 Giorgia Querin
Giorgia Querin Peter Bede
Peter Bede Veronique Marchand-Pauvert1
Veronique Marchand-Pauvert1 Pierre-Francois Pradat
Pierre-Francois Pradat- 1Laboratoire d'Imagerie Biomédicale, CNRS, INSERM, Sorbonne Université, Paris, France
- 2APHP, Département de Neurologie, Centre Référent SLA, Hôpital Pitié-Salpêtrière, Paris, France
- 3Computational Neuroimaging Group, Academic Unit of Neurology, Trinity College Dublin, Dublin, Ireland
- 4Northern Ireland Centre for Stratified Medicine, Biomedical Sciences Research Institute Ulster University, C-TRIC, Altnagelvin Hospital, Londonderry, United Kingdom
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease, is a rare, X-linked, late onset neuromuscular disorder. The disease is caused by a CAG trinucleotide repeat expansion in the first exon of the androgen receptor gene. It is characterized by slowly progressive lower motor neurons degeneration, primary myopathy and widespread multisystem involvement. Respiratory involvement is rare, and the condition is associated with a normal life expectancy. Despite a plethora of therapeutic studies in mouse models, no effective disease-modifying therapy has been licensed for clinical use to date. The development of sensitive monitoring markers for the particularly slowly progressing pathology of SBMA is urgently required to aid future clinical trials. A small number of outcome measures have been proposed recently, including promising biochemical markers, which show correlation with clinical disability and disease-stage and progression. Nevertheless, a paucity of SBMA-specific biomarker studies persists, delaying the development of monitoring markers for pharmaceutical trials. Collaborative efforts through international consortia and multicenter registries are likely to contribute to the characterization of the natural history of the condition, the establishment of disease-specific biomarker panels and ultimately contribute to the development of disease-modifying drugs.
Introduction
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease, is a rare, X-linked, adult onset, neuromuscular disorder (1) characterized by slowly progressive lower motor neuron (LMN) degeneration, skeletal muscle pathology and by a spectrum of multi-organ involvement (2–4). The disease is caused by a CAG repeat expansion in the first exon of the androgen receptor (AR) gene encoding for a poly-glutamine (polyQ) tract. A repeat number higher than 38 is considered pathogenic (5). PolyQ-AR toxicity is hormone-dependent and CAG repeat size inversely correlates with age of symptom onset but not with disease progression rates (6, 7). Heterozygous female carriers of the mutation only present subtle signs of neuromuscular involvement such as muscle cramps and hand tremor (8, 9). The disease is rare, with an estimated prevalence of 3.5/100,000 male inhabitants in southern Europe (10, 11) but the presence of a founder effect is retained to cause considerable differences in the distribution of the disease in various geographical regions (12, 13). Subjects with minimal symptoms and the relatively limited awareness of the condition make it likely that the real prevalence of SBMA is underestimated.
Despite several promising therapeutic studies (14), no disease-modifying treatment currently exists for SBMA. Similarly to SMA, the lack of sensitive monitoring markers for the slow progression rates of SBMA is one of main the barriers to successful clinical trials (15, 16). The objective of this work is the systematic review of candidate biomarkers in SBMA and the appraisal of their potential in clinical management and pharmaceutical trials.
The Neurological Presentation
Limb weakness is present in 97% of SBMA cases. It usually appears at the of age of 35–40 and starts typically proximally in the lower limbs (2, 3, 6, 17). However, tremors, muscle cramps, myalgia, gynecomastia, and exercise intolerance are often reported long before the onset of frank limb weakness (17, 18). Clinical signs of LMN involvement, such as fasciculations, muscle cramps, and atrophy are invariably present. Proximal muscles are predominantly affected, leading to difficulties in climbing stairs and getting up from a sitting position. Motor impairment is usually slowly progressive (19) and survival is only slightly reduced (6, 17). In addition to limb muscle wasting, fasciculations, and decreased deep tendon reflexes, clinical features often include a high-frequency postural hand tremor and postural leg tremor (20).
