 Ping Yin
Ping Yin Yafen Wei
Yafen Wei Xu Wang
Xu Wang Mingqin Zhu
Mingqin Zhu Jiachun Feng1*
Jiachun Feng1*- 1Department of Neurology and Neuroscience Center, First Hospital of Jilin University, Changchun, China
- 2First Department of Neurology and Neuroscience Center, Heilongjiang Provincial Hospital, Harbin, China
Ischemic stroke contributes to ~80% of all stroke cases. Recanalization with thrombolysis or endovascular thrombectomy are currently critical therapeutic strategies for rebuilding the blood supply following ischemic stroke. However, recanalization is often accompanied by cerebral ischemia reperfusion injury that is mediated by oxidative stress and inflammation. Resolution of inflammation belongs to the end stage of inflammation where inflammation is terminated and the repair of damaged tissue is started. Resolution of inflammation is mediated by a group of newly discovered lipid mediators called specialized pro-resolving lipid mediators (SPMs). Accumulating evidence suggests that SPMs decrease leukocyte infiltration, enhance efferocytosis, reduce local neuronal injury, and decrease both oxidative stress and the production of inflammatory cytokines in various in vitro and in vivo models of ischemic stroke. In this review, we summarize the mechanisms of reperfusion injury and the various roles of SPMs in stroke therapy.
Overview of Cerebral Ischemic Reperfusion Injury
Stroke is one of the most common causes that leads to morbidity and mortality all over the world. In the acute phase of ischemic stroke, treatments should focus on saving penumbral tissue as much as possible. There are two main recanalization methods: (1) intravascular thrombolysis with recombination tissue-type plasminogen activator (rt-PA) (1, 2), and (2) endovascular thrombectomy (3). It has been demonstrated that recanalization improves the clinical prognosis in acute stroke patients. However, in some cases, severe side effects, such as fatal edema or intracranial hemorrhage, are reported after ischemia reperfusion (4). Moreover, animal studies showed that reperfusion after a long ischemic period could result in a larger damage size than the volume that corresponds to the permanently occluded vessel (5, 6). Therefore, even though reperfusion may reduce infarct size and improve clinical outcomes, it may also exacerbate cerebral injury via a process named “cerebral ischemia reperfusion injury (CIRI)” (4). Many studies support the idea that oxidative stress and inflammation are involved in CIRI, and ultimately lead to apoptosis and cell death (7, 8).
Cellular Events Involved in CIRI
Endothelial Cells (ECs)
CIRI is characterized by increased blood-brain barrier (BBB) permeability. ECs are critical structures that make up the BBB, a strong mechanical barrier in the brain that prohibits the transcellular movement of large molecules. ECs are tightly interconnected through specific proteins called tight junctions, which function to seal the inter-endothelial spaces, thereby inhibiting the delivery of immune cells and hydrophilic molecules. Under ischemic conditions, excessive accumulation of reactive oxygen metabolites may cause ECs to swell and detach, leading to impaired BBB function, protein extravasation, and interstitial edema (9). Furthermore, BBB breakdown induces ECs to produce selectins, integrins, and pro-inflammatory cytokines and chemokines, such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-6, and prostaglandins. The released pro-inflammatory mediators affects the microvasculature in the injury site, where leukocytes attach to the ECs and cause reduced blood supply and hypoxia (9). Leukocytes activation including the production of cytokines, which then further upregulating the expression of adhesion molecules on the surface of both ECs and leukocytes. The increased expression of leukocyte adhesion molecules and the increased production of reactive oxygen species, leading to the peroxidation of cellular membrane components, further lead to the element of vascular permeability and vasogenic edema (10). Therefore, a vicious cycle of inflammation finally occurs.
Microglial Cells
Microglial cells are resident macrophages in the brain. Traditionally, activated microglia are classified into two subtypes: M1 and M2 phenotypes, with M1 phenotypes having pro-inflammatory and M2 phenotypes playing anti-inflammatory roles (11, 12). Evidence shows that microglial cells are activated quickly after ischemia onset and produce a series of pro-inflammatory mediators, such as reactive oxygen species (ROS) (13), IL-1, IL-6, TNF-α, and matrix metalloproteinase (MMP)-9, which can be toxic to neurons. It has been found that these events precede leukocyte infiltration into the brain. Thus, early microglial activation plays a detrimental role by increasing BBB permeability, therefore, contributing to the increased number of leukocytes infiltration to the brain (14, 15). In fact, inhibiting microglial activation in the early phase of CIRI protects the brain against injury by maintaining BBB integrity (14). Furthermore, inhibiting M1 microglial migration in the hyper-acute phase of transient middle cerebral artery occlusion (MCAO) significantly attenuates infarct volumes and improves neurological outcomes (16). In later phases of CIRI, blood monocytes/macrophages infiltrate the brain. Microglia and macrophages share some common features, but many investigations have shown that there are markers that distinguish microglia from hematogenous infiltrating macrophages (17). Activated microglia/macrophages could play a protective role in later phases of CIRI by triggering neutrophil apoptosis and phagocytosis of apoptotic neutrophils, thereby preventing the release of toxic molecules into the nearby tissue. This is a crucial step in resolution of inflammation and preventing further tissue damage of CIRI (18). The protective role of microglia/macrophages may also be mediated by their ability to produce various neurotrophic factors and anti-inflammatory cytokines, such as fibroblast growth factor, transforming growth factor (TGF)-β1, IL-4, IL-10, and IL-13, which are involved in ending inflammation and initiating tissue repair (12, 19, 20). A recent study found that microglia depletion exacerbated post-ischemia inflammation and brain injury via augmented inflammatory mediators produced by astrocytes. Microglia can restrict the ischemia-induced astrocytic response (21).
Astrocytes
After cerebral ischemia, astrocytes undergo reactive astrogliosis, characterized by swelling in morphology, enhanced expression of glial fibrillary acidic protein, and upregulated Calcium signaling (22). Astrocytes upregulate major histocompatibility complexes and inflammatory mediators, such as pro-inflammatory cytokines (IL-6 and IL-1β), chemokines [CXC-chemokine ligand (CXCL) 1, CXCL10, CXCL12, and monocyte chemoattractant protein 1 (MCP-1)], and inducible nitric oxide synthase (iNOS). Although astrocytes may be harmful due to their hyper reactivity to toxic injuries and glial scar, astrocytes also perform multiple homeostatic functions for neurovascular unit survival and maintenance (23). For instance, astrocytic glycogen stores maintain neuronal metabolism under hypoglycemia (24, 25). Furthermore, astrocytes modulate the ion buffering (26), uptake toxic neurotransmitters (27, 28), synthesize neuroprotective mediators (29), control cerebral blood flow (CBF) via the release of vasoactive substances, such as prostaglandin E2 and epoxyeicosatrienoic acids (30), transport water, release antioxidant molecules (31), and likely participate in adult neurogenesis (32).
Neutrophils
Neutrophils infiltrate the injury site early following CIRI (33). They are recruited from the blood to injury site through specific bindings with chemokines, for instance CXCL1, CXCL2, and CXCL3. A recent study has shown that blocking neutrophil-specific chemokine receptors, such as CXC-chemokine receptor (CXCR), reduces ischemic brain injury (34). Scavenger receptors, such as cluster of differentiation 36 (CD36), may also play important roles in neutrophil accumulation after ischemic CNS injury (35). Neutrophils respond to damage-associated molecular patterns (DAMPs) via binding to toll-like receptors (TLRs). Once activated by inflammatory mediators, neutrophils upregulate their surface adhesion receptors, such as CD15 and CD11b, to promote their adherence to ECs and their migration into inflamed tissue (36). The mechanisms underlying neutrophil-mediated damage in CIRI include neutrophil sludging, causing microvascular hypoperfusion, excessive production of ROS, release of pro-inflammatory cytokines and chemokines, elastase, and MMPs, and increased adhesion molecule expression. In one study, there were three-fold more neutrophils in permanent MCAO compared to transient MCAO, suggesting that ongoing hypoxia is a major stimulus for immune cell infiltration (37). Although evidences have suggested that preventing neutrophil infiltration into the ischemic tissue inside the brain can minimize infarct size (38, 39), some studies also suggest that the number of neutrophils in an ischemic brain does not predict stroke severity (40). Hence, the role of neutrophils in stroke is more complicated than what we currently know.
Platelets
The inflammatory roles of platelets have recently been highlighted (41, 42). During inflammation, ECs express intercellular adhesion molecule (ICAM)-1, platelet selectin (P-selectin), endothelial selectin (E-selectin) as well as vascular cell adhesion molecule (VCAM) (43), causing platelets to adhere to and activate ECs. Platelets express CD154 (also known as CD40L) on their surface and secrete IL-1β. In turn, platelet CD154 can cause ECs to secrete adhesion molecules including E-selectin, ICAM-1, and VCAM-1, as well as chemokines such as MCP-1 and anti-inflammatory cytokines IL-8 (44). IL-1β release further increases EC permeability and recruits leukocytes to attach to ECs (45). Together, this augments inflammation and activates inflammatory cells, such as monocytes and neutrophils. Platelet P-selectin and CD154 are respectively recognized by P-selectin glycoprotein ligand 1 of monocytes and CD40 of neutrophils, leading to the formation of platelet-leukocyte aggregates and thereby contributing to the innate immune response (46). In addition, platelet activation can release serotonin and chemokine ligand (CCL) 5, which are known to mediate T cell activation and differentiation. Platelets also produce many pro-inflammatory chemokines and cytokines, and may also promote inflammation by releasing microparticles (47–49). Platelets are major sources of circulating microparticles (50). Microparticles contain proteins including P-selectin, chemokines, such as CCL5, and cytokines, such as IL-1β (51). Microparticles also contain different forms of RNAs, such as microRNA (miRNA), small non-coding RNAs that play important roles in post-transcriptional regulation of gene expression. Platelets may affect the nearby cells by transferring miRNA (52, 53). For example, miR-320b transferred into ECs can decrease ICAM-1 expression (52). In the same way, miR-126-3p, can be taken up by macrophages, resulting in an increased phagocytic phenotype (53).
T Cells and B Cells
As BBB permeability increases, DAMPs may invade the brain and function as antigenic molecules for the immune system. After stroke onset, an infiltration of lymphocytes may also directly contribute to brain injury (54). Studies utilizing ischemic stroke mice models have demostrated that the numbers of both T cells and antigen-presenting cells increase in delayed phase (3–7 days) of post-reperfusion, suggesting that antigen presentation and T cell responses may play a role in the pathophysiology of ischemic stroke (55, 56). CD4+ helper, CD8+ cytotoxic, and γδT cells play detrimental roles in various stroke models (57, 58). CD4+ T cells produce pro-inflammatory cytokines, such as interferon (IFN)-γ, and CD8+ T cells secrete perforin/granzymes (59, 60), all of which could cause neuronal death and worsen stroke outcomes. It is thus unsurprising that, in mice, CD4+ helper/CD8+ cytotoxic T cell deficiency results in smaller infarct size, less infiltrating leukocytes, and better prognosis after transient MCAO (57). γδT lymphocytes are involved in ischemic brain injury by producing the pro-inflammatory molecule IL-17 (58, 61). In stroke, IL-17 and TNF-α synergistically stimulate astrocytes to secrete chemokines that are attractive for neutrophils, such as CXCL-1, leading to the infiltration of neutrophil, and thus increased damage of brain tissue (62). Furthermore, T regulatory cells (Tregs) might play a protective role in ischemic brain injury by producing the anti-inflammatory cytokines IL-10 as well as TGF-β (63) and down-regulating the neutrophil production of MMP-9, protecting the BBB (64, 65). In addition, the anti-CD28 antibody CD28SA was proved to improve neurological function after experimental ischemic stroke by increasing the number of Treg cells (66). On the contrary, there have been suggestions that Treg cells could be harmful in the early phase of ischemic brain injury for the reason that they may cause dysregulation of immune system and vascular malfunction (67, 68).
In the weeks to months after stroke, immunoglobulin synthesis was found in the cerebrospinal fluid (CSF) of ischemic stroke patients (69), suggesting B cell were activated as a result of brain damage. Consistent with this finding, it has also been found that activated B cells could have an impact on the cognitive function and recovery (70).
