 Xue Zhang
Xue Zhang Fei Gao
Fei Gao Dongdong Wang
Dongdong Wang Jianmin Zhang
Jianmin Zhang- Department of Immunology, Research Center on Pediatric Development and Diseases, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and School of Basic Medicine, Peking Union Medical College, State Key Laboratory of Medical Molecular Biology, Beijing, China
Tau protein—a member of the microtubule-associated protein family—is a key protein involved in many neurodegenerative diseases. Tau pathology in neurodegenerative diseases is characterized by pathological tau aggregation in neurofibrillary tangles (NFTs). Diseases with this typical pathological feature are called tauopathies. Parkinson's disease (PD) was not initially considered to be a typical tauopathy. However, recent studies have demonstrated increasing evidence of tau pathology in PD. A genome-wide association (GWA) study indicated a potential association between tauopathy and sporadic PD. The aggregation and deposition of tau were also observed in ~50% of PD brains, and it seems to be transported from neuron to neuron. The aggregation of NFTs, the abnormal hyperphosphorylation of tau protein, and the interaction between tau and alpha-synuclein may all contribute to the cell death and poor axonal transport observed in PD and Parkinsonism.
Introduction
Tau protein is produced from a single human gene named microtubule-associated protein tau (MAPT), which is located on chromosome 17 and encodes a cytoskeletal protein that stabilizes microtubules (1). Although tau is widely distributed in neurons of the central nervous system (CNS), its levels in CNS astrocytes and oligodendrocytes are low (2). Tau proteins play a role in stabilizing microtubules, binding to membrane, and regulating axonal transport (3–5). Under physiological conditions, tau is highly soluble and unfolded. However, with changes in isoforms or phosphorylation patterns in pathological states, tau proteins become insoluble and misfolded, causing damage to neurons and axonal transport (6, 7). Protein misfolding, accumulation, and aggregation have been observed in many neurodegenerative diseases (8), which may contribute to neuron damage and neurological disorders. The pathological aggregation of tau or neurofibrillary tangles are known as tauopathy, an important hallmark of many human neurodegenerative disease, such as Alzheimer's disease (AD) and Parkinson's disease (PD) (9, 10).
Parkinson's disease, named after Dr. James Parkinson, is a major neurodegenerative disease that primarily affects motor systems but can also be accompanied by cognitive and behavioral problems (11). There is a widespread neuron degeneration in PD brains, affecting up to 70% of dopaminergic neurons in the substantia nigra (SN) by the time of death (12, 13). The neuropathological hallmarks of PD include Lewy bodies (LBs) in the SN, brainstem, and rostral and forebrain regions and the selective deletion of dopaminergic neurons in the SN (14, 15). Cell-death induced damage in SN may be the source of patient movement disorders. Although the causes of this cell death are generally unclear, researchers have observed an enrichment of tau protein and alpha-synuclein in neuronal Lewy bodies, which may be related to tauopathy in PD (16). Immunohistochemistry with anti-tau antibodies showed high level of NFTs in the substantia nigra from post-mortem human brain tissue (17). Researchers have also reported that tauopathies in PD and PD with dementia (PDD) were only observed in DA neurons of the nigrostriatal region, which contrasts with the wide-spread expression pattern of tau throughout the entire brain in AD (18).
Although tau pathology in AD and other tau-associated neurodegenerative diseases have been previously described, the importance of tau pathology in PD has been undervalued. Therefore, we reviewed the tau pathologies that might be involved in PD (Figure 1), seeking to identify tau as a potential therapeutic target.
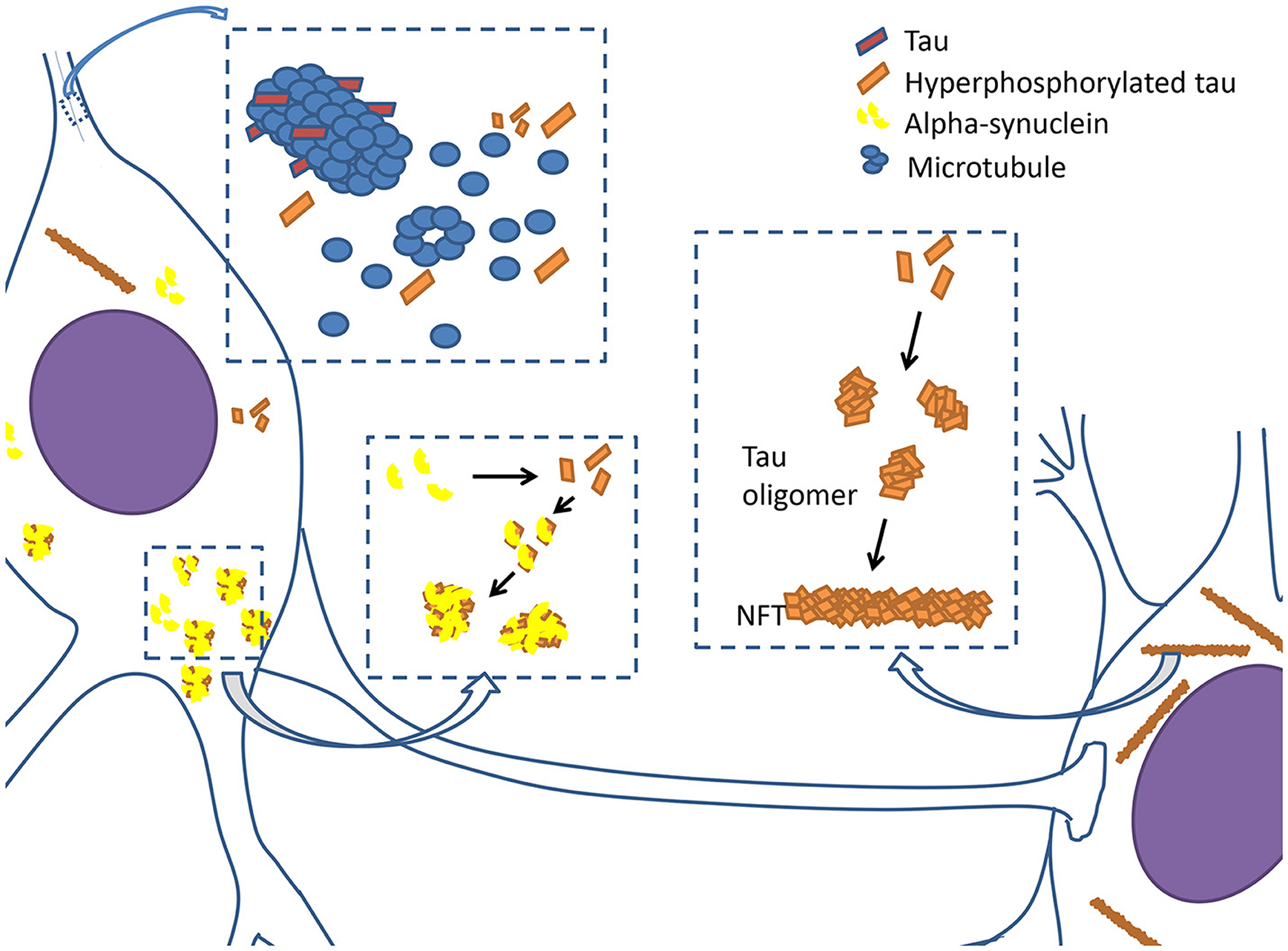
Figure 1. Tauopathy in PD. Tau proteins are integrated with microtubules and stabilize microtubules in physiological condition. However, when tau proteins are hyperphosphorylated, they begin to disintegrate from microtubules, causing neuronal dysfunction. Hyperphosphorylated tau proteins prone to assemble together to form oligomers and develop into filamentous neurofibrillary tangles (NFTs). Hyperphosphorylated tau proteins also interact with α-synuclein to promote aggregation and fibrilization each other, and subsequently cause the formation of lewy bodies and axonal transport dysfunction.
Assembled Tau in PD
Structure of Normal TAU
There are six different isoforms of tau protein in the human brain, with the differences among them resulting from alternative mRNA splicing of a single gene located on chromosome 17 (19). The microtubule-binding domains of the protein consist of adjoining sequence and repeat sequences. The six isoforms are divided into two categories based on the number of these repeats, namely, 3R and 4R. The 3R tau isoform has three repeats, while 4R tau has four repeats (20). Each of the repeats is able to bind to microtubules, and the more repeats the protein has, the stronger affinity it will have with them (21). Therefore, when compared with 3R tau, 4R tau is considerably easier to bundle with and polymerize microtubules. In different tauopathies, the pathological tau protein has different isoforms and conformations. Progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) are both associated with Parkinson's disease and are associated with 4R tau deposits in neurons and microglia (22). A study of a multigenerational family suffering from X-linked parkinsonian syndrome also showed a strong 4R tauopathy in the striatum (23).
Assembled TAU in PD and Other Neurodegenerative Diseases
Tau protein is soluble and unfolded under physiological conditions; however, in many neurodegenerative diseases, tau appears to be insoluble and assembled (24). The most probable mechanism of tau assembly involves mutations of the microtubule-associated protein tau (MAPT). MAPT is a single gene located on chromosome 17q21, containing 16 exons (25). A genome-wide association (GWA) study for sporadic PD cases in Europe confirmed that MAPT is closely linked to sporadic PD (26). MAPT is divided into two haplogroups called H1 and H2 based on whether the gene is in the inverted orientation (27). A meta-analysis in Caucasian populations reported that the H2 haplotype is more relevant to PD than the H1 haplotype, as the risk of suffering from PD is lower in Caucasians with the H2 haplotype (28). Certain FTDP-17 mutations, including missense mutations, deletions in the coding region, and intronic mutations, result in tau aggregation. This aggregation can cause dominantly inherited frontotemporal dementia and Parkinsonism linked to chromosome 17 (29). Most missense mutations in the coding region tend to affect how well tau can associate with microtubules (30). However, studies also show that some missense mutations such as G272V, P301L, and P301S may play an important role in filament assembly because they markedly facilitated the propensity for tau to assemble (31). If the mutation is in the repeat region or if changes in the relative amounts of 3R tau or 4R tau could lead to overproduction of 4R tau, the filament morphology tends to be incorrectly over-folded (32). Tau deposition and assembled filaments are observed in many neurodegenerative diseases and are considered a typical neuropathological hallmark. Significantly lower levels of soluble tau and a lower 3R-tau to 4R-tau ratio has been shown in the SN of patients with PD (33), indicating tauopathy similarity between PD and AD. PSP and CBD are subtypes of Parkinson's disease known as Parkinson-plus syndromes, and both are associated with the formation of tau deposits. Filamentous tau deposits can be observed in neurons and microglia in these diseases (34).
Prion-Like Pathological TAU Spreading in Animal Models and Patients With PD
Increasing evidence shows that tau aggregation and deposition contribute to PD pathology. Thus, to best understand the mechanisms underlying PD pathogenesis, early diagnosis, and treatment, determining how tau aggregation spreads to other areas is imperative. Researchers have observed Lewy bodies in grafted neurons that patients with PD received as transplants (35). The assumption was that neurofibrillary lesions spread along the neuronal pathways in the brain. Recent evidence shows that misfolded tau can move from cell to cell, similar to prion disease (36). Clavaguera and other researchers inserted a mutant human tau transgene into mice to show that human tau can be transported from neuron to neuron (37). Another study has shown that the spreading of the tau inclusions depended on the initial injection site of synthetic tau fibrils. The pathological tau were more likely to spread through functionally connected neuroanatomical pathways rather than through adjacent anatomical locations (38).
After showing prion-like transmission and spreading of tauopathy by injecting pathological tau from the human brain into transgenic ALZ17 mice, researchers assessed the role that different tau strains play in this pathological process. By separately injecting human brain homogenates from patients who suffered from argyrophilic grain disease (AGD), PSP, and CBD into different ALZ17 mice, researchers demonstrated that the different tau isoforms may induce different tauopathies. Mice receiving CBD or AGD tau differentially displayed silver-positive or silver-negative astrocytic plaques that matched the injection patterns and that were highly similar to the types of tau-related pathological damage typically found in the brains of patients suffering from the respective diseases (39). Furthermore, 12 months after injecting ALZ17 mice with brain homogenate from mice that had been injected with the human tau P301S transgene 18 months earlier, the ALZ17 mice showed fewer tau inclusions than those of mice that had been injected with AGD brain extracts (40). Similar phenomena were observed in mice injected with filament 4R-tau strains formed in HEK293T cells (41). Further, in patients with PD who received cell-replacement therapy to repair brain damage, hyperphosphorylated tau such as phospho-tau Ser202 and Thr205 were found in grafted neurons years after transplantation (42). Taken together, these findings demonstrate tau strain-specific prion-like transmission and spreading in the disease state, including in PD. Additionally, the specific strain plays an important role in causing distinct pathologies.
Abnormal Hyperphosphorylation of the TAU Protein
Hyperphosphorylation of the tau protein is another mechanism through which tau might accumulate and form filaments, which might also influence the ability of tau to bind to microtubules, possibly limiting how microtubules can be combined and resulting in their aggregation into NFTs (43). In this way, the microtubules might disintegrate, eventually leading to the impaired transport capacity of axon microtubules. Tau protein appears to be easily phosphorylated because of its 85 potential phosphorylation sites, and researchers have characterized over 20 kinases that may be related to the phosphorylation of tau protein after its transcription (44). With respect to the healthy human brain, there are only two or three phosphorylated amino acid residues in tau protein, while there may be considerably more in brains exhibiting tauopathy (45). Additional research indicates that the most likely mechanism underlying the hyperphosphorylation is either upregulated protein-kinase activity or downregulated protein-phosphatase activity (46). Among protein kinases, GSK-3β (a proline-directed protein kinases) and CDK5 (a non-PDPK non-proline-directed protein kinase) are probably the two most important kinases in tauopathy. Using neuronal stem/progenitor cells and transgenic mice, researchers have demonstrated a pivotal role for GSK-3β in the interaction between DA neurons and astrocytes during damage and recovery (47), which might be related to the death of DA neurons in PD. Further, the application of CDK5 in cortex suffering from Lewy body disease was reported in 2000, indicating that CDK5 may participate in the formation of Lewy bodies (48). In contrast, unlike GSK-3β and CDK5, a series of protein phosphatases (PP-2A, PP-2B, and PP-1) can dephosphorylate protein tau in vitro and in vivo, which may act to protect against tauopathy (49). Reduced activity levels of PP-2A in the brains of patients with PD and AD indirectly confirmed this inference (50, 51).
Hyperphosphorylation of tau protein is an important step in tau aggregation and the formation of neurofibrillary tangles (52). Antibodies targeting p-taus were able to detect tau isoforms in brain tissue suffering from sporadic PD or dementia with Lewy bodies, indicating the existence of hyperphosphorylated tau protein in NFTs (53). Tau aggregation more easily begins from the C-terminus of the protein (54). Therefore, kinases that phosphorylate at the C-terminus might be crucial for the formation of tau filaments and aggregates. For example, an in vitro study indicated that the GSK-3β associated with the phosphorylation of tau at the C-terminal had an ability to promote the fibrillation of the protein, while the level of microtubule assembly stayed low due to DYRK1A (55). A study on the structure and dynamics of phosphorylated tau filaments using computer simulations indicated that the all the masses and charges had changed because of phosphorylation at regions associated with microtubules, resulting in further aggregation of tau (56).
Phosphorylated tau is also proved to be related to the N-methyl-D-aspartic receptor (NMDAR) at postsynaptic sites (57), which suggests that the toxic pathology of tau phosphorylation is associated with the synapse. Meanwhile, the FTDP-17 tau mutant, which is known to be associated with PDD, was shown to interfere with synaptic vesicles in presynaptic terminals, causing the dysfunction of vesicle traffic and presynaptic activity (58). There are also a number of studies showing that the hyperphosphorylation of tau protein may depolymerize microtubules, causing their dysfunction, impaired axonal transport, and ultimately cell death (59). Okadaic acid, an inhibitor of tau phosphorylation, was used to investigate the synaptic structure of neurons in rats (60). This study highlights the potential relationship between phosphorylated tau and the loss of synaptic function. Similar results have been shown in animal models. Transgenic mice expressing human tau P301L show Parkinsonism as early as 6.5 months (61), while a similar phenotype occurs in the K3 mouse model that expresses human tau with the K396I mutation. In this latter case, Parkinsonism symptoms can be improved using L-dopa (62).
P-TAU Associated With Alpha-Synuclein Leads to Toxic Injury in PD
One of the key proteins involved in PD pathology is alpha-synuclein, a highly soluble neuronal cytoplasmic protein that is localized to presynaptic elements in the CNS (63). Under certain conditions, such as missense mutations, post-translational modifications (e.g., phosphorylation and C-terminal truncation), and peroxynitrite stimulation, alpha-synuclein is prone to being fibrillated and to residing in Lewy bodies with other proteins (64), which is a feature of PD that occurs along with Lewy neurites.
Researchers found that tau protein, especially phosphorylated tau, existed in Lewy bodies along with alpha-synuclein and that neurofibrillary tangles could be observed around Lewy bodies (53, 65, 66). This phenomenon led researchers to hypothesize a positive interaction between tau and alpha-synuclein. A transgenic mouse model of PD showed increased levels of p-tau and the co-localization and overexpression of alpha-synuclein and p-tau, which were deposited in large inclusion bodies that are considered similar to Lewy bodies in PD (67). A series of experiments in vitro indicate that tau incubated with synthesized alpha-synuclein oligomers can induce all forms of tau, including the assembly of toxic tau forms (68). Furthermore, studies using QBI293 cells demonstrated that alpha-synuclein induces tangles of tau and promotes phosphorylation of tau in cells (69). Reports also indicate that the nucleus of neurons were surrounded by alpha-synuclein and human tau with the P301L mutation, which may be to blame for the loss of neuronal function (70). Researchers have been able to successfully detect the aggregation of the two proteins in brains of patients with PD using two novel antibodies specific to oligomeric tau and alpha-synuclein (71). Similar results were shown in a transgenic mouse model that was inoculated with alpha-synuclein supplied from preformed fibrils, indicating that the existence of both alpha-synuclein and tau promotes fibrillation, and this phenomenon is also confirmed in human brain (72).
Studies on the genetics of brain tissue indicate that an interaction between tau and alpha-synuclein in (PDD) with Lewy bodies (73).
As mentioned above, specific protein kinases may hyperphosphorylate tau protein at certain sites, causing toxic isoforms of tau. Among these kinases, protein kinases A can be stimulated by alpha-synuclein, resulting in tau phosphorylation at Ser262/356 (74). Studies focused on GSK-3β, which is associated with the toxic p-tau isoform in AD, also indicate that there may be an interaction between alpha-synuclein and accumulated p-tau (75). Another study using transgenic mice that overexpressed or lacked alpha-synuclein demonstrated that alpha-synuclein is indispensable for the activation of GSK-3β. A co-IP experiment in SH-SY5Y cells also demonstrated the existence of an alpha-synuclein, p-tau, and p-GSK-3β (76) complex. Furthermore, researchers have developed a mouse model with a S9A-point mutation of human GSK-3β to investigate the relationship between alpha-synuclein and p-tau, showing a positive association between the two proteins in vitro, as well as in behavioral, and biochemical experiments (77, 78). Additionally, p-GSK-3β-Y216, the kinase-active form of GSK-3β, is co-localized with both p-tau and alpha-synuclein and is broadly expressed in the whole brain, while p-tau, and alpha-synuclein are expressed in TH+ DA neurons of the midbrain (78).
After showing the positive relationship between p-tau and alpha-synuclein, researchers are still investigating the mechanism underlying the toxic interaction between the two proteins. Several studies indicate that neurotoxic MPP+ induces the abnormal hyperphosphorylation of tau along with alpha-synuclein in vitro (79) and in vivo (80). Studies in a drosophila model demonstrated that tau interacting with alpha-synuclein ruined the organization of the cytoskeleton, leaving low-functioning axonal transport and structural abnormalities in neuronal synapses that resulted in PD-related cell death (81). However, the precise relationship between tau and alpha-synuclein and the molecular mechanisms responsible for PD are still unclear. There might be a cascade reaction in which the accumulation of alpha-synuclein in synapses recruits tau (82) and induces damage; the resulting low-functioning axonal transport will further promote the accumulation of tau and alpha-synuclein, and therefore, more fibrillation will be present in neurons, which will eventually lead to cell death.
Conclusions
Tau is a key protein in many neurodegenerative diseases; however, its importance has been underestimated preoperatively in PD and PDD. Soluble, unfolded tau, after being phosphorylated or mutated, becomes insoluble and misfolded, resulting in conformational changes in microtubules and the aggregation of NFTs. The mobility of abnormal tau through brain tissue in PD is similar to prion-like diseases. The accumulation of hyperphosphorylated tau also affects axonal transport and appears to work with alpha-synuclein to contribute to tauopathy in PD and AD.
Although there is no effective treatment or drug therapy for PD and other similar neurodegenerative diseases, understanding the structure, function, and mechanism of tau and tau pathology might be helpful for early diagnosis and treatment of PD in the future.
Author Contributions
XZ and FG wrote the manuscript. DW, CL, and YF helped to edit the manuscript. JZ and WH edited the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31471016), the CAMS Initiative for Innovative Medicine (2016-I2 M-1-008), and the National Key Research and Development Program of China (2016YFA0101001).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Shaw-Smith C, Pittman A, Willatt L, Martin H, Rickman L, Gribble S, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. (2006) 38:1032–7. doi: 10.1038/ng1858
2. Shin RW, Iwaki T, Kitamoto T, Tateishi J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer's disease brain tissues. Lab Invest. (1991) 64:693–702.
3. Gauthierkemper A, Weissmann C, Golovyashkina N, Sebölemke Z, Drewes G, Gerke V, et al. The frontotemporal dementia mutation R406W blocks tau's interaction with the membrane in an annexin A2–dependent manner. J Cell Biol. (2011) 192:647–61. doi: 10.1083/jcb.201007161
4. Chen J, Kanai Y, Cowan NJ, Hirokawa N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature (1992) 360:674–7. doi: 10.1038/360674a0
5. Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. (1995) 131:1327–40. doi: 10.1083/jcb.131.5.1327
6. Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. Natl Acad Sci USA. (2001) 98:6923–8. doi: 10.1073/pnas.121119298
7. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del TK. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. (2006) 112:389–404. doi: 10.1007/s00401-006-0127-z
8. Jones DR, Moussaud S, McLean P. Targeting heat shock proteins to modulate α-synuclein toxicity. TherAdv Neurol Disord. (2014) 7:33. doi: 10.1177/1756285613493469
9. Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick's disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. (2001) 101:167–73.
10. Armstrong RA. Cairns NJ. Spatial patterns of the tau pathology in progressive supranuclear palsy. Neurol Sci. (2013) 34:337–44. doi: 10.1007/s10072-012-1006-0
11. Jenner P, Morris HR, Robbins TW, Goedert M, Hardy J, Benshlomo Y, et al. Parkinson's disease – the debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J Parkinsons Dis. (2013) 3:1. doi: 10.3233/JPD-130175
12. Cookson MR. THE BIOCHEMISTRY OF PARKINSON'S DISEASE*. Ann Rev Biochem. (2005) 74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400
13. Davie CA. A review of Parkinson's disease. Br Med Bull. (2008) 86:109–27. doi: 10.1093/bmb/ldn013
14. Braak, H. Braak E. Pathoanatomy of Parkinson's disease. J Neurol. (2000) 247:II3–II10. doi: 10.1007/PL00007758
15. Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson's disease. Neurosci Lett. (1997) 239:45–8. doi: 10.1016/S0304-3940(97)00891-4
16. Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. (2003) 62:389–97. doi: 10.1093/jnen/62.4.389
17. Schneider JA, Li JL, Li Y, Wilson RS, Kordower JH, Bennett DA. Substantia nigra tangles are related to gait impairment in older persons. Digest World Core Med J. (2006) 59:166–73. doi: 10.1002/ana.20723
18. Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson's disease brains with and without dementia. Exp Neurol. (2010) 225:210–8. doi: 10.1016/j.expneurol.2010.06.017
19. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. (1989) 3:519.
20. Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, et al. The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol. (2003) 106:323–36. doi: 10.1007/s00401-003-0734-x
21. Kumaran R, Kingsbury A, Coulter I, Lashley T, Williams D, de Silva R, et al. DJ-1 (PARK7) is associated with 3R and 4R tau neuronal and glial inclusions in neurodegenerative disorders. Neurobiol Dis. (2007) 28:122. doi: 10.1016/j.nbd.2007.07.012
22. Flament S, Delacourte A, Verny M, Hauw JJ, Javoyagid F. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. (1991) 81:591–6. doi: 10.1007/BF00296367
23. Poorkaj P, Raskind WH, Leverenz JB, Matsushita M, Zabetian CP, Samii A, et al. A novel X-linked 4-repeat tauopathy with parkinsonism and spasticity. Move Disord. (2010) 25:1409–17. doi: 10.1002/mds.23085.
24. King ME, Gamblin TC, Kuret J, Binder LI. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. (2000) 74:1749–57. doi: 10.1046/j.1471–4159.2000.0741749.x
25. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. (1988) 85:4051–5. doi: 10.1073/pnas.85.11.4051
26. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93. doi: 10.1038/ng.3043
27. Almos PZ, Horváth S, Czibula A, Raskó I, Sipos B, Bihari P, et al. H1 tau haplotype-related genomic variation at 17q21.3 as an Asian heritage of the European Gypsy population. Heredity (2008) 101:416–9. doi: 10.1038/hdy.2008.70
28. Zhang CC, Zhu JX, Wan Y, Tan L, Wang HF, Yu JT, et al. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget (2017) 8:44994–5007. doi: 10.18632/oncotarget.16690
29. Goedert, M. Spillantini MG. Tau mutations in frontotemporal dementia FTDP-17 and their relevance for Alzheimer's disease. Biochimica Et Biophysica Acta (2000) 1502:110–21.
30. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann Rev Neurosci. (2001) 24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121
31. Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. Febs Lett. (1999) 447:195.
32. Peck A, Sargin ME, Lapointe NE, Rose K, Manjunath BS, Feinstein SC, et al. Tau isoform-specific modulation of kinesin-driven microtubule gliding rates and trajectories as determined with tau-stabilized microtubules. Cytoskeleton (2011) 68:44–55. doi: 10.1002/cm.20494
33. Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. (2012) 18:291–5. doi: 10.1038/nm.2613
34. Ouchi H, Toyoshima Y, Tada M, Oyake M, Aida I, Tomita I, et al. Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Move Disord. (2014) 29:238–44. doi: 10.1002/mds.25746
35. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. (2008) 14:501–3. doi: 10.1038/nm1746
36. Clavaguera F, Tolnay M, Goedert M. The prion-like behavior of assembled Tau in transgenic mice. Cold Spring Harb Perspect Med. (2017) 7:a024372. doi: 10.1101/cshperspect.a024372
37. Lasagnareeves CA, Castillocarranza DL, Sengupta U, Guerreromunoz MJ, Kiritoshi T, Neugebauer V, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. (2012) 2:700. doi: 10.1038/srep00700
38. Stancu IC, Vasconcelos B, Ris L, Wang P, Villers A, Peeraer E, et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. (2015) 129:875–94. doi: 10.1007/s00401-015-1413-4
39. Tolnay, M. Clavaguera F. Argyrophilic grain disease: a late-onset dementia with distinctive features among tauopathies. Neuropathology (2004) 24:269–83. doi: 10.1111/j.1440-1789.2004.00591.x
40. Clavaguera, F, Akatsu, H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA. (2013) 110:9535–40. doi: 10.1073/pnas.1301175110
41. Sanders DW, Kaufman SK, Devos SL, Sharma AM, Mirbaha H, A. Li, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron (2014) 82:1271–88. doi: 10.1016/j.neuron.2014.04.047
42. Cisbani G, Maxan A, Kordower JH, Planel E, Freeman TB, Cicchetti F. Presence of tau pathology within foetal neural allografts in patients with Huntington's and Parkinson's disease. Brain A J Neurol. (2017) 140:2982–92. doi: 10.1093/brain/awx255
43. Chohan MO, Haque N, Alonso A, Elakkad E, Grundkeiqbal I, Grover A, et al. Hyperphosphorylation-induced self assembly of murine tau: a comparison with human tau. J Neural Trans. (2005) 112:1035–47. doi: 10.1007/s00702-004-0241-9
44. Duka V, Lee JH, Credle J, Wills J, Oaks A, Smolinsky C, et al. Identification of the sites of tau hyperphosphorylation and activation of tau kinases in synucleinopathies and Alzheimer's diseases. PLoS ONE (2013) 8:e75025. doi: 10.1371/journal.pone.0075025
45. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. (2009) 15:112–9. doi: 10.1016/j.molmed.2009.01.003
46. Noble DW, Pooler AM, Hanger DP. Advances in tau-based drug discovery. Expert Opin Drug Discovery (2011) 6:797–810. doi: 10.1517/17460441.2011.586690
47. L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Serapide MF, et al. Wnt/beta-catenin signaling is required to rescue midbrain dopaminergic progenitors and promote neurorepair in ageing mouse model of Parkinson's disease. Stem Cells (2014) 32:2147–63. doi: 10.1002/stem.1708
48. Takahashi M, Iseki E, Kosaka K. Cyclin-dependent kinase 5 (Cdk5) associated with Lewy bodies in diffuse Lewy body disease. Brain Res. (2000) 862:253–6.
49. Sun XY, Wei YP, Xiong Y, Wang XC, Xie AJ, Wang XL, et al. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J Biol Chem. (2012) 287:11174–82. doi: 10.1074/jbc.M111.309070
50. Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. (2005) 22:1942–5. doi: 10.1111/j.1460-9568.2005.04391.x
51. Park HJ, Lee KW, Park ES, S. Oh, Yan R, Zhang J, et al. Dysregulation of protein phosphatase 2A in parkinson disease and dementia with lewy bodies. Ann Clin Transl Neurol. (2016) 3:769–80. doi: 10.1002/acn3.337
52. Alonso AC, Grundkeiqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. (1996) 2:783–7.
53. Arima K, Hirai S, Sunohara N, Aoto K, Izumiyama Y, Uéda K, et al. Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in lewy bodies in sporadic Parkinson's disease and in dementia with Lewy bodies. Brain Res. (1999) 843:53–61.
54. Yanagawa H, Chung SH, Ogawa Y, Sato K, Shibata-Seki T, Masai J, et al. Protein anatomy: C-tail region of human tau protein as a crucial structural element in Alzheimer's paired helical filament formation in vitro. Biochemistry (1998) 37:1979–88. doi: 10.1021/bi9724265
55. Liu F, B. Li, Tung EJ, Grundkeiqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci (2007) 26:3429–36.doi: 10.1111/j.1460-9568.2007.05955.x
56. Xu L, Zheng J, Margittai M, Nussinov R, Ma B. How does hyperphopsphorylation promote tau aggregation and modulate filament structure and stability? Acs Chem Neurosci. (2016) 7:565–75. doi: 10.1021/acschemneuro.5b00294
57. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, Van EJ, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell (2010) 142:387–97. doi: 10.1016/j.cell.2010.06.036
58. Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun. (2017) 8:1–13. doi: 10.1038/ncomms15295
59. Jaworski T, Kügler S, Leuven FV. Modeling of tau-mediated synaptic and neuronal degeneration in alzheimer's disease. Int J Alzheimer's Dis. (2010) 2010:573138. doi: 10.4061/2010/573138
60. Malchiodi-Albedi F, Petrucci TC, Picconi B, Iosi F, Falchi M. Protein phosphatase inhibitors induce modification of synapse structure and tau hyperphosphorylation in cultured rat hippocampal neurons. J Neurosci Res. (2015) 48:425–38. doi: 10.1002/(SICI)1097-4547(19970601)48:5<425::AID-JNR4>3.0.CO;2-G
61. Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van SM, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. (2000) 25:402–5. doi: 10.1038/78078
62. Ittner LM, Fath T, Ke YD, Bi M, Eersel JV, Li KM, et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci USA. (2008) 105:15997–6002. doi: 10.1073/pnas.0808084105
63. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-Synuclein locus triplication causes Parkinson's disease. Science (2003) 302:841.doi: 10.1126/science.1090278
64. Engelender S. Ubiquitination of alpha-synuclein and autophagy in Parkinson's disease. Autophagy (2008) 4:372–4.
65. Yamaguchi K, Cochran EJ, Murrell JR, Polymeropoulos MH, Shannon KM, Crowther RA, et al. Abundant neuritic inclusions and microvacuolar changes in a case of diffuse Lewy body disease with the A53T mutation in the alpha-synuclein gene. Acta Neuropathol. (2005) 110:298–305. doi: 10.1007/s00401-005-1042-4
66. Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM, et al. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. (2002) 104:7–11. doi: 10.1007/s00401-002-0563-3
67. Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, et al. Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson's disease. Eur J Neurosci. (2011) 33:1598–610. doi: 10.1111/j.1460-9568.2011.07660.x
68. Lasagnareeves CA, Castillocarranza DL, Guerreromuñoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry (2010) 49:10039–41. doi: 10.1021/bi1016233
69. Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds, and provides novel insights into the hyperphosphorylation of tau. J Neurosci Offic J Soc Neurosci. (2011) 31:7604–18. doi: 10.1523/JNEUROSCI.0297-11.2011
70. Benussi L, Ghidoni R, Paterlini A, Nicosia F, Alberici AC, Signorini S, et al. Interaction between tau and alpha-synuclein proteins is impaired in the presence of P301L tau mutation. Exp Cell Res. (2005) 308:78–84. doi: 10.1016/j.yexcr.2005.04.021
71. Sengupta U, Guerrero-Muñoz MJ, Castillo-Carranza DL, Lasagna-Reeves CA, Gerson JE, Paulucci-Holthauzen AA, et al. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol Psychiatry (2015) 78:672–83. doi: 10.1016/j.biopsych.2014.12.019
72. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell (2013) 154:103–17. doi: 10.1016/j.cell.2013.05.057
73. Simónsánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-Wide Association Study reveals genetic risk underlying Parkinson's disease. Nat Genet. (2009) 41:1308–12. doi: 10.1038/ng.487
74. Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. Alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem. (1999) 274:25481–9.
75. Wills J, Credle J, Haggerty T, Lee JH, Oaks AW, Sidhu A. Tauopathic changes in the striatum of A53T α-synuclein mutant mouse model of Parkinson's Disease. PLoS ONE (2011) 6:e17953. doi: 10.1371/journal.pone.0017953
76. Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson's disease models. FASEB J. (2009) 23:2820–30. doi: 10.1096/fj.08-120410
77. Li B, Ryder J, Su Y, Moore SA Jr, Liu F, Solenberg P, et al. Overexpression of GSK3betaS9A resulted in tau hyperphosphorylationand morphology reminiscent of pretangle-like neurons in the brainof PDGSK3beta transgenic mice. Transgen Res. (2004) 13:385–96. doi: 10.1023/B:TRAG.0000040039.44899.6f
78. Credle JJ, George JL, Wills J, Duka V, Shah K, Lee YC, et al. GSK-3beta dysregulation contributes to parkinson's-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and alpha-synuclein. Cell Death Differentiat. (2015) 22:838–51. doi: 10.1038/cdd.2014.179
79. Duka, T. and Sindhu A. The neurotoxin, MPP +, induces hyperphosphorylation of Tau, in the presence of alpha- Synuclein, in SH-SY5Y neuroblastoma cells. Neurotoxicity Res. (2006) 10:1–10. doi: 10.1007/BF03033329
80. Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, et al. Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J. (2006) 20:2302–12. doi: 10.1096/fj.06-6092com
81. Roy, B. Jackson GR. Interactions between Tau and alpha-synuclein augment neurotoxicity in a Drosophila model of Parkinson's disease. Hum Mol Genet. (2014) 23:3008–23. doi: 10.1093/hmg/ddu011
Keywords: tauopathy, Parkinson's disease (PD), hyperphosphorylation, alpha-synuclien, tau protein
Citation: Zhang X, Gao F, Wang D, Li C, Fu Y, He W and Zhang J (2018) Tau Pathology in Parkinson's Disease. Front. Neurol. 9:809. doi: 10.3389/fneur.2018.00809
Received: 27 November 2017; Accepted: 07 September 2018;
Published: 02 October 2018.
Edited by:
Graziella Madeo, National Institutes of Health (NIH), United StatesReviewed by:
Giuseppe Sciamanna, Università degli Studi di Roma Tor Vergata, ItalyFrancisco José Pan-Montojo, Ludwig-Maximilians-Universität München, Germany
Copyright © 2018 Zhang, Gao, Wang, Li, Fu, He and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei He, aGV3ZWluZ2RAMTI2LmNvbQ==
Jianmin Zhang, anpoYW5nNDJAMTYzLmNvbQ==