilepsy. Obtaining a correct genetic diagnosis is important as it may alter the clinical decision on epilepsy surgery, selection of antiepileptic drugs and reproductive counseling (28). In routine clinical care, careful selection of patients with specific phenotypes/syndromes or positive family histories, adopting a broader panel with more genes, or using WES or even whole genome sequencing are likely to further increase the diagnostic yield.
Ethic Approval and Consents to Participate
This study was approved by the local human research ethics committees (Chang Gung Medical Foundation Institutional Reviewer Board 104-2308B and University Malaya Medical Centre Medical Research Ethics Committee 944.3) and written consents were obtained from all subjects.
Author Contributions
C-KC, Y-CC, C-HL, C-WL, W-NC, C-CN, and K-SL contributed to the acquisition and interpretation of the data and revising the manuscript for intellectual content. M-HT and D-YH contributed to the design and conceptualization of the study; analysis and interpretation of the data; drafting, revising and final approval of the manuscript for intellectual content.
Funding
The study was supported by grants from Chang Gung Medical Foundation (CMRPG8F0701, CMPRG8G0251) and the Ministry of Science and Technology, Taiwan (MOST103-2314-B-182A-025-MY2, 105-2314-B-182A-124, 106-2314-B-182A-077) to M-HT, KMUH104-4T06, KMU-TP104A37, KMUH105-5T03, KMUHS10601 to D-YH, and the University of Malaya under High Impact Research Grant from the governmental research fund (UM.C/625/1/HIR/MOHE/MED/45).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past coauthorship with one of the authors M-HT.
Acknowledgments
The authors wish to express their gratitude to the patients and their families for participating in this study. The authors also wish to thank the following facilities for technical support: The Genomic and Proteomic Core Laboratory and The Biobank, Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.00515/full#supplementary-material
References
1. Hauser WA, Kurland LT. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia (1975) 16:1–66.
2. King MA, Newton MR, Jackson GD, Fitt GJ, Mitchell LA, Silvapulle MJ, et al. Epileptology of the first-seizure presentation: a clinical, electroencephalographic, and magnetic resonance imaging study of 300 consecutive patients. Lancet (1998) 352:1007–11.
3. Berg AT, Testa FM, Levy SR, Shinnar S. Neuroimaging in children with newly diagnosed epilepsy: a community-based study. Pediatrics (2000) 106:527–32. doi: 10.1542/peds.106.3.527
4. Von Oertzen J, Urbach H, Jungbluth S, Kurthen M, Reuber M, Fernandez G, et al. Standard magnetic resonance imaging is inadequate for patients with refractory focal epilepsy. J Neurol Neurosurg Psychiatry (2002) 73:643–7. doi: 10.1136/jnnp.73.6.643
5. Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. (2015) 21:395–400. doi: 10.1038/nm.3824
6. Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. (2014) 75:782–7. doi: 10.1002/ana.24126
7. Colosimo E, Gambardella A, Mantegazza M, Labate A, Rusconi R, Schiavon E, et al. Electroclinical features of a family with simple febrile seizures and temporal lobe epilepsy associated with SCN1A loss-of-function mutation. Epilepsia (2007) 48:1691–6. doi: 10.1111/j.1528-1167.2007.01153.x
8. Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain J Neurol. (2007) 130:843–52. doi: 10.1093/brain/awm002
9. Ishida S, Picard F, Rudolf G, Noe E, Achaz G, Thomas P, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 45:552–5. doi: 10.1038/ng.2601
10. Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. (2013) 45:546–51. doi: 10.1038/ng.2599
11. Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. (2013) 45:1067–72. doi: 10.1038/ng.2728
12. Carvill GL, Regan BM, Yendle SC, O'Roak BJ, Lozovaya N, Bruneau N, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. (2013) 45:1073–6. doi: 10.1038/ng.2727
13. Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Boneschi FM, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. (2002) 30:335–41. doi: 10.1038/ng832
14. Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain J Neurol. (2007) 130:100–9. doi: 10.1093/brain/awl272
15. Zhang YH, Burgess R, Malone JP, Glubb GC, Helbig KL, Vadlamudi L, et al. Genetic epilepsy with febrile seizures plus: refining the spectrum. Neurology (2017) 89:1210–9. doi: 10.1212/WNL.0000000000004384
16. Steinlein OK, Magnusson A, Stoodt J, Bertrand S, Weiland S, Berkovic SF, et al. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet. (1997) 6:943–7.
17. Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. (2012) 44:1188–90. doi: 10.1038/ng.2440
18. Phillips HA, Favre I, Kirkpatrick M, Zuberi SM, Goudie D, Heron SE, et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. (2001) 68:225–31. doi: 10.1086/316946
19. Aridon P, Marini C, Di Resta C, Brilli E, De Fusco M, Politi F, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. (2006) 79:342–50. doi: 10.1086/506459
20. de Kovel CG, Brilstra EH, van Kempen MJ, Van't Slot R, Nijman IJ, Afawi Z, et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med. (2016) 4:568–80. doi: 10.1002/mgg3.235
21. Moller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. (2016) 7:210–9. doi: 10.1159/000448369
22. Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia (2012) 53:1387–98. doi: 10.1111/j.1528-1167.2012.03516.x
23. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia (2015) 56:707–16. doi: 10.1111/epi.12954
24. Della Mina E, Ciccone R, Brustia F, Bayindir B, Limongelli I, Vetro A, et al. Improving molecular diagnosis in epilepsy by a dedicated high-throughput sequencing platform. Eur J Hum Genet. (2015) 23:354–62. doi: 10.1038/ejhg.2014.92
25. Wang J, Gotway G, Pascual JM, Park JY. Diagnostic yield of clinical next-generation sequencing panels for epilepsy. JAMA Neurol. (2014) 71:650–1. doi: 10.1001/jamaneurol.2014.405
26. Kodera H, Kato M, Nord AS, Walsh T, Lee M, Yamanaka G, et al. Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia (2013) 54:1262–9. doi: 10.1111/epi.12203
27. Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. (2013) 45:825–30. doi: 10.1038/ng.2646
28. Perucca P, Scheffer IE, Harvey AS, James PA, Lunke S, Thorne N, et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. (2017) 131:1–8. doi: 10.1016/j.eplepsyres.2017.02.001
29. Hildebrand MS, Myers CT, Carvill GL, Regan BM, Damiano JA, Mullen SA, et al. A targeted resequencing gene panel for focal epilepsy. Neurology (2016) 86:1605–12. doi: 10.1212/WNL.0000000000002608
30. Tsai MH, Chan CK, Chang YC, Yu YT, Chuang ST, Fan WL, et al. DEPDC5 mutations in familial and sporadic focal epilepsy. Clin Genet. (2017) 92:397–404. doi: 10.1111/cge.12992
31. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
32. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
33. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. (2011) 43:491–8. doi: 10.1038/ng.806
34. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics (2013) 11:11 10 1–33. doi: 10.1002/0471250953.bi1110s43
35. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arVix (2012):1207.3907.
36. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (2010) 26:841–2. doi: 10.1093/bioinformatics/btq033
37. Bennett CA, Petrovski S, Oliver KL, Berkovic SF. ExACtly zero or once: a clinically helpful guide to assessing genetic variants in mild epilepsies. Neurol Genet. (2017) 3:e163. doi: 10.1212/NXG.0000000000000163
38. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
39. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7:248–9. doi: 10.1038/nmeth0410-248
40. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods (2014) 11:361–2. doi: 10.1038/nmeth.2890
41. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5 doi: 10.1038/ng.2892
42. MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature (2014) 508:469–76. doi: 10.1038/nature13127
43. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
44. R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2014). Available online at: http://www.R-project.org/
45. Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology (2011) 76:594–600. doi: 10.1212/WNL.0b013e31820c309b
Keywords: focal epilepsy, multigene panel, targeted resequencing, NGS, multiplex PCR
Citation: Tsai M-H, Chan C-K, Chang Y-C, Lin C-H, Liou C-W, Chang W-N, Ng C-C, Lim K-S and Hwang D-Y (2018) Molecular Genetic Characterization of Patients With Focal Epilepsy Using a Customized Targeted Resequencing Gene Panel. Front. Neurol. 9:515. doi: 10.3389/fneur.2018.00515
Received: 08 March 2018; Accepted: 11 June 2018;
Published: 06 July 2018.
Edited by:
Adam Strzelczyk, Universitätsklinikum Frankfurt, GermanyReviewed by:
Sara Baldassari, INSERM Délégation Paris 6, FranceYvonne Weber, Universität Tübingen, Germany
Copyright © 2018 Tsai, Chan, Chang, Lin, Liou, Chang, Ng, Lim and Hwang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng-Han Tsai, bWVuZ2hhbkBjZ21oLm9yZy50dw==
Daw-Yang Hwang, OTEwMjA4QGttdWgub3JnLnR3