Dendritic Cells (DCs)
DCs serve as antigen presenting cells in the brain that process antigenic materials and present them on their surface to immune cells. Under normal conditions, DCs cannot be found inside the brian (71). It has been demonstrated that DCs are present in the parenchyma of inflamed brains as early as 1 h after ischemia in an MCAO rat model (72). In line with this findings, Gelderblom et al. demonstrated that DCs were observed inside the brain together with other type of immune cells after CIRI (55). Importantly, results from experimental studies prove that DC infiltration after ischemia stroke worsens the clinical outcomes (73). Granulocyte-colony stimulating factor (G-CSF) mediates the migration of DCs after transient MCAO and, thus, inhibiting G-CSF could attenuate cerebral infarct volume and inflammation (73). Furthermore, DCs that present in the ischemic brain injury site can activate T cells, initiating a long-lasting immune response and worsening the clinical outcomes.
Mast Cells (MCs)
MCs locate in various parts of in the brain, including the cerebral cortex, thalamus, and diencephalic parenchyma. Importantly, MCs that locate in the perivascular positions have the capability to quickly respond to stimuli, and produce of a series mediator such as vasodilatory, pro-inflammatory mediators and proteolytic molecules. Therefore, MCs can play an important role in the defense of various stimuli. Several mediators synthesized by MCs can affect stroke outcomes. TNF-α which comprises 25% of the MC granule, enhances BBB permeability and increases T cell infiltration, proliferation, function, and cytokine production (74). MC-derived endothelin, endoglin, and MMP-9 increase neutrophil infiltration, BBB leakage, and edema after reperfusion in a transient MCAO mouse model (75). MC-derived gelatinase contributes to early ischemic BBB disruption and edema formation (76). MC-derived IL-6 contributes to increased brain granulocytes, macrophage activation, infarct size exacerbation, and brain swelling (77).
Molecular Events Involved in CIRI
Free radicals and pro-inflammatory mediators are quickly produced from the damaged tissue after ischemic stroke. These mediators can cause cerebral ECs to synthesize adhesion molecules, increase adhesion and trans-endothelial migration of leukocytes from the periphery. Infiltrating leukocytes can then release more cytokines and chemokines, which produce free radicals that complement and activate MMPs. This further amplifies inflammation by promoting local immune cell activation and leukocyte infiltration, eventually leading to BBB disruption, brain edema, and neuronal death.
The Role of Free Radicals in CIRI
There are two major classes of free radicals: ROS and reactive nitrogen species (RNS). During thrombolysis, reperfusion may cause increased ROS and RNS production, both of which are mediators of neurotoxicity and BBB breakdown.
ROS
ROS consists of active species, including hydroxyl radicals, superoxide anion radicals, peroxide (H2O2), hydrogen, and singlet oxygen, among others. Under normal conditions, ROS have critical biological functions and function as redox signaling molecules. However, under pathological conditions, such as CIRI, excessive ROS are produced. In various ischemia/reperfusion (I/R) organs, xanthine oxidase, ROS are synthesized by the enzymatic activity of NOS and nicotinamide adenine dinucleotide phosphate (NADPH). In CIRI, the mitochondria are the main source of ROS (78). I/R cause electron leakage and excessive production of free radical in the mitochondrion. Over abundant free radicals can cause oxidative injury to the mitochondrial respiratory chain, metabolizing enzymes further increase the electron leakage and free radical release (79, 80). Moreover, free radicals damage mitochondrial membrane structures (81), and increase mitochondrial permeability transition pore opening (82), leading to impairment of membrane potential and more oxidative stress (80). Increased mitochondrial permeability also increases the release of pro-apoptotic mediators into the cytoplasm (83). Moreover, I/R-induced damage also impairs mitochondrial dynamics and mitophagy, thereby affecting the quality control of the mitochondrial network (84, 85). Eventually, mitochondrial dysfunction leads to increased apoptosis. ROS also directly contribute to neuronal death by oxidizing proteins, damaging DNA, and inducing lipid peroxidation.
RNS
Two common RNS that are well documented in CIRI are nitric oxide (NO) and peroxynitrite (ONOO−). A low-concentration of NO produced by endothelial NOS (eNOS) acts as an indispensable messenger to regulate pre- and/or post-synaptic activities (86), as well as maintaining vascular physiological functions (87), CBF (88), and inflammatory responses (89), whereas high-concentrations of NO produced from iNOS and neuronal NOS can trigger cell death and BBB leakage during I/R injury (90). iNOS activation upregulates autophagy of vascular ECs and increases apoptosis during CIRI (91). During CIRI, NO is synthesized together with superoxide (O2−·) and quickly interacts with O2−· at a diffusion-limited rate to produce ONOO−. In physiological conditions, ONOO− can directly react with thiols, or the radical products of ONOO− decomposition may indirectly oxidize other cellular components, such as lipids, proteins, and DNA. However, excessive ONOO− triggers inflammation, lipid membrane peroxidation, and mitochondrial dysfunction (92), subsequently exacerbating BBB disruption and brain dysfunction (93). Plasma 3-nitrotyrosine, which is often used as an ONOO− footprint, has a positive correlation with brain ischemic severity in stroke patients. RNS-mediated autophagy/mitophagy may play an indispensable role in CIRI. ONOO−-mediated autophagy could also induce tight junction protein degradation (94).
The Role of Complement Proteins in CIRI
Complement proteins play a key role in the inflammatory reaction following CIRI. In ischemic stroke, BBB disruption allows circulating complement proteins to enter the CNS; further, complement proteins can also be locally produced by CNS resident cells. Due to excessive glutamate and ionic imbalance, CIRI results in a depletion of cellular energy resources, as well as an increase in ROS release, apoptotic and necrotic cell death, and excitotoxic insults. These pathological processes result in the activation of complement proteins via different pathways including the classical, alternative and extrinsic protease pathways. All of these pathways are involved in the formation of membrane attack complexes (MACs). The other two products that emerge following complement protein activation are opsonins (C1q, mannose-binding lectin (MBL) and C3b/i C3b/C3d) and anaphylatoxins (C3a and C5a). The alternative pathway is consistently active, and hydrolyzed C3 can be placed on the surface of immune cells that can lead to subsequently C3 activation. The classical pathway is activated by apoptotic cells or through recognition by Clq. The extrinsic protease pathway is activated through the specific binding of carbohydrate molecules by ficolins or MBL. Activation of these pathways leads to activation of C3. C3 cleavage generates C3a and C3b, the latter is cleaved further to generate membrane-bound iC3b, C3d, C3dg and opsonins, which are recognized by their specific receptors on the immune cells. C3 cleavage also result in C5 cleavage, generating soluble C5a and membrane-bound C5b, the later starts the terminal pathway and leads to the formation of MAC that directly lyse the cells and stimulates cells to produce pro-inflammatory mediators. The anaphylatoxins C3a and C5a recruit and activate leukocytes. The complement opsonins promote microglial phagocytosis and release IFN-γand other cytokines (95). Signaling of C3d through complement receptor 2 inhibits neuronal progenitor cell proliferation (96).
Infarct development was strongly correlated with robust complement protein activation (97). The presence complement molecules including Clq, MBL, C3, C4d, C5a, and C9 has been observed in ischemic brain sites (98, 99) and increased levels of C3, C4d, C5a, and C5b-9 have also been found in the plasma of stroke patients (100). CD59, expressed by astrocytes and microglia, is a MAC inhibitory protein. Exacerbated pathology and behavior deficits have been found in CD59 knockout ischemic stroke mice (101). In line with these findings, experiments using other complement related genetic models proved the pathological roles of Clq (102), C3 (103, 104), and C5 (105) in stroke. Genetic MBL deficiency is protective in clinical and experimental severe cerebral I/R injuries (106), but not in mild cerebral ischemia (107). It has been demonstrated that intravenous rt-PA dramatically upregulates complement cascade activation in ischemic brains, and pharmacologic inhibition of complement proteins protects brains against the adverse effects of rt-PA thrombolysis in stroke (108). These studies have led many researchers to focus on inhibiting complement proteins to alleviate stroke injuries (109–112), including depletion of complement proteins, suppressing complement-driven cellular recruitment, neutralizing MBL, and inhibiting complement protein activation, among others. Other investigations have targeted down-stream mediators of the activated complement system. Anaphylatoxin C3a was thought to have neuroprotective and neurotrophic properties (113). C3a regulated astrocytes response to ischemia and increased their capacity to react to ischemic stress (114). Studies have also shown that applying specific C3a receptor agonists protects against intestinal I/R injury by inhibiting neutrophil mitigation (115). In subacute to chronic phases of ischemic stroke, C3a–C3a receptor signaling stimulated post-stroke synaptogenesis and axonal plasticity, and intranasal treatment with C3a receptor agonists improved functional recovery (116). C5a interacts with its canonical receptor C5aR1 and initiate the complement-mediated inflammatory processes. Its inhibition is generally thought to mitigate acute cerebral I/R injury (117). Since complement proteins are also important for immune surveillance and homeostatic activities, many therapies that systemically inhibit complement systems have potential risks. Nonetheless, complement inhibitors that target specific sites are under investigation (118).
The Role of MMPs in CIRI
MMPs are responsibe for protein digestion and extracellular matrix (ECM) turnover and belong to a group of zinc-binding endopeptidases. There are 23 different types of MMPs in humans, which are classified according to their structural similarity (119). Microglia are major sources of MMPs (39). Activated MMPs can be found immediately after ischemia onset and last for 5 days (39, 119). MMPs mediate basal lamina protein disruption by causing BBB hyperpermeability, leukocyte extravasation, cerebral edema, and hemorrhagic transformation (HT). Although different types of MMPs act differently, there is accumulating evidence that MMP-2, MMP-9, and MMP-12 are involved in the pathogenesis of BBB disruption and cerebral edema formation during ischemic stroke.
MMP-9-deficient mice had smaller ischemic volumes compared with controls in a permanent focal cerebral ischemia mouse model (120). Some clinical studies have suggested that the levels of circulating MMP-9 are significantly correlated to disease severity and infarct volume in the hyperacute phase (121, 122), as well as late hemorrhagic infarction incidence between 5 and 7 days after stroke onset (123). A recent study showed that higher MMP-9 levels in the serum of patients with acute ischemic stroke is correlated with the severity of clinical symptoms; Therefore, MMP-9 may serve as an important biomarker for ischemic stroke patent (124). Furthermore, rt-PA was shown to upregulate MMP-9 levels in vivo in a rat model of stroke (125), emphasizing the impact of MMP-9 activation in rt-PA-associated side effects, such as HT.
The role of MMP-2 in ischemic stroke is more complex. It has been shown previously that the levels of MMP-2 are lower in the acute phase of stroke mice model. However, the levels of MMP-2 in the serum of stroke patients are not correlated to infarct size or stroke disability (126). Actually, MMP-2 activity has been shown increased in the latter phase of in ischemic mice model (119) and the MMP-2 levels are higher in ischemic stroke patients in the recovery phase (126). A recent study using a non-human primate MCAO model sampled CSF and found that MMP-2 and MMP-13, but not MMP-9, correlate to the CSF/serum albumin ratio, a sign of BBB permeability (122).
MMP-12 expression increases as early as 1 h after permanent focal cerebral ischemia (127) and can persist for 14 days after I/R (128). Elevated MMP-12 after ischemic stroke disrupts the BBB by degrading tight junction proteins (127). MMP-12 also contributes to ischemic brain cell apoptosis, infarct volume enlargement, myelin basic protein degradation, and demyelination (128). Additionally, MMP-12 can activate other MMPs, such as pro-MMP-1 and pro-MMP-9 (129). Thus, MMP-12 enhances the proteolysis cascade. Studies show that plasma MMP-12 levels are associated with stroke severity and elevated MMP-12 might predict poor outcomes (130).
The inflammatory response after cerebral ischemia is described in Figure 1.
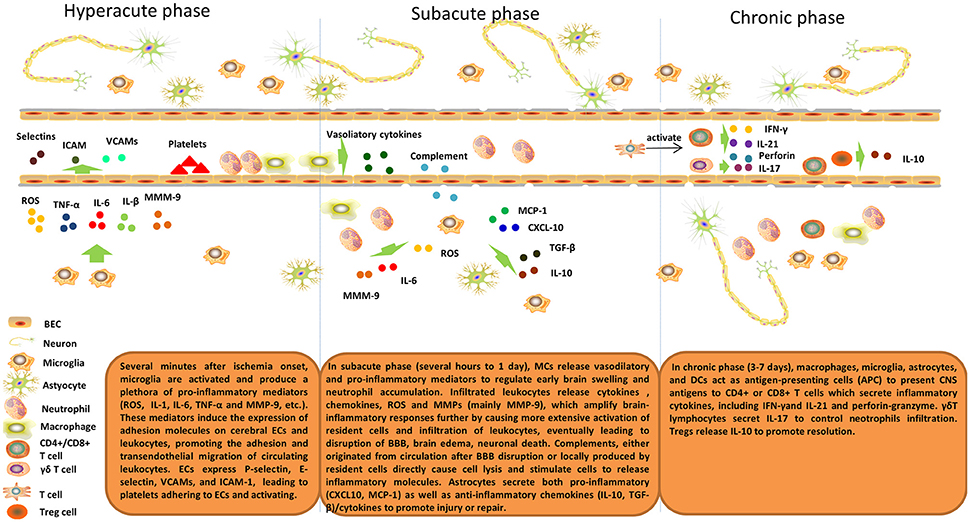
Figure 1. Inflammatory response after cerebral ischemia. Brain post-ischemic inflammatory responses are characterized by innate immune activation followed by adaptive immune activation. Microglial cells are activated within minutes of ischemia onset and produce a plethora of pro-inflammatory mediators (ROS, IL-1, IL-6, TNF-α, and MMP-9, etc.). These mediators induce expression of adhesion molecules on cerebral ECs and leukocytes and, thus, promote adhesion and transendothelial migration of circulating leukocytes. ECs express P-selectin, E-selectin, VCAMs, and ICAM-1, which lead to platelets adhering to and activating ECs. In the subacute phase (hours to 1 day), MCs release vasodilatory and pro-inflammatory mediators to regulate early brain swelling and neutrophil accumulation. Infiltrating leukocytes release cytokines, chemokines, ROS and MMPs (mainly MMP-9), which amplify brain-inflammatory responses further by causing more extensive activation of resident cells and infiltration of leukocytes, eventually leading to disruption of the BBB, brain edema, and neuronal death. Complements, either originating from the circulation after BBB disruption or locally produced by resident cells directly cause cell lysis and stimulate cells to release inflammatory molecules. Astrocytes secrete both pro-inflammatory (CXCL10, MCP-1) and anti-inflammatory chemokines (IL-10, TGF-β)/cytokines to promote injury or repair. In the delayed phase (3–7 days), macrophages, microglia, astrocytes, and DCs act as antigen-presenting cells (APCs) to present CNS antigens to CD4+ or CD8+ T cells that secrete inflammatory cytokines, including IFN-γ and IL-21 and perforin-granzyme. γδT lymphocytes secrete IL-17 to control neutrophil infiltration. Tregs release IL-10 to promote resolution.
Resolution of Inflammation in CIRI
A transient inflammatory reaction exists within the first week after ischemic stroke (131). Ultimately, the inflammation subsides, and inflamed brain tissue returns to homeostasis. Resolution of inflammation is an active and highly regulated biochemical process mediated by a group of lipid mediators, termed specialized pro-resolving mediators (SPMs). Four classes of SPMs have been discovered, including resolvins, lipoxins, protectins, and maresins. The complete stereochemistry of each SPM is known (132). The biological functions of SPMs include cessation of neutrophil infiltration, decreased production of pro-inflammatory mediators, increased uptake of apoptotic neutrophils and cellular debris, and promotion of macrophage transformation from the M1 to M2 phenotype, among others (49, 133).
Synthesis of SPMs
In the acute phase of the inflammatory response, free polyunsaturated fatty acids (PUFAs) were released from the membrane by the activity of phospholipase enzymes [e.g., cytosolic phospholipase A2 (cPLA2)]. SPM are derived from PUFAs, which including arachidonic acid (AA) that belongs to omega-6 fatty acid and eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) that belong to omega-3 fatty acids. The crucial enzymes involved in this synthesis are lipoxygenases (LOX), cyclooxygenases (COX), and cytochrome P450 monooxygenases (CYP450). SPMs are generated by leukocytes, structural cells, some organs, and tissue during the resolution of inflammation (134) and their production is ultimately controlled by the vagus nerve (135, 136).
Lipoxins are biosynthesized from AA. Three main lipoxin synthesis pathways have been reported. In the first pathway, Lipoxin A4 (LXA4) and lipoxin B4 (LXB4) are produced by the oxygenation of AA by 15-LOX and 5-LOX, and then by enzymatic hydrolysis in mucosal tissues (137). In the second pathway, LXA4 is produced by 5-LOX in blood vessels and LXB4 is synthesized by 12-LOX in platelets. The third pathway is aspirin-triggered pathway, aspirin changes COX-2 activity by increasing the acetylation of COX-2, leading to the production of a new lipoxin analog termed aspirin-triggered lipoxins (138). Inactivation of lipoxin catalysis by the enzyme 15-prostaglandin dehydrogenase can result in the synthesis of a series of stable lipoxin analogs (139) that retain all of the biological functions of lipoxins.
Two groups of resolvins exist: the the D-series, derived from DHA and E-series, derived from EPA (140). Two resolvin E1 (RvE1) formation pathways have been reported. The first is the acetylation of COX-2 at Ser516 by aspirin and 5-LOX (141). The second is oxidation by CYP450 followed by 5-LOX oxidation (142). RvE2 is derived from EPA via the 18R-hydroxyeicosapentaenoic acid and 5S-hydroperoxide, which is 5-LOX dependent (143). Afterwards, the 5S-hydroperoxide is directly converted to RvE2 without epoxidation process. Unlike RvE1 and RvE2, the formation of a third member, RvE3, does not require 5-LOX (144). DHA can be converted to 17-hydroperoxide intermediate (17-H(p)DHA) in the presence of 15-LOX, which can further give rise to D-series resolvins (RvDs). The hydroperoxide 5-LOX can be converted to 7S, 8S-epoxide by epoxidation. 7S, 8S-epoxide can then further converted to RvD1 or RvD2 enzymatic hydrolyzation (145). RvD3–D4 as well as aspirin-triggered 17R D-series resolvins (AT-RvDs) have also been discovered and their stereochemical structures have been reported recently (146, 147).
Protectins are also biosynthesized from DHA. DHA is firstly to a 17S-hydroxyperoxide-containing intermediate converted by lipoxygenase, and then the intermediate is engulfed by leukocytes to give rise to protectin D1 (PD1 or NPD1) (148). Besides, aspirin-triggered protectin D1 (AT-NPD1), has been uncovered as well (149).
The maresins (MaRs) synthesis pathway is started by the 14-lipoxygenation of DHA to generate a 14S-hydro(peroxy)-4Z,7Z,10Z,12E,14S,16Z,19Z-docosahexaenoic acid intermidate and then 13S,14S-e MaR by the enzymatic activity of 12-LOX. 13S,14S-e MaR converted to MaR1 by enzymatic hydrolyzation or epoxy hydration, to MaR2.
The Roles of SPMs in CIRI
As discussed above, inflammation plays an important role in CIRI, so it is plausible that resolving inflammation would be a promising therapeutic target for CIRI. In animal models of ischemic stroke, SPMs and their aspirin-triggered forms, are endogenously produced (150–152) and have protective roles.
The Effects of SPMs on ECs
Vascular ECs play an important role in modulating the passage of molecules and cells from the circulation cross the vessel wall during the inflammation. Therefore, vascular permeability and EC activation is critical to resolve inflammation in CIRI. Moreover, ECs are involved in SPM production and can express receptors that SPMs can directly interact with during acute phase of inflammatory response (153). For example, ECs express ALX/FPR2 (formyl-peptide receptor type 2 or LXA4 receptor; termed Fpr2/3 in mice) and G-protein coupled receptor (GPR) 32, which are receptors both for LXA4 and RvD1. Upon binding to these receptors, SPMs can regulate leukocyte recruitment and adhesion. It has been demonstrated that LXA4 downregulates VCAM-1, E-selectin (154), and ICAM-1 (155), and increases prostacyclin production, which then counter-regulates leukocyte–EC interactions and platelet activation (156). Moreover, LXA4 has further EC-specific effects, such as downregulating NADPH oxidase, which decreases ROS generation (157). An LXA4 analog reduces brain injury by improving BBB function, inhibiting MMP-9 expression, and upregulating tissue inhibitor of metalloproteinase-1 (TIMP-1) protein (158). Resolvins also have the ability to regulate microvasculature permeability. RvD1 protects the human EC barrier against endotoxin-induced impairment (159) and aspirin-triggered-RvD1, reducing the endothelial barrier permeability in an acute lung injury mice model (160).
The Effects of SPMs on Polymorphonuclear Leukocytes (PMNs)
After ischemia, leukocyte infiltration, as well as ROS and pro-inflammatory mediator production, results in tissue damage. Therefore, inhibiting leukocyte accumulation or removing leukocytes altogether, is crucial for brain protection during ischemia. Various ischemia models in different organs have supported the idea that SPMs inhibit leukocyte accumulation and promote phagocytosis of apoptotic leukocytes. In a mesenteric artery I/R mouse model, an LXA4 analog detached leukocytes from endothelium cells by binding to FPR (161) and also diminished PMN infiltration into lungs after hind limb I/R (162–164). RvD1 has been reported to reduce fibrosis and the accumulation of leukocytes, as well as enhancing cardiac function after in experimental mice model of myocardial infarction (MI) (152). Biosynthesis of RvDs and protectins has been found after I/R in the kidneys. Furthermore, treatment with RvDs prior to ischemia decreases leukocyte infiltration and tissue fibrosis (151). In the context of CIRI, RvD1 and PD1 are also protective, as they reduce the number of infiltrating leukocytes and prevent NF-κB and COX-2 activation in neurons (150). In a mouse hind limb I/R model, resolvins protect against leukocyte recruitment into the lungs (165, 166). RvEl has potent effects on leukocytes by decreasing migration, inhibiting rolling, enhancing CCR5 expression, and downregulating NF-κB pathway signaling (167). In a mouse renal pedicle I/R model, PD1 reduces kidney PMN influx and amplifies renoprotective heme-oxygenase(HO)-1 protein and mRNA expression (168).
The Effects of SPMs on Macrophages
Efferocytosis, or the clearing of dead cells, is essential for tissue to successfully resolve the inflammatory response. Cardiac function impairment has been found in acute MI mice deficient in Mertk and/or Mfge8, which are the receptors of efferocytosis. This is caused by an abrogation of efferocytosis rather than a change in circulating or tissue-resident macrophage/monocyte infiltration (169, 170). A study using a bilateral common carotid artery occlusion (BCCAO)/reperfusion mouse model has demonstrated that LXA4 analogs can enhance efferocytosis, thereby promoting a resolution of inflammation (171). Macrophage/microglia survival and phagocytosis in a highly oxidative environment after CIRI is of critical importance. It has been demonstrated that RvD1, upon binding to ALX/FPR2, has protective effects on macrophages during efferocytosis from apoptosis induced by oxidative stress, by down-regulating NADPH oxidase and the reducing the levels of apoptotic proteins such as Bcl-x and Bcl-2 (172). During efferocytosis, SPM production increases, further enhancing debris and apoptotic cell clearance (173).
SPMs are also thought to modify macrophage phenotype from pro-inflammatory M1 to anti-inflammatory M2 by binding to GPR32 (174). The shift in phenotype toward M2 is associated with reparative and anti-inflammatory functions. RvD1 has been proved protective I/R injury model of liver by promoting M2 polarization of macrophage and promoting efferocytosis (175). It has also been shown to induce M2 macrophage phenotypes to resolve adipose tissue inflammation (176). It has been demonstrated that RvD2 administration increases macrophage polarization to the M2 phenotype in the arterial walls of a murine model of arterial neointima formation (177). MaR1 has also been shown to stimulate the M1 to M2 phenotype transformation besides its pro-resolving bio functions and tissue restorative abilities (178).
As we know, ECs, PMNs, and microglia/macrophages are primary sources of inflammatory molecules during CIRI. The pro-inflammatory mediators such as cytokines, chemokines as well as adhesion molecules, which contribute to leukocyte infiltration, oxidative stress, BBB disruption, and neurotoxic substance release, among others. Anti-inflammatory cytokines inhibit pro-inflammatory cytokines and stimulate tissue repair during inflammation resolution. SPMs can increase anti-inflammatory mediators and decrease pro-inflammatory mediators. In a superior mesenteric artery I/R mouse model, an LXA4 analog decreases vascular permeability, leukocyte influx, and hemorrhage in the intestines, as well as inhibiting reperfusion-induced remote injury to the lungs by activating ALX/FPR2 for IL-10 production (179). In a bilateral kidney I/R mouse model, an LXA4 analog alleviates I/R injury by reducing PMN infiltration and pro-inflammatory cytokine (IL-1, IL-6) and chemokine (growth regulated oncogene-1 [GRO1]) mRNA levels, and increasing renal mRNA levels of (SOCS)-1 and SOCS-2 that are suppressors of cytokine signaling (180). Further, by using oligonucleotide microarray and bioinformatics, Kieran et al. found that the LXA4 analog is renoprotective since it modifies changes in the mRNA levels of many families of harmful molecules, including pro-inflammatory cytokines, cellular adhesion molecules, and proteases (181).
The Effects of SPMs on Oxidative Stress and Metabolism
As discussed above, oxidative stress plays an important role in the pathogenesis of CIRI. SPMs can attenuate CIRI by acting as potent antioxidants and mediators. In a spinal cord I/R rabbit model, LXA4 improves neurological function and reduces cell apoptosis, partly by decreasing MDA levels and increasing superoxide dismutase (SOD) activity (182). In different I/R experimental models, the mechanisms by which LXA4 attenuates oxidative stress includes downregulating GRP-78 (78 kDa glucose-regulated protein) and caspase-12 (183), activating the Keap1/Nrf2 (Ketch-like ECH-associated protein 1/nuclear respiratory factor 2) (184) and Nrf2 (185) pathways, and acting as an agonist of peroxisome proliferator-activated receptor (PPAR)γ (186). MaR1 suppresses free radicals by activating HO-1 pathway mediated by Nrf-2 in a left pulmonary hilum I/R mouse model (187).
In CIRI, SPMs may improve energy metabolism disorders to protect tissue from injury. RvD1 has been shown protective for mitochondrial in I/R lung model, by increasing Na+-K+-ATPase activity, retune the balance of ATP/ADP ratio, and decreasing apoptosis (188). LXA4 attenuates metabolic disturbance in MI by upregulating Na+-K+-ATPase expression (189). LXA4 also upregulates Cx43 expression to prevent arrhythmogenesis (189). The roles of each SPM on cells and moleculars in CIRI are summarized in Figure 2.
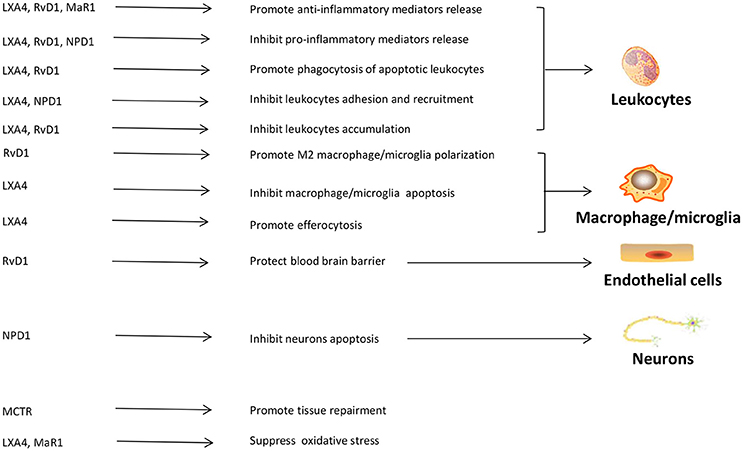
Figure 2. The roles of SPMs in cerebral ischemia reperfusion injury. SPMs can display their protective roles through interacting with leukocytes, macrophages/microglia, vascular endothelial cells, and neurons in cerebral ischemic reperfusion injury. The main functions of SPMs in cerebra ischemic reperfusion injury include regulating leukocytes adhesion, infiltration and apoptosis, mediating inflammatory mediators release, adjusting macrophages/microglia polarization, protecting blood brain barrier, inhibiting neuronal apoptosis, promoting tissue restitution, suppressing oxidative stress. LXA4, lipoxin A4; RvD1, resolving D1; MaR1, maresin1; NPD1, protectin D1; MCTR, maresin conjugates in tissue regeneration.
Promoting Inflammation Resolution to Treat I/R Injury
The discovery that the resolution of inflammation is a highly regulated process is crucial, because this knowledge has led to investigations of new therapeutics with selective targets that will safely resolve inflammation without compromising host defense (190). Each SPM has potent pro-resolving functions that are fundamental to inflammation resolution. Current experiments are investigating if targeting these pro-resolution functions can be useful in the treatment of CIRI (Table 1).
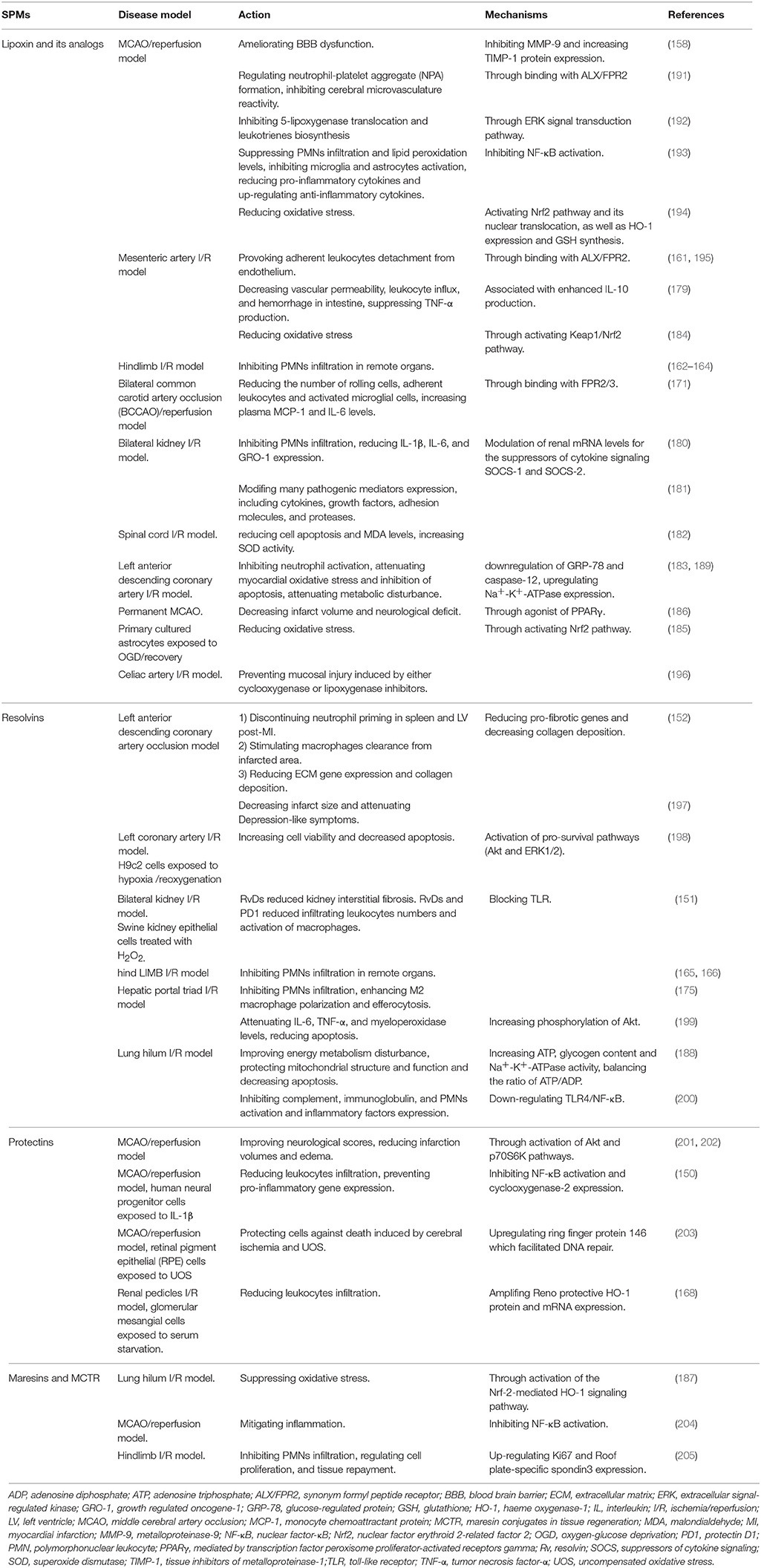
Table 1. Studies of SPMs in I/R injury experiment models.
LXA4
LXA4 binds with high affinity to the GPR termed ALX/FPR2, which is expressed on various cellular types, such as leukocytes, ECs, fibroblasts, microlgia, macrophage, and neuronal cells (206). Activation of this receptor could promote a resolution of inflammation and tissue repair. Exogenous introduction of LXA4 in Fpr2/3+/+mice attenuates inflammation in I/R injury, while this result cannot be observed in Fpr2/3−/− mice, demonstrating the protective role of Fpr2/3 in I/R injury modles (191, 195). Consistent with these findings, studies in CIRI rats have found that LXA4 is also neuroprotective, reducing leukocyte infiltration and astrocyte activation, decreasing TNF-α and IL-1β production, and reducing leukotrienes, as well as extracellular signal-regulated kinase (ERK) phosphorylation (192, 193). It has been demonstrated that LXA4 reduces infarct size and improves neurological scores, partially by inducing Nrf2 synthesis and its translocation into the nucleus, as well as increasing HO-1 and glutathione (GSH) synthesis; thus, combating increased oxidative stress (194). In addition, the PPAR agonist rosiglitazone has been shown to be neuroprotective by increasing LXA4 and reducing leukotriene B4 (LTB4) in experimental stroke (186). Besides its neuroprotective effects, LXA4 may also attenuate the side effects of drugs that treat cerebrovascular diseases. In a celiac artery I/R rat model, LXA4 prevents against cyclooxygenase or lipoxygenase inhibitors induced injury of celiac mucosal (196).
Resolvins
RvD1 binds to and activates human GPR32 and ALX/FPR2, while recombinant GPR32 can be active by RvD3 and RvD5 (207). RvD2 activates a novel GPR termed DRV2/GPR18, and RvD2-DRV2 interactions could stimulate macrophage phagocytosis (166, 208). RvDs may inhibit PMN activation and decrease their infiltration, as well as enhancing macrophage phagocytosis of apoptotic cells/bacteria (190). RvD1 is endogenously produced in response to I/R injury in kidneys, inhibition of leukocyte accumulation, and TLR-mediated macrophage activation (151). Moreover, RvD1 resolves inflammation and enhances ventricular function after MI (152). In rats, RvD1 inhibits inflammatory responses following liver I/R injury (199) and this protective effect is related to a shift to M2 phenotype of macrophage and enhanced efferocytosis induced by ALX/FPR2 activation. The underlying mechanisms mediating the protection of I/R injury might involve the inhibition of complements, activation of immunoglobulin and neutrophils, downregulation of TLR4/NF-κB, and decreased expression of pro-inflammatory factors (200). Inflammation resolution involves a specific miRNA, regulated by SPM receptors (209, 210). RvD1-GPR32 increases miR-208 and the anti-inflammatory cytokine IL-10, and downregulates miR-219, decreasing LTB4 via the regulation of 5-LOX (209). In a left anterior descending coronary artery I/R rat model, RvD1 not only decreases infarct size, but also attenuates depression-like symptoms, suggesting that RvD1 might be involved in neurotransmitter secretion (197).
RvE1 binds to GPR ChemR23 and LTB4 receptor 1 (BLT1). ChemR23 is highly expressed in dendrites and macrophages (141). RvE1 has been shown to decrease infarct size in a rat model of MI/reperfusion injury by decreasing apoptosis in cells induced by hypoxia (198). RvE1 stimulates macrophage phagocytosis of apoptotic leukocytes, decreases the pro-inflammatory cytokines IFN-γ as well as IL-6 production (211), inhibits DC migration, and reduces pro-inflammatory cytokine release (141) via phosphoprotein-mediated signaling (212). The pro-inflammatory lipid mediator LTB4 binds to BLT1 and promotes leukocyte survival. RvE1 competes with LTB4 and blocks the binding of LTB4 to BLT1 to increase the apoptosis of leukocytes and efferocytosis (213). It has also been reported that ChemR23 expression is restricted to naive M1, but not M2, macrophages (214). The authors also found that treating M1 macrophages that express ChemR23 with RvE1 enhanced the phagocytosis of microbial particles and increased IL-10 transcription, resulting in a resolution-type macrophage that is different from the anti-inflamatory M2 phenotype.
NPD1
NPD1 is generated in rodents after ischemia and plays an important role in PMN decrement (150). Bazan and colleagues have demonstrated the beneficial roles of NPD1 in the CNS. NPD1 reduces oxidative stress, thus improving cell survival. NPD1 also has effect on microglia cells in ocular disease models (215). AT-NPD1 is endogenously generated by aspirin and DHA treatment in the cerebral stroke models. Synthetic 17R-NPD1 has been shown reduced the brain edema and the infarct size and reduces the neurological deficiency (201). 17R-NPD1 derived from aspirin-triggered DHA reduces human PMN infiltration, enhances microphage efferocytosis (216), improves neurological scores, and reduces total lesion volume and brain edema, as computed by T2-wighed image (T2WI) (201). In an MCAO/reperfusion rat model, NPD1 significantly reduces the infarct sizes in aged rats via activation of the Akt and p70S6K pathways (202). Furthermore, NPD1 up-regulates ring finger protein 146, which facilitates DNA repair and protects cells against death induced by cerebral ischemia (203).
Maresin
Intracerebroventricular injection of MaR1 has been shown to significantly reduce infarct volumes and neurological defects, protecting the neurons from injury in a CIRI mouse model, by reducing the pro-inflammatory responses and inhibiting the activation of NF-κB p65 and its translocation to the nucleus (204). In a hindlimb I/R mouse model, maresin conjugates in tissue regeneration (MCTR) protects the lungs and spleen from tissue damage mediated by leukocytes and increases cell growth and tissue restoration in the lungs (205). A recent study has found that platelets express the SPM receptors GPR32 and ALX. MaR1 could enhance platelet aggregation, spreading and suppressing the release of pro-inflammatory and pro-thrombotic mediators; therefore, MaR1 may be used as a new class of antiplatelet agents (217).
Conclusions and Future Perspectives
CIRI can worsen patient outcomes through excessive inflammation. However, neuronal damage following CIRI is often irreversible; therefore, there is an unmet clinical need to explore novel therapeutic options to resolve the aggressive inflammatory state after cerebral ischemic insults to limit the irreversible neural damage. Dietary n-3 supplements are widely used, but clinical trials with n-3 fatty acids have yielded mixed results (218). The lack of success may be because aging patients and those with obesity-associated diseases may have difficulty converting PUFAs into SPMs (219). The biosynthesis pathways for both omega-3 and omega-6 fatty acids are complex and involve competition for enzymes to produce various bioactive mediators without pro-resolving actions. Thus, SPMs may be more potent and biologically relevant than fatty acids, their nutritional precursors (220, 221). The new challenge ahead is if the novel SPMs that stimulate inflammation resolution can be harnessed to treat I/R injury. Stable synthetic mimetics to endogenous SPMs and synthetic SPM receptor agonists are under development for various chronic inflammatory disease models (133); however, the side effects accompanying these need to be investigated further (222). It is our hope that the results from these studies will provide new stroke treatments by controlling resolution and downstream mechanisms of inflammatory responses.
Author Contributions
PY, XW, and YW wrote the manuscript. MZ and JF reviewed the topic and established a basic framework for the manuscript.
Funding
The authors are grateful for the support from grants from the National Science Foundation of China (No. 31600820, NO. 81701158); The Health and Family Planning Commission of Jilin Province (No. 2016Q036); The Science and technology planning project of Jilin Province (No. 20180520110JH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DP-G and handling Editor declared their shared affiliation.
References
1. Albers GW, Bates VE, Clark WM, Bell R, Verro P, Hamilton SA. Intravenous tissue-type plasminogen activator for treatment of acute stroke: the Standard Treatment with Alteplase to Reverse Stroke (STARS) study. JAMA (2000) 283:1145–50. doi: 10.1001/jama.283.9.1145
2. Lees KR, Bluhmki E, von Kummer R, Brott TG, Toni D, Grotta JC, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet (2010) 375:1695–703. doi: 10.1016/S0140-6736(10)60491-6
3. Albers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med. (2018) 378:708–18. doi: 10.1056/NEJMoa1713973
4. Molina CA. Reperfusion therapies for acute ischemic stroke: current pharmacological and mechanical approaches. Stroke (2011) 42:S16–19. doi: 10.1161/STROKEAHA.110.598763
5. Zhang L, Zhang ZG, Liu X, Hozeska A, Stagliano N, Riordan W, et al. Treatment of embolic stroke in rats with bortezomib and recombinant human tissue plasminogen activator. Thromb Haemost. (2006) 95:166–73. doi: 10.1160/TH05-07-0477
6. Yamashita T, Deguchi K, Nagotani S, Abe K. Vascular protection and restorative therapy in ischemic stroke. Cell Transplant. (2011) 20:95–7. doi: 10.3727/096368910X532800
7. Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res. (2016) 119:142–58. doi: 10.1161/CIRCRESAHA.116.308022
8. Mizuma A, Yenari MA. Anti-inflammatory targets for the treatment of reperfusion injury in stroke. Front Neurol. (2017) 8:467. doi: 10.3389/fneur.2017.00467
9. Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. (2008) 30:783–93. doi: 10.1179/174313208x341085
10. Yilmaz G, Granger DN. Leukocyte recruitment and ischemic brain injury. Neuromol Med. (2010) 12:193–204. doi: 10.1007/s12017-009-8074-1
11. Perego C, Fumagalli S, De Simoni MG. Three-dimensional confocal analysis of microglia/macrophage markers of polarization in experimental brain injury. J VisExp. (2013) 79:e50605. doi: 10.3791/50605
12. Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation (2014) 11:98. doi: 10.1186/1742-2094-11-98
13. Pun PBL, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. (2009) 43:348–64. doi: 10.1080/10715760902751902
14. Yenari MA, Xu LJ, Tang XN, Qiao YL, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents - Improvement by minocycline in vivo and in vitro. Stroke (2006) 37:1087–93. doi: 10.1161/01.STR.0000206281.77178.ac
15. del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang XY, Berg GI, et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke (2007) 38:646–51. doi: 10.1161/01.STR.0000254477.34231.cb
16. Huang M, Wan Y, Mao L, He QW, Xia YP, Li M, et al. Inhibiting the Migration of M1 Microglia at Hyperacute Period Could Improve Outcome of tMCAO Rats. CNS Neurosci Ther. (2017) 23:222–32. doi: 10.1111/cns.12665
17. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
18. Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. (2006) 20:714–6. doi: 10.1096/fj.05-4882fje
19. Perez-de Puig I, Miro F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Marquez-Kisinousky L, et al. IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J Cereb Blood Flow Metab. (2013) 33:1955–66. doi: 10.1038/jcbfm.2013.155
20. Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci. (2015) 9:147. doi: 10.3389/fnins.2015.00147
21. Jin WN, Shi SXY, Li ZG, Li MS, Wood K, Gonzales RJ, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. (2017) 37:2224–36. doi: 10.1177/0271678x17694185
22. Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia (2005) 50:287–98. doi: 10.1002/glia.20181
23. Becerra-Calixto A, Cardona-Gomez GP. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci. (2017) 10:88. doi: 10.3389/fnmol.2017.00088
24. Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, et al. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. (2005) 79:74–80. doi: 10.1002/jnr.20335
25. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell (2011) 144:810–23. doi: 10.1016/j.cell.2011.02.018
26. Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. (2000) 36:291–300. doi: 10.1016/S0197-0186(99)00137-0
27. Zou J, Wang YX, Dou FF, Lu HZ, Ma ZW, Lu PH, et al. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem Int. (2010) 56:577–84. doi: 10.1016/j.neuint.2009.12.021
28. Kaczor P, Rakus D, Mozrzymas JW. Neuron-astrocyte interaction enhance GABAergic synaptic transmission in a manner dependent on key metabolic enzymes. Front Cell Neurosci. (2015) 9:120. doi: 10.3389/fncel.2015.00120
29. Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia (2006) 53:688–95. doi: 10.1002/glia.20328
30. Newman EA. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140195. doi: 10.1098/rstb.2014.0195
31. Pentreath VW, Slamon ND. Astrocyte phenotype and prevention against oxidative damage in neurotoxicity. Hum Exp Toxicol. (2000) 19:641–9. doi: 10.1191/096032700676221595
32. Duan CL, Liu CW, Shen SW, Yu Z, Mo JL, Chen XH, et al. Striatal astrocytes transdifferentiate into functional mature neurons following ischemic brain injury. Glia (2015) 63:1660–70. doi: 10.1002/glia.22837
33. Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, et al. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. (2013) 23:34–44. doi: 10.1111/j.1750-3639.2012.00614.x
34. Herz J, Sabellek P, Lane TE, Gunzer M, Hermann DM, Doeppner TR. Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke (2015) 46:2916–25. doi: 10.1161/Strokeaha.115.010620
35. Garcia-Bonilla L, Racchumi G, Murphy M, Anrather J, Iadecola C. Endothelial CD36 contributes to postischemic brain injury by promoting neutrophil activation via CSF3. J Neurosci. (2015) 35:14783–93. doi: 10.1523/Jneurosci.2980-15.2015
36. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
37. Chu HX, Kim HAH, Lee S, Moore JP, Chan CT, Vinh A, et al. Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab. (2014) 34:450–9. doi: 10.1038/jcbfm.2013.217
38. Sladojevic N, Stamatovic SM, Keep RF, Grailer JJ, Sarma JV, Ward PA, et al. Inhibition of junctional adhesion molecule-A/LFA interaction attenuates leukocyte trafficking and inflammation in brain ischemia/reperfusion injury. Neurobiol Dis. (2014) 67:57–70. doi: 10.1016/j.nbd.2014.03.010
39. Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. (2015) 22:1258–77. doi: 10.2174/0929867322666150209154036
40. Enzmann G, Mysiorek C, Gorina R, Cheng YJ, Ghavampour S, Hannocks MJ, et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. (2013) 125:395–412. doi: 10.1007/s00401-012-1076-3
41. Mancuso ME, Santagostino E. Platelets: much more than bricks in a breached wall. Br J Haematol. (2017) 178:209–19. doi: 10.1111/bjh.14653
42. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
43. Khlgatian M, Nassar H, Chou HH, Gibson FC, Genco CA. Fimbria-dependent activation of cell adhesion molecule expression in Porphyromonas gingivalis-infected endothelial cells. Infect Immun. (2002) 70:257–67. doi: 10.1128/Iai.70.1.257-267.2002
44. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature (1998) 391:591–4. doi: 10.1038/35393
45. Puhlmann M, Weinreich DM, Farma JM, Carroll NM, Turner EM, Alexander HR. Interleukin-1 beta induced vascular permeability is dependent on induction of endothelial Tissue Factor (TF) activity. J Transl Med. (2005) 3:37. doi: 10.1186/1479-5876-3-37
46. Semple JW, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
47. Mause SF, Weber C. Microparticles protagonists of a novel communication network for intercellular information exchange. Circ Res. (2010) 107:1047–57. doi: 10.1161/Circresaha.110.226456
48. Vasina E, Heemskerk JW, Weber C, Koenen RR. Platelets and platelet-derived microparticles in vascular inflammatory disease. Inflamm Allergy Drug Targets (2010) 9:346–54. doi: 10.2174/187152810793938008
49. Dalli J, Serhan CN. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front Immunol. (2017) 8:1400. doi: 10.3389/fimmu.2017.01400
50. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood (2014) 123:2759–67. doi: 10.1182/blood-2013-11-462432
51. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles - A transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arteriosc Thromb Vasc Biol. (2005) 25:1512–8. doi: 10.1161/01.Atv.0000170133.43608.37
52. Gidlof O, van der Brug M, Ohman J, Gilje P, Olde B, Wahlestedt C, et al. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood (2013) 121:3908–17. doi: 10.1182/blood-2012-10-461798
53. Laffont B, Corduan A, Rousseau M, Duchez AC, Lee CHC, Boilard E, et al. Platelet microparticles reprogram macrophage gene expression and function. Thromb Haemost. (2016) 115:311–23. doi: 10.1160/Th15-05-0389
54. Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood (2010) 115:3835–42. doi: 10.1182/blood-2009-10-249078
55. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke (2009) 40:1849–57. doi: 10.1161/Strokeaha.108.534503
56. Petrovic-Djergovic D, Hyman MC, Ray JJ, Bouis D, Visovatti SH, Hayasaki T, et al. Tissue-Resident Ecto-5 ' Nucleotidase (CD73) regulates leukocyte trafficking in the ischemic brain. J Immunol. (2012) 188:2387–98. doi: 10.4049/jimmunol.1003671
57. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation (2006) 113:2105–12. doi: 10.1161/Circulationaha.105.593046
58. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gamma delta T cells in the delayed phase of ischemic brain injury. Nat Med. (2009) 15:946–U150. doi: 10.1038/nm.1999
59. Clarkson BDS, Ling CY, Shi YJ, Harris MG, Rayasam A, Sun DD, et al. T cell-derived interleukin (IL)-21 promotes brain injury following stroke in mice. J Exp Med. (2014) 211:595–604. doi: 10.1084/jem.20131377
60. Mracsko E, Liesz A, Stojanovic A, Lou WPK, Osswald M, Zhou W, et al. Antigen dependently activated cluster of differentiation 8-positive T cells cause perforin-mediated neurotoxicity in experimental stroke. J Neurosci. (2014) 34:16784–95. doi: 10.1523/Jneurosci.1867-14.2014
61. Gelderblom M, Arunachalam P, Magnus T. Gamma delta T cells as early sensors of tissue damage and mediators of secondary neurodegeneration. Front Cell Neurosci. (2014) 8:368. doi: 10.3389/fncel.2014.00368
62. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood (2012) 120:3793–802. doi: 10.1182/blood-2012-02-412726
63. Bonaventura A, Liberale L, Vecchie A, Casula M, Carbone F, Dallegri F, et al. Update on inflammatory biomarkers and treatments in ischemic stroke. Int J Mol Sci. (2016) 17:E1967. doi: 10.3390/ijms17121967
64. Li PY, Gan Y, Sun BL, Zhang F, Lu BF, Gao YQ, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. (2013) 74:458–71. doi: 10.1002/ana.23815
65. Li PY, Mao LL, Liu XR, Gan Y, Zheng J, Thomson AW, et al. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke (2014) 45:857–64. doi: 10.1161/Strokeaha.113.004100
66. Na SY, Mracsko E, Liesz A, Hunig T, Veltkamp R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke (2015) 46:212–20. doi: 10.1161/Strokeaha.114.007756
67. Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Gobel K, Schuhmann MK, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood (2013) 121:679–91. doi: 10.1182/blood-2012-04-426734
68. Xu XM, Li M, Jiang YJ. The paradox role of regulatory T cells in ischemic stroke. Sci World J. (2013) 2013:174373. doi: 10.1155/2013/174373
69. Pruss H, Iggena D, Baldinger T, Prinz V, Meisel A, Endres M, et al. Evidence of intrathecal immunoglobulin synthesis in stroke a cohort study. Arch Neurol. (2012) 69:714–7. doi: 10.1001/archneurol.2011.3252
70. Doyle KP, Quach LN, Sole M, Axtell RC, Nguyen TVV, Soler-Llavina GJ, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. (2015) 35:2133–45. doi: 10.1523/Jneurosci.4098-14.2015
71. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. (2012) 122:1164–71. doi: 10.1172/Jci58644
72. Kostulas N, Li HL, Xiao BG, Huang YM, Kostulas V, Link H. Dendritic cells are present in ischemic brain after permanent middle cerebral artery occlusion in the rat. Stroke (2002) 33:1129–34. doi: 10.1161/hs0402.105379
73. Dietel B, Cicha I, Kallmunzer B, Tauchi M, Yilmaz A, Daniel WG, et al. Suppression of dendritic cell functions contributes to the anti-inflammatory action of granulocyte-colony stimulating factor in experimental stroke. Exp Neurol. (2012) 237:379–87. doi: 10.1016/j.expneurol.2012.06.019
74. Mekori YA, Metcalfe DD. Mast cell-T cell interactions. J Allergy Clin Immunol. (1999) 104(3 Pt 1), 517–23.
75. McKittrick CM, Lawrence CE, Carswell HV. Mast cells promote blood brain barrier breakdown and neutrophil infiltration in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab. (2015) 35:638–47. doi: 10.1038/jcbfm.2014.239
76. Mattila OS, Strbian D, Saksi J, Pikkarainen TO, Rantanen V, Tatlisumak T, et al. Cerebral mast cells mediate blood-brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke (2011) 42:3600–5. doi: 10.1161/STROKEAHA.111.632224
77. Arac A, Grimbaldeston MA, Nepomuceno AR, Olayiwola O, Pereira MP, Nishiyama Y, et al. Evidence that meningeal mast cells can worsen stroke pathology in mice. Am J Pathol (2014) 184:2493–504. doi: 10.1016/j.ajpath.2014.06.003
78. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. (2015) 6:524–51. doi: 10.1016/j.redox.2015.08.020
79. Musatov A, Robinson NC. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic Res. (2012) 46:1313–26. doi: 10.3109/10715762.2012.717273
80. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
81. Tajeddine N. How do reactive oxygen species and calcium trigger mitochondrial membrane permeabilisation? Biochim Biophys Acta (2016) 1860:1079–88. doi: 10.1016/j.bbagen.2016.02.013
82. Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, et al. SPG7 Is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell (2015) 60:47–62. doi: 10.1016/j.molcel.2015.08.009
83. Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, et al. Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. (2017) 6:e005328. doi: 10.1161/JAHA.116.005328
84. Anzell AR, Maizy R, Przyklenk K, Sanderson TH. Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol Neurobiol. (2017) 55:2547–64. doi: 10.1007/s12035-017-0503-9
85. Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta (2017) 1864:1260–73. doi: 10.1016/j.bbamcr.2017.03.006
86. Bradley SA, Steinert JR. Nitric Oxide-Mediated Posttranslational Modifications: Impacts at the Synapse. Oxid Med Cell Longev. (2016) 2016:5681036. doi: 10.1155/2016/5681036
87. Li Q, Atochin D, Kashiwagi S, Earle J, Wang A, Mandeville E, et al. Deficient eNOS phosphorylation is a mechanism for diabetic vascular dysfunction contributing to increased stroke size. Stroke (2013b) 44:3183–8. doi: 10.1161/STROKEAHA.113.002073
88. Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KT. The role of the nitric oxide pathway in brain injury and its treatment–from bench to bedside. Exp Neurol. (2015) 263:235–43. doi: 10.1016/j.expneurol.2014.10.017
89. Zanini GM, Cabrales P, Barkho W, Frangos JA, Carvalho LJ. Exogenous nitric oxide decreases brain vascular inflammation, leakage and venular resistance during Plasmodium berghei ANKA infection in mice. J Neuroinflammation (2011) 8:66. doi: 10.1186/1742-2094-8-66
90. Chen XM, Chen HS, Xu MJ, Shen JG. Targeting reactive nitrogen species: a promising therapeutic strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol Sin. (2013) 34:67–77. doi: 10.1038/aps.2012.82
91. Zhu T, Yao Q, Wang W, Yao H, Chao J. iNOS Induces Vascular Endothelial Cell Migration and Apoptosis Via Autophagy in Ischemia/Reperfusion Injury. Cell Physiol Biochem. (2016) 38:1575–88. doi: 10.1159/000443098
92. Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ. Nitrotyrosine as a marker for peroxynitrite-induced neurotoxicity: the beginning or the end of the end of dopamine neurons? J Neurochem. (2004) 89:529–36. doi: 10.1111/j.1471-4159.2004.02346.x
93. Ding R, Chen Y, Yang S, Deng X, Fu Z, Feng L, et al. Blood-brain barrier disruption induced by hemoglobin in vivo: Involvement of up-regulation of nitric oxide synthase and peroxynitrite formation. Brain Res. (2014) 1571:25–38. doi: 10.1016/j.brainres.2014.04.042
94. Han F, Chen YX, Lu YM, Huang JY, Zhang GS, Tao RR, et al. Regulation of the ischemia-induced autophagy-lysosome processes by nitrosative stress in endothelial cells. J Pineal Res. (2011) 51:124–35. doi: 10.1111/j.1600-079X.2011.00869.x
95. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
96. Moriyama M, Fukuhara T, Britschgi M, He Y, Narasimhan R, Villeda S, et al. Complement receptor 2 is expressed in neural progenitor cells and regulates adult hippocampal neurogenesis. J Neurosci. (2011) 31:3981–9. doi: 10.1523/JNEUROSCI.3617-10.2011
97. Szeplaki G, Szegedi R, Hirschberg K, Gombos T, Varga L, Karadi I, et al. Strong complement activation after acute ischemic stroke is associated with unfavorable outcomes. Atherosclerosis (2009) 204:315–20. doi: 10.1016/j.atherosclerosis.2008.07.044
98. Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, et al. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. (2000) 161:373–82. doi: 10.1006/exnr.1999.7273
99. Pedersen ED, Loberg EM, Vege E, Daha MR, Maehlen J, Mollnes TE. In situ deposition of complement in human acute brain ischaemia. Scand J Immunol. (2009) 69:555–62. doi: 10.1111/j.1365-3083.2009.02253.x
100. Mocco J, Wilson DA, Komotar RJ, Sughrue ME, Coates K, Sacco RL, et al. Alterations in plasma complement levels after human ischemic stroke. Neurosurgery (2006)59:28–33; discussion 28–33. doi: 10.1227/01.NEU.0000219221.14280.65
101. Harhausen D, Khojasteh U, Stahel PF, Morgan BP, Nietfeld W, Dirnagl U, et al. Membrane attack complex inhibitor CD59a protects against focal cerebral ischemia in mice. J Neuroinflammation (2010) 7:15. doi: 10.1186/1742-2094-7-15
102. Ten VS, Sosunov SA, Mazer SP, Stark RI, Caspersen C, Sughrue ME, et al. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke (2005) 36:2244–50. doi: 10.1161/01.STR.0000182237.20807.d0
103. Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. (2006) 99:209–17. doi: 10.1161/01.RES.0000232544.90675.42
104. Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, et al. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J Neuroinflammation (2015) 12:247. doi: 10.1186/s12974-015-0464-8
105. Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci USA. (2007) 104:14104–9. doi: 10.1073/pnas.0700506104
106. Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS ONE (2010) 5:e8433. doi: 10.1371/journal.pone.0008433
107. Morrison H, Frye J, Davis-Gorman G, Funk J, McDonagh P, Stahl G, et al. The contribution of mannose binding lectin to reperfusion injury after ischemic stroke. Curr Neurovasc Res. (2011) 8:52–63. doi: 10.2174/156720211794520260
108. Zhao XJ, Larkin TM, Lauver MA, Ahmad S, Ducruet AF. Tissue plasminogen activator mediates deleterious complement cascade activation in stroke. PLoS ONE (2017) 12:e0180822. doi: 10.1371/journal.pone.0180822
109. Alawieh A, Elvington A, Tomlinson S. Complement in the Homeostatic and Ischemic Brain. Front Immunol. (2015) 6:417. doi: 10.3389/fimmu.2015.00417
110. Brennan FH, Lee JD, Ruitenberg MJ, Woodruff TM. Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Semin Immunol. (2016) 28:292–308. doi: 10.1016/j.smim.2016.03.015
111. Fumagalli S, De Simoni MG. Lectin complement pathway and its bloody interactions in brain ischemia. Stroke (2016) 47:3067–73. doi: 10.1161/STROKEAHA.116.012407
112. Chen X, Arumugam TV, Cheng YL, Lee JH, Chigurupati S, Mattson MP, et al. Combination therapy with low-dose IVIG and a C1-esterase inhibitor ameliorates brain damage and functional deficits in experimental ischemic stroke. NeuromolMed. (2018) 20:63–72. doi: 10.1007/s12017-017-8474-6
113. Jarlestedt K, Rousset CI, Stahlberg A, Sourkova H, Atkins AL, Thornton C, et al. Receptor for complement peptide C3a: a therapeutic target for neonatal hypoxic-ischemic brain injury. FASEB J. (2013) 27:3797–804. doi: 10.1096/fj.13-230011
114. Shinjyo N, de Pablo Y, Pekny M, Pekna M. Complement peptide C3a promotes astrocyte survival in response to ischemic stress. Mol Neurobiol. (2016) 53:3076–87. doi: 10.1007/s12035-015-9204-4
115. Wu MC, Brennan FH, Lynch JP, Mantovani S, Phipps S, Wetsel RA, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA. (2013) 110:9439–44. doi: 10.1073/pnas.1218815110
116. Stokowska A, Atkins AL, Moran J, Pekny T, Bulmer L, Pascoe MC, et al. Complement peptide C3a stimulates neural plasticity after experimental brain ischaemia. Brain (2017) 140:353–69. doi: 10.1093/brain/aww314
117. Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, et al. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery (2008) 63:122–5; discussion 125–6. doi: 10.1227/01.NEU.0000335079.70222.8D
118. Alawieh A, Tomlinson S. Injury site-specific targeting of complement inhibitors for treating stroke. Immunol Rev. (2016) 274:270–80. doi: 10.1111/imr.12470
119. Chang JJ, Stanfill A, Pourmotabbed T. The role of matrix metalloproteinase polymorphisms in ischemic stroke. Int J Mol Sci. (2016) 17:1323. doi: 10.3390/ijms17081323
120. Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. (2000) 20:1681–9. doi: 10.1097/00004647-200012000-00007
121. Demir R, Ulvi H, Ozel L, Ozdemir G, Guzelcik M, Aygul R. Relationship between plasma metalloproteinase-9 levels and volume and severity of infarct in patients with acute ischemic stroke. Acta Neurol Belg. (2012) 112:351–6. doi: 10.1007/s13760-012-0067-4
122. Zhang Y, Fan F, Zeng G, Zhou L, Zhang Y, Zhang J, et al. Temporal analysis of blood-brain barrier disruption and cerebrospinal fluid matrix metalloproteinases in rhesus monkeys subjected to transient ischemic stroke. J Cereb Blood Flow Metab. (2017) 37:2963–74. doi: 10.1177/0271678X16680221
123. Montaner J, Alvarez-Sabin J, Molina CA, Angles A, Abilleira S, Arenillas J, et al. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke (2001) 32:2762–7.
124. Zhong C, Yang J, Xu T, Xu T, Peng Y, Wang A, et al. Serum matrix metalloproteinase-9 levels and prognosis of acute ischemic stroke. Neurology (2017) 89:805–12. doi: 10.1212/WNL.0000000000004257
125. Tsuji K, Aoki T, Tejima E, Arai K, Lee SR, Atochin DN, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke (2005) 36:1954–9. doi: 10.1161/01.STR.0000177517.01203.eb
126. Lucivero V, Prontera M, Mezzapesa DM, Petruzzellis M, Sancilio M, Tinelli A, et al. Different roles of matrix metalloproteinases-2 and−9 after human ischaemic stroke. Neurol Sci. (2007) 28:165–70. doi: 10.1007/s10072-007-0814-0
127. Chelluboina B, Klopfenstein JD, Pinson DM, Wang DZ, Vemuganti R, Veeravalli KK. Matrix Metalloproteinase-12 Induces Blood-Brain Barrier Damage After Focal Cerebral Ischemia. Stroke (2015) 46:3523–31. doi: 10.1161/STROKEAHA.115.011031
128. Chelluboina B, Warhekar A, Dillard M, Klopfenstein JD, Pinson DM, Wang DZ, et al. Post-transcriptional inactivation of matrix metalloproteinase-12 after focal cerebral ischemia attenuates brain damage. Sci Rep. (2015) 5:9504. doi: 10.1038/srep09504
129. Matsumoto S, Kobayashi T, Katoh M, Saito S, Ikeda Y, Kobori M, et al. Expression and localization of matrix metalloproteinase-12 in the aorta of cholesterol-fed rabbits: relationship to lesion development. Am J Pathol. (1998) 153:109–19.
130. Ma F, Rodriguez S, Buxo X, Morancho A, Riba-Llena I, Carrera A, et al. Plasma matrix metalloproteinases in patients with stroke during intensive rehabilitation therapy. Arch Phys Med Rehabil. (2016) 97:1832–40. doi: 10.1016/j.apmr.2016.06.007
131. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
132. Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med. (2017) 58:114–29. doi: 10.1016/j.mam.2017.03.005
133. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. (2017) 31:1273–88. doi: 10.1096/fj.201601222R
134. Arnardottir H, Orr SK, Dalli J, Serhan CN. Human milk proresolving mediators stimulate resolution of acute inflammation. Mucosal Immunol. (2016) 9:757–66. doi: 10.1038/mi.2015.99
135. Mirakaj V, Dalli J, Granja T, Rosenberger P, Serhan CN. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J Exp Med. (2014) 211:1037–48. doi: 10.1084/jem.20132103
136. Dalli J, Colas RA, Arnardottir H, Serhan CN. Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity (2017) 46:92–105. doi: 10.1016/j.immuni.2016.12.009
137. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. (2001) 2:612–9. doi: 10.1038/89759
138. Serhan CN, Chiang N. Novel endogenous small molecules as the checkpoint controllers in inflammation and resolution: entree for resoleomics. Rheum Dis Clin North Am. (2004) 30:69–95. doi: 10.1016/S0889-857X(03)00117-0
139. Petasis NA, Keledjian R, Sun YP, Nagulapalli KC, Tjonahen E, Yang R, et al. Design and synthesis of benzo-lipoxin A4 analogs with enhanced stability and potent anti-inflammatory properties. Bioorg Med Chem Lett. (2008) 18:1382–7. doi: 10.1016/j.bmcl.2008.01.013
140. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. (2005) 174:4345–55. doi: 10.4049/jimmunol.174.7.4345
141. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. (2005) 201:713–22. doi: 10.1084/jem.20042031
142. Arita M, Clish CB, Serhan CN. The contributions of aspirin and microbial oxygenase to the biosynthesis of anti-inflammatory resolvins: novel oxygenase products from omega-3 polyunsaturated fatty acids. Biochem Biophys Res Commun. (2005) 338:149–57. doi: 10.1016/j.bbrc.2005.07.181
143. Tjonahen E, Oh SF, Siegelman J, Elangovan S, Percarpio KB, Hong S, et al. Resolvin E2: identification and anti-inflammatory actions: pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem Biol. (2006) 13:1193–202. doi: 10.1016/j.chembiol.2006.09.011
144. Isobe Y, Arita M, Matsueda S, Iwamoto R, Fujihara T, Nakanishi H, et al. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J Biol Chem. (2012) 287:10525–34. doi: 10.1074/jbc.M112.340612
145. Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. (2007) 282:9323–34. doi: 10.1074/jbc.M609212200
146. Winkler JW, Orr SK, Dalli J, Cheng CY, Sanger JM, Chiang N, et al. Resolvin D4 stereoassignment and its novel actions in host protection and bacterial clearance. Sci Rep. (2016) 6:18972. doi: 10.1038/srep18972
147. Dalli J, Winkler JW, Colas RA, Arnardottir H, Cheng CY, Chiang N, et al. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem Biol. (2013) 20:188–201. doi: 10.1016/j.chembiol.2012.11.010
148. Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, et al. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. (2006) 176:1848–59. doi: 10.4049/jimmunol.176.3.1848
149. Shinohara M, Mirakaj V, Serhan CN. Functional Metabolomics Reveals Novel Active Products in the DHA Metabolome. Front Immunol. (2012) 3:81. doi: 10.3389/fimmu.2012.00081
150. Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. (2003) 278:43807–17. doi: 10.1074/jbc.M305841200
151. Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol. (2006) 177:5902–11. doi: 10.4049/jimmunol.177.9.5902
152. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, et al. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol. (2015) 84:24–35. doi: 10.1016/j.yjmcc.2015.04.003
153. Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med. (2015) 21:1071–5. doi: 10.1038/nm.3911
154. Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Villela CG, Fierro IM. Novel lipid mediator aspirin-triggered lipoxin A4 induces heme oxygenase-1 in endothelial cells. Am J Physiol Cell Physiol. (2005) 289:C557–563. doi: 10.1152/ajpcell.00045.2005
155. Souza MC, Padua TA, Torres ND, Souza Costa MF, Candea AP, Maramaldo T, et al. Lipoxin A4 attenuates endothelial dysfunction during experimental cerebral malaria. Int Immunopharmacol. (2015) 24:400–7. doi: 10.1016/j.intimp.2014.12.033
156. Brezinski ME, Gimbrone MAJr, Nicolaou KC, Serhan CN. Lipoxins stimulate prostacyclin generation by human endothelial cells. FEBS Lett. (1989) 245:167–72.
157. Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Fierro IM. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb Haemost. (2007) 97:88–98. doi: 10.1160/TH06-06-0315
158. Wu Y, Wang YP, Guo P, Ye XH, Wang J, Yuan SY, et al. A lipoxin A4 analog ameliorates blood-brain barrier dysfunction and reduces MMP-9 expression in a rat model of focal cerebral ischemia-reperfusion injury. J Mol Neurosci. (2012) 46:483–91. doi: 10.1007/s12031-011-9620-5
159. Zhang X, Wang T, Gui P, Yao C, Sun W, Wang L, et al. Resolvin D1 reverts lipopolysaccharide-induced TJ proteins disruption and the increase of cellular permeability by regulating IkappaBalpha signaling in human vascular endothelial cells. Oxid Med Cell Longev. (2013) 2013:185715. doi: 10.1155/2013/185715
160. Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. (2013) 6:256–66. doi: 10.1038/mi.2012.66
161. Gavins FN, Yona S, Kamal AM, Flower RJ, Perretti M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood (2003) 101:4140–7. doi: 10.1182/blood-2002-11-3411
162. Sun YP, Tjonahen E, Keledjian R, Zhu M, Yang R, Recchiuti A, et al. Anti-inflammatory and pro-resolving properties of benzo-lipoxin A(4) analogs. Prostaglandins Leukot Essent Fatty Acids (2009) 81:357–66. doi: 10.1016/j.plefa.2009.09.004
163. Chiang N, Gronert K, Clish CB, O'Brien JA, Freeman MW, Serhan CN. Leukotriene B4 receptor transgenic mice reveal novel protective roles for lipoxins and aspirin-triggered lipoxins in reperfusion. J Clin Invest. (1999) 104:309–16. doi: 10.1172/JCI7016
164. Bannenberg G, Moussignac RL, Gronert K, Devchand PR, Schmidt BA, Guilford WJ, et al. Lipoxins and novel 15-epi-lipoxin analogs display potent anti-inflammatory actions after oral administration. Br J Pharmacol. (2004) 143:43–52. doi: 10.1038/sj.bjp.0705912
165. Kasuga K, Yang R, Porter TF, Agrawal N, Petasis NA, Irimia D, et al. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J Immunol. (2008) 181:8677–87. doi: 10.4049/jimmunol.181.12.8677
166. Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med. (2015) 212:1203–17. doi: 10.1084/jem.20150225
167. Herrera BS, Hasturk H, Kantarci A, Freire MO, Nguyen O, Kansal S, et al. Impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infect Immun. (2015) 83:792–801. doi: 10.1128/IAI.02444-14
168. Hassan IR, Gronert K. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J Immunol. (2009) 182:3223–32. doi: 10.4049/jimmunol.0802064
169. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. (2013) 113:1004–12. doi: 10.1161/CIRCRESAHA.113.301198
170. Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, et al. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation (2016) 133:826–39. doi: 10.1161/CIRCULATIONAHA.115.020857
171. Smith HK, Gil CD, Oliani SM, Gavins FN. Targeting formyl peptide receptor 2 reduces leukocyte-endothelial interactions in a murine model of stroke. FASEB J. (2015) 29:2161–71. doi: 10.1096/fj.14-263160
172. Lee HN, Surh YJ. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem Pharmacol. (2013) 86:759–69. doi: 10.1016/j.bcp.2013.07.002
173. Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood (2012) 120:e60–72. doi: 10.1182/blood-2012-04-423525
174. Schmid M, Gemperle C, Rimann N, Hersberger M. Resolvin D1 polarizes primary human macrophages toward a proresolution phenotype through GPR32. J Immunol. (2016) 196:3429–37. doi: 10.4049/jimmunol.1501701
175. Kang JW, Lee SM. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim Biophys Acta (2016) 1861(9 Pt A), 1025–35. doi: 10.1016/j.bbalip.2016.06.002
176. Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol. (2011) 187:5408–18. doi: 10.4049/jimmunol.1100225
177. Akagi D, Chen M, Toy R, Chatterjee A, Conte MS. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. (2015) 29:2504–13. doi: 10.1096/fj.14-265363
178. Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrom JZ, Petasis NA, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. (2013) 27:2573–83. doi: 10.1096/fj.13-227728
179. Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, et al. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. (2007) 179:8533–43. doi: 10.4049/jimmunol.179.12.8533
180. Leonard MO, Hannan K, Burne MJ, Lappin DW, Doran P, Coleman P, et al. 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J Am Soc Nephrol. (2002) 13:1657–62. doi: 10.1097/01.ASN.0000015795.74094.91
181. Kieran NE, Doran PP, Connolly SB, Greenan MC, Higgins DF, Leonard M, et al. Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. (2003) 64:480–92. doi: 10.1046/j.1523-1755.2003.00106.x
182. Liu ZQ, Zhang HB, Wang J, Xia LJ, Zhang W. Lipoxin A4 ameliorates ischemia/reperfusion induced spinal cord injury in rabbit model. Int J Clin Exp Med. (2015) 8:12826–33.
183. Zhao Q, Hu X, Shao L, Wu G, Du J, Xia J. LipoxinA4 attenuates myocardial ischemia reperfusion injury via a mechanism related to downregulation of GRP-78 and caspase-12 in rats. Heart Vessels (2014) 29:667–78. doi: 10.1007/s00380-013-0418-y
184. Han X, Yao W, Liu Z, Li H, Zhang ZJ, Hei Z, et al. Lipoxin A4 preconditioning attenuates intestinal ischemia reperfusion injury through Keap1/Nrf2 pathway in a lipoxin A4 receptor independent manner. Oxid Med Cell Longev. (2016) 2016:9303606. doi: 10.1155/2016/9303606
185. Wu L, Li HH, Wu Q, Miao S, Liu ZJ, Wu P, et al. Lipoxin A4 activates Nrf2 pathway and ameliorates cell damage in cultured cortical astrocytes exposed to oxygen-glucose deprivation/reperfusion insults. J Mol Neurosci. (2015) 56:848–57. doi: 10.1007/s12031-015-0525-6
186. Sobrado M, Pereira MP, Ballesteros I, Hurtado O, Fernandez-Lopez D, Pradillo JM, et al. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARgamma-dependent, neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci. (2009) 29:3875–84. doi: 10.1523/JNEUROSCI.5529-08.2009
187. Sun Q, Wu Y, Zhao F, Wang J. Maresin 1 ameliorates lung ischemia/reperfusion injury by suppressing oxidative stress via activation of the Nrf-2-mediated HO-1 signaling pathway. Oxid Med Cell Longev. (2017) 2017:9634803. doi: 10.1155/2017/9634803
188. Zhao Q, Wu J, Hua Q, Lin Z, Ye L, Zhang W, et al. Resolvin D1 mitigates energy metabolism disorder after ischemia-reperfusion of the rat lung. J Transl Med. (2016) 14:81. doi: 10.1186/s12967-016-0835-7
189. Zhao Q, Shao L, Hu X, Wu G, Du J, Xia J, et al. Lipoxin a4 preconditioning and postconditioning protect myocardial ischemia/reperfusion injury in rats. Mediators Inflamm. (2013) 2013:231351. doi: 10.1155/2013/231351
190. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi: 10.1038/nature13479
191. Vital SA, Becker F, Holloway PM, Russell J, Perretti M, Granger DN, et al. Formyl-peptide receptor 2/3/lipoxin A4 receptor regulates neutrophil-platelet aggregation and attenuates cerebral inflammation: impact for therapy in cardiovascular disease. Circulation (2016) 133:2169–79. doi: 10.1161/CIRCULATIONAHA.115.020633
192. Wu L, Miao S, Zou LB, Wu P, Hao H, Tang K, et al. Lipoxin A4 inhibits 5-lipoxygenase translocation and leukotrienes biosynthesis to exert a neuroprotective effect in cerebral ischemia/reperfusion injury. J Mol Neurosci. (2012) 48:185–200. doi: 10.1007/s12031-012-9807-4
193. Ye XH, Wu Y, Guo PP, Wang J, Yuan SY, Shang Y, et al. Lipoxin A4 analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Res. (2010) 1323:174–83. doi: 10.1016/j.brainres.2010.01.079
194. Wu L, Liu ZJ, Miao S, Zou LB, Cai L, Wu P, et al. Lipoxin A4 ameliorates cerebral ischaemia/reperfusion injury through upregulation of nuclear factor erythroid 2-related factor 2. Neurol Res. (2013) 35:968–75. doi: 10.1179/1743132813Y.0000000242
195. Brancaleone V, Gobbetti T, Cenac N, le Faouder P, Colom B, Flower RJ, et al. A vasculo-protective circuit centered on lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 operative in murine microcirculation. Blood (2013) 122:608–17. doi: 10.1182/blood-2013-04-496661
196. Peskar BM, Ehrlich K, Schuligoi R, Peskar BA. Role of lipoxygenases and the lipoxin A(4)/annexin 1 receptor in ischemia-reperfusion-induced gastric mucosal damage in rats. Pharmacology (2009) 84:294–9. doi: 10.1159/000244017
197. Gilbert K, Bernier J, Godbout R, Rousseau G. Resolvin D1, a metabolite of omega-3 polyunsaturated fatty acid, decreases post-myocardial infarct depression. Mar Drugs (2014) 12:5396–407. doi: 10.3390/md12115396
198. Keyes KT, Ye Y, Lin Y, Zhang C, Perez-Polo JR, Gjorstrup P, et al. Resolvin E1 protects the rat heart against reperfusion injury. Am J Physiol Heart Circ Physiol. (2010) 299:H153–164. doi: 10.1152/ajpheart.01057.2009
199. Zhang T, Shu HH, Chang L, Ye F, Xu KQ, Huang WQ. Resolvin D1 protects against hepatic ischemia/reperfusion injury in rats. Int Immunopharmacol. (2015) 28:322–7. doi: 10.1016/j.intimp.2015.06.017
200. Zhao Q, Wu J, Lin Z, Hua Q, Zhang W, Ye L, et al. Resolvin D1 alleviates the lung ischemia reperfusion injury via complement, immunoglobulin, TLR4, and inflammatory factors in rats. Inflammation (2016) 39:1319–33. doi: 10.1007/s10753-016-0364-9
201. Bazan NG, Eady TN, Khoutorova L, Atkins KD, Hong S, Lu Y, et al. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp Neurol. (2012) 236:122–30. doi: 10.1016/j.expneurol.2012.04.007
202. Eady TN, Belayev L, Khoutorova L, Atkins KD, Zhang C, Bazan NG. Docosahexaenoic acid signaling modulates cell survival in experimental ischemic stroke penumbra and initiates long-term repair in young and aged rats. PLoS ONE (2012) 7:e46151. doi: 10.1371/journal.pone.0046151
203. Belayev L, Mukherjee PK, Balaszczuk V, Calandria JM, Obenaus A, Khoutorova L, et al. Neuroprotectin D1 upregulates Iduna expression and provides protection in cellular uncompensated oxidative stress and in experimental ischemic stroke. Cell Death Differ. (2017) 24:1091–9. doi: 10.1038/cdd.2017.55
204. Xian W, Wu Y, Xiong W, Li L, Li T, Pan S, et al. The pro-resolving lipid mediator Maresin 1 protects against cerebral ischemia/reperfusion injury by attenuating the pro-inflammatory response. Biochem Biophys Res Commun. (2016) 472:175–81. doi: 10.1016/j.bbrc.2016.02.090
205. Dalli J, Chiang N, Serhan CN. Identification of 14-series sulfido-conjugated mediators that promote resolution of infection and organ protection. Proc Natl Acad Sci USA. (2014) 111:E4753–4761. doi: 10.1073/pnas.1415006111
206. Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. (2010) 24:4240–9. doi: 10.1096/fj.10-159913
207. Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature (2012) 484:524–8. doi: 10.1038/nature11042
208. Chiang N, de la Rosa X, Libreros S, Serhan CN. Novel resolvin D2 receptor axis in infectious inflammation. J Immunol. (2017) 198:842–51. doi: 10.4049/jimmunol.1601650
209. Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. Self-limited versus delayed resolution of acute inflammation: temporal regulation of pro-resolving mediators and microRNA. Sci Rep. (2012) 2:639. doi: 10.1038/srep00639
210. Li Y, Dalli J, Chiang N, Baron RM, Quintana C, Serhan CN. Plasticity of leukocytic exudates in resolving acute inflammation is regulated by MicroRNA and proresolving mediators. Immunity (2013) 39:885–98. doi: 10.1016/j.immuni.2013.10.011
211. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature (2007) 447:869–74. doi: 10.1038/nature05877
212. Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, Serhan CN. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem. (2010) 285:3451–61. doi: 10.1074/jbc.M109.044131
213. El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci USA. (2012) 109:14983–8. doi: 10.1073/pnas.1206641109
214. Herova M, Schmid M, Gemperle C, Hersberger M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J Immunol. (2015) 194:2330–7. doi: 10.4049/jimmunol.1402166
215. Sheets KG, Jun B, Zhou Y, Zhu M, Petasis NA, Gordon WC, et al. Microglial ramification and redistribution concomitant with the attenuation of choroidal neovascularization by neuroprotectin D1. Mol Vis. (2013) 19:1747–59.
216. Serhan CN, Fredman G, Yang R, Karamnov S, Belayev LS, Bazan NG, et al. Novel proresolving aspirin-triggered DHA pathway. Chem Biol. (2011) 18:976–87. doi: 10.1016/j.chembiol.2011.06.008
217. Lannan KL, Spinelli SL, Blumberg N, Phipps RP. Maresin 1 induces a novel pro-resolving phenotype in human platelets. J Thromb Haemost. (2017) 15:802–13. doi: 10.1111/jth.13620
218. Ramsden CE. Breathing easier with fish oil - a new approach to preventing asthma? N Engl J Med. (2016) 375:2596–8. doi: 10.1056/NEJMe1611723
219. Halade GV, Kain V, Black LM, Prabhu SD, Ingle KA. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging (2016) 8:2611–34. doi: 10.18632/aging.101077
220. Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R, et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol. (2010) 177:2116–23. doi: 10.2353/ajpath.2010.091082
221. Fredman G, Serhan CN. Specialized proresolving mediator targets for RvE1 and RvD1 in peripheral blood and mechanisms of resolution. Biochem J. (2011) 437:185–97. doi: 10.1042/BJ20110327
Keywords: cerebral ischemia reperfusion, inflammation, resolution, specialized pro-resolving lipid mediators, stroke
Citation: Yin P, Wei Y, Wang X, Zhu M and Feng J (2018) Roles of Specialized Pro-Resolving Lipid Mediators in Cerebral Ischemia Reperfusion Injury. Front. Neurol. 9:617. doi: 10.3389/fneur.2018.00617
Received: 27 March 2018; Accepted: 10 July 2018;
Published: 31 July 2018.
Edited by:
Pierangelo Geppetti, Università degli Studi di Firenze, ItalyReviewed by:
Domenico Pellegrini-Giampietro, Università degli Studi di Firenze, ItalyShekher Mohan, College of Pharmacy, Natural and Health Sciences, Manchester University, United States
Copyright © 2018 Yin, Wei, Wang, Zhu and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingqin Zhu, bWluZ3Fpbi56aHVAaG90bWFpbC5jb20=
Jiachun Feng, ZmVuZ2pjZnJhbmtAcXEuY29t