Bulbar impairment occurs in about 10–30% of patients at the onset of the disease (17), but it is present in the majority of the patients at later stages. It slowly progresses over time and may lead to aspiration pneumonia, which is a frequent cause of death in SBMA (6). Dysphagia is due to impaired oro-pharyngeal phase of deglutition (21), and is associated with tongue's muscles weakness, fasciculations, and atrophy (21). Dysarthria is characterized by hypernasality secondary to incomplete soft palate elevation and is associated with dysphonia. Speech impairment can evolve into markedly reduced intelligibility. Facial weakness and asymmetry, perioral fasciculations, myokymia, and jaw drop are also common clinical features (21–23). Recurrent laryngospasms have been noted in up to 47% of SBMA patients (24).
The presence of a distal sensory neuropathy is a hallmark feature of the disease (25) which has been described in post-mortem studies (26), sural nerve biopsies (27), and neurophysiology (28). The sensory neuropathy may be asymptomatic or manifests in distal numbness and paraesthesia in the lower limbs and reduced sensation for vibration. Neurophysiological examination readily detects reduced or absent sensory action potentials (SAPs) (28, 29). Degeneration of small myelinated and unmyelinated fibers may explain the high incidence of neuropathic pain (30) in SBMA.
Multisystem Involvement
Complex multi-organ involvement is a hallmark feature of SBMA. The core non-neurological features of SBMA include gynecomastia, testicular atrophy, reduced fertility and erectile dysfunction. Dysfunction of the AR protein leads to partial androgen insensitivity (31), manifesting in erectile dysfunction (3), gynecomastia and reduced fertility (31, 32). Testosterone and dehydro-epiandrosterone sulfate (DHEAS) are elevated in up to 38% of patients (32). The Androgen Sensitivity Index (ASI) (LH × testosterone), which reflects androgen resistance, is found to be increased in almost half of the patients (3, 32). DHEAS is thought to correlate with CAG repeat number as well as disease duration (32). Metabolic syndrome with increased BMI, elevated serum cholesterol, triglycerides, and fasting glucose is also a key feature of the disease (3, 31–33) and insulin resistance is associated with disease severity (34). Liver involvement with steatosis and sometimes inflammation has been described (33), but the risk of progression to liver fibrosis is unclear. Recurrent urinary symptoms and incomplete bladder emptying may affect more than the third of male SBMA patients even in the absence of benign prostatic hyperplasia, which is likely to be explained by pelvic floor and bulbuocanvernosus muscle dysfunction (3). While there is no evidence of a primary cardiomyopathy in SBMA (35), Brugada-like ECG abnormalities have been reported in almost half of the patients in a large Japanese cohort (36). Obstructive sleep apnea (OSA), poor sleep quality and periodic limb movements in sleep have also been reported (37).
Biomarkers in SBMA
A biomarker is a parameter that can be measured accurately and reproducibly and used as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention (WHO definition, 1998). An ideal biomarker should have a predictive value and capture subtle changes over relatively short periods of time. Additional requirements to biomarkers include cost-effectiveness, non-invasiveness, and reproducibility (38, 39). It is generally agreed that no single biomarker is suitable for diagnostic, prognostic and monitoring roles and a panel of several markers may be better suited as multirole indicators (40). SBMA is a rare and slowly progressing condition, therefore the development of sensitive outcome measures would enable smaller sample-size and shorter duration of pharmaceutical trials (41, 42).
Biomarkers of Neurological Involvement in SBMA
In recent years, an unprecedented interest has developed in the standardized assessment of neuromuscular performance in SBMA, evaluation of novel therapeutic strategies (14) and in the launch of national SBMA registries (42, 43). Many of the commonly used instruments, such as the MRC score, respiratory function parameters, the modified Norris scale, ALSFRS-r, Quantitative Myasthenia Gravis Score etc. are non-specific to SBMA, yet remain widely utilized. As these tools have been developed for other conditions, new batteries of tests have been recently proposed to specifically appraise disability in SBMA (Table 1).

Table 1. Research studies considering motor and bulbar skills-related outcome measures.
6-Minute-Walk-Test (6MWT)
The 6-minute-walk-test (6MWT) was proposed as an accurate marker of disease progression (44). It measures the distance a person can walk within 6 min and is regarded as a composite proxy of cardiopulmonary and neuromuscular abilities (61). Due to its relative simplicity and cost-effectiveness it has been widely adopted as an outcome measure in several neuromuscular conditions, such SMA and myopathies (62, 63). The 6MWT is traditionally considered the most reliable marker of motor impairment in SBMA, it reliably captures a 10% decline over 1 year (44) and has been used as a primary outcome measure in clinical trials (45, 57). A shorter version of the test, the “2-MWT,” also exists and is thought to be reliable (63).
Adult Myopathy Assessment Tool (AMAT)
The Adult myopathy assessment tool (AMAT) is a performance-based instrument composed of functional and endurance subscales (46). AMAT provides a comprehensive evaluation of motor function, and muscle fatigue, which is a key facet of disability in SBMA (64). One of the strengths of AMAT is that it can also be applied to non-ambulatory patients. It is widely used in both SBMA registers (43) and in clinical trials (47, 52).
SBMA Functional Rating Scale (SBMAFRS)
The SBMA functional rating scale (SBMAFRS) SBMAFRS is a recently validated scale (48, 49), which has been developed from the ALSFRS-r (65) and specifically adapted for the disability profile of SBMA. It is a questionnaire-based scale that measures physical function in activities of daily living (ADL) and consists of five main domains measuring bulbar, upper-limb, lower-limb, truncal, and respiratory function. The SBMAFRS has proven to be more sensitive than the ALSFRS-r in evaluating SBMA patients with moderate motor deficits (48).
1234-Scale
The 1234-scale is another questionnaire-based scale based on the ALSFRS-r, which focuses on SBMA-associated motor disability (50). It includes items such as the ability to do push-ups, ability to run and to stand up from a squatting position. The 1234-scale has shown good internal validity and high reliability (50), but its sensitivity as a monitoring marker has not been confirmed.
Quantitative Muscle Strength Assessment (QMA)
Manual muscle testing (MMT) is commonly used to describe muscle weakness in neuromuscular conditions even though it is highly evaluator-dependent (66). A number of more objective techniques are available to evaluate muscle strength quantitatively in the four limbs (67). Grip strength measured by a handheld dynamometer is one of the simplest and most reproducible QMA parameters. Significant changes in grip strength have been observed in a 3-year longitudinal study of SBMA (19), but progressive changes have not been captured over a 1-year follow-up (44). QMA of maximal voluntary isometric muscle strength has been repeatedly proposed as an outcome measure for clinical trials (46, 47, 52, 54), but its efficacy as a biomarker is limited by considerable inter-centers variability.
Videofluoroscopy (VF)
Videofluoroscopy (VF) is routinely used to evaluate dysphagia in a range of neurological conditions. In SBMA, VF can reliably detect the impairment of the oral phase of deglutition confirming large amount of oral barium residue (56). VF has been previously used in clinical trials (51, 55, 68), but the lack of standardization makes it less suitable for robust multicenter studies.
Fiber Endoscopic Evaluation of Swallowing
Fiber endoscopic evaluation of swallowing has also been assessed as a candidate biomarker of bulbar impairment, but the diagnostic and prognostic value of the technique is yet to be validated (21).
Tongue Pressure
Tongue pressure measurements using an electronic device has been proposed as a biomarker of dysphagia in SBMA, and has been shown to be a low-cost and reliable way of detecting tongue weakness early in the course of the disease (54). An important limitation is that it is susceptible to a ceiling effect in subjects with severe bulbar impairment. Nevertheless, it has been used successfully in a trial of head-lift exercises as a possible rehabilitation strategy in SBMA-associated dysphagia (55).
Electrophysiology
Standard electrophysiology measures are routinely used in the diagnostic work-up of SBMA, but they exhibit limited sensitivity to longitudinal changes (28). This is somewhat unexpected given the correlation between CAG repeat numbers and electrophysiological parameters (29). Quantitative Motor Unit Number Estimation (MUNE) techniques have emerged as a promising way of quantifying motor neuron loss in a number of motor neuron diseases (69, 70). Significant MUNE reductions have been shown in SBMA patients both in cross-sectional and longitudinal study designs, making it one of the most promising candidate outcome measures (58, 59). MUNIX is a more recent, non-invasive method of quantifying motor neuron loss, that has already been utilized in ALS (71), peripheral neuropathies (72), and more recently in adult SMA patients (16). The motor unit size index (MUSIX) (CMAP amplitude/MUNIX) is increasingly accepted as a measure of compensatory collateral sprouting. This technique has not been tested in SBMA yet, but is likely be a promising tool in the evaluation of longitudinal motor neurons loss.
Quantitative Muscle MRI
While quantitative muscle MRI would be an obvious candidate marker of disease progression in SBMA, there is a surprising scarcity of such studies. Existing studies have shown that muscle imaging can effectively detect muscle pathology in distal leg muscles which is less obvious on clinical assessment (60).
Spinal Cord Imaging
Spinal cord imaging has seen unprecedented advances in recent years and has been applied successfully to other motor neuron diseases such as ALS (73–75), and SMA (15) to characterize gray (76) and white matter pathology (77). There is an ongoing study to test its efficacy in SBMA patients (NCT02885870).
Quantitative Brain Imaging
Quantitative brain imaging studies demonstrated white matter alterations in the corticospinal tracts (CST), limbic system (78, 79), brainstem and cerebellum (80). Voxel-based morphometry (VBM) of SBMA cohorts revealed gray matter atrophy in the frontal lobes and in the brainstem (78–81). Frontal hypometabolism has been detected by positron-emission-tomography (PET) (82). These studies confirm the multisystem nature of SBMA-associated pathology, and that neurodegeneration is not limited to LMNs but involve the CSTs and widespread cerebral regions. Despite imaging evidence of extra-motor involvement, neuropsychological studies have only detected subtle frontal dysfunction in small study populations (83, 84) which were not confirmed in larger cohorts (85, 86).
Biomarkers of Multisystem Involvement in SBMA
Increased Serum CK Levels
Increased serum CK levels have been reported by almost every SBMA study and support the hypothesis of a primary myopathy in SBMA (87, 88). Elevated serum CK levels can be detected prior to symptom onset (89) and may be most marked around disease manifestation (18, 19). Nevertheless, no correlation was found between serum CK levels and age of onset, CAG repeat numbers, disease duration or rate of progression (6, 19). As a result, CK levels are thought to be useful as part of the diagnostic workup, but of limited use in monitoring disease progression.
Transaminases Levels
Transaminases levels have also consistently been shown to be raised in SBMA including the pre-symptomatic phase of the disease (89), but they do not correlate with the progression of the neurological symptoms. The clinical significance of raised transaminases in SBMA is a topic of debate and its prognostic value remains to be established (33).
Serum Creatinine Level
Serum creatinine level has also been proposed as a potential biomarker (90) despite its lack of specificity to SBMA. It tends to be reduced in the pre-symptomatic and symptomatic phases of the disease (91) and correlate well with parameters of motor impairment (6, 19, 91).
Proxies of Metabolic Syndrome and Insulin Resistance
Proxies of metabolic syndrome and insulin resistance are considered closely associated with primary molecular disease mechanisms. The homeostasis model assessment of insulin resistance (HOMA-IR) index correlated significantly with motor function parameters in one study (34), but this relationship has not been confirmed by others (32). Hormones levels and ASI (Androgen Sensitivity Index) have also been repeatedly proposed as markers of SBMA. Free testosterone levels correlate with muscle strength in one study (2) but it does not correlate with CAG repeat numbers or disease progression according to others (57). DHEAS levels have been linked to disease duration (91).
Skin Biopsies
Skin biopsies have been performed in some clinical trials to evaluate changes in the frequency of anti-polyQ antibody-positive cells after treatment (57). This index may be sensitive to changes during pharmacological treatment but the methodology is inherently invasive and poorly harmonized across different centers.
Adipose Tissue Quantification
A recent study proposed adipose tissue quantification using whole-body MRI and reported significant subcutaneous fat accumulation in SBMA patients. This correlated both with CAG repeat lengths, disease duration and progression rates (32). These data suggest that adipose tissue MRI may be an additional marker of multisystem involvement in SBMA.
Discussion and Future Perspectives
Interest in SBMA biomarkers has grown steadily in recent years, fuelled both by accruing knowledge about pathogenesis and novel therapeutic strategies (14, 42). SBMA is now widely recognized as a multisystem syndrome (3). A multitude of studies focus on multi-organ involvement, and the systemic phenotype is now considered just as relevant as the neurological manifestations. It is increasingly recognized that non-neurological features of the disease have an equally important impact on the patients' quality of life (3, 31–34, 87, 88, 91, 92). Until now, clinical trials on SBMA focused almost exclusively on the treatment of motor symptoms (14, 45, 47, 51–53, 55, 57, 68, 92, 93), but a shift to targeted molecular therapies (94) and focus on systemic processes are likely to be witnessed in the near future. From a clinical trial perspective, ideal biomarkers should undergo robust validation, sensitivity and specificity profiling, and sampling and measurement harmonization across different centers. Crucially, candidate markers should be able to detect the subtle changes expected after the administration of a specific treatment (95). Given the particularly slow progression rates observed in SBMA, the definition of an effective outcome measures is challenging. The integration of neurological, metabolic, and endocrine indicators seems essential into composite biomarker panels in addition to functional scales. Serum creatinine levels appear to correlate strongly with motor impairment and HOMA-IR index with disease duration (34). The convincing validation of these parameters and their use as effective outcome measures in clinical trials will require robust multicenter study designs (96) (Figure 1).
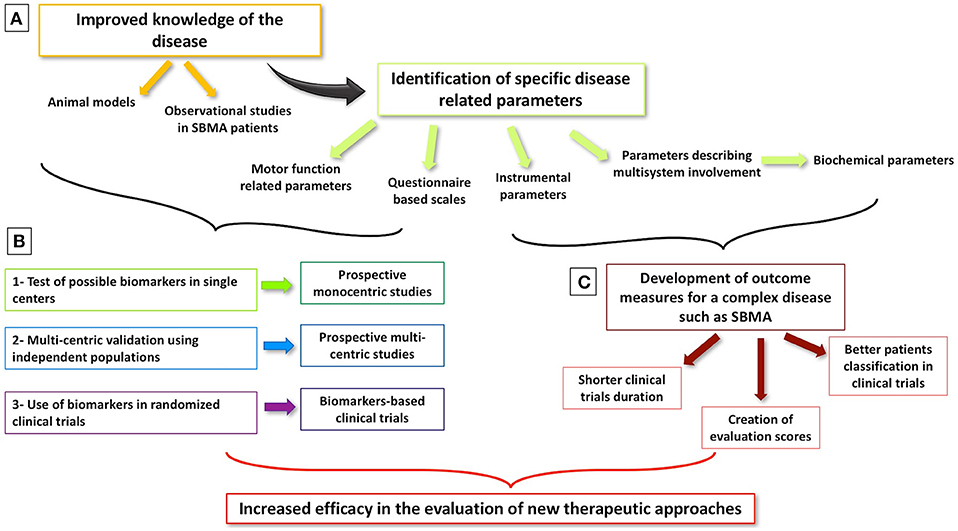
Figure 1. Milestones of biomarker development in SBMA. (A) Better knowledge of SBMA through animal models and observational studies allows the identification of possible biomarkers of disease status and of its progression. (B) Different steps are needed to develop and validate a biomarker in order to make it a reliable outcome measure in clinical trials. (C) Considered the complexity of SBMA and its multi-system presentation, the development of global biomarkers, including both motor function and biochemical parameters, is warranted with the aim of improving the efficacy of upcoming clinical trials.
Furthermore, the comparison of the specificity profile of candidate biomarkers seems essential to define their roles in clinical applications. The establishment of national and international SBMA registers is a clear priority which will be an invaluable resource for future SBMA research (42). As in other neurodegenerative conditions (95, 96), the integration of clinical, molecular, imaging and neurophysiological markers may be required for assessing the efficacy of disease-modifying interventions (95, 96). To conclude, we underline the relevance of considering both motor (muscle force evaluation, questionnaire based scales, and performed tasks) and biochemical parameters as possible outcome measures for a multi-system and complex pathology as SBMA. Beyond their monitoring roles, validated biomarkers will also aid patient stratification upon entry into pharmacological trials (97).
Author Contributions
The paper was drafted by GQ and PB and has been reviewed for intellectual content by VM-P and P-FP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the generosity and kindness of our patients for participating in research on SBMA worldwide. Peter Bede is supported by the Health Research Board (HRB—Ireland; HRB EIA-2017-019), the Irish Institute of Clinical Neuroscience IICN—Novartis Ireland Research Grant, the Iris O'Brien Foundation, the Perrigo Clinician-Scientist Research Fellowship, and the Research Motor Neuron (RMN-Ireland) Foundation. Pierre-Francois Pradat is supported by the French Association for Myopathies (AFM-Telephon), the Institute for Research in Brain and Spinal Cord (IRME) the French Association for Research in ALS (ARSLA) and the Target ALS Foundation.
References
1. Fischbeck KH. Kennedy disease. J Inherit Metab Dis. (1997) 20:152–8. doi: 10.1023/A:1005344403603
2. Rhodes LE, Freeman BK, Auh S, Kokkinis AD, La Pean A, Chen C, et al. Clinical features of spinal and bulbar muscular atrophy. Brain (2009) 132:3242–51. doi: 10.1093/brain/awp258
3. Querin G, Bertolin C, da Re E, Volpe M, Zara G, Pegoraro E, et al. Non-neural phenotype of spinal and bulbar muscular atrophy: results from a large cohort of Italian patients. J Neurol Neurosurg Psychiatry (2016) 87:810–6. doi: 10.1136/jnnp-2015-311305
4. Querin G, Sorarù G, Pradat PF. Kennedy disease (X-linked recessive bulbospinal neuronopathy): a comprehensive review from pathophysiology to therapy. Rev Neurol (Paris) (2017) 173:326–37. doi: 10.1016/j.neurol.2017.03.019
5. La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature (1991) 352:77–9. doi: 10.1038/352077a0
6. Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M, et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain (2006) 129:1446–55. doi: 10.1093/brain/awl096
7. Chahin N, Klein C, Mandrekar J, Sorenson E. Natural history of spinal-bulbar muscular atrophy. Neurology (2008) 70:1967–71. doi: 10.1212/01.wnl.0000312510.49768.eb
8. Sorarù G, D'Ascenzo C, Polo A, Palmieri A, Baggio L, Vergani L, et al. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci. (2008) 264:100–5. doi: 10.1016/j.jns.2007.08.012
9. Ishihara H, Kanda F, Nishio H, Sumino K, Chihara K. Clinical features and skewed X-chromosome inactivation in female carriers of X-linked recessive spinal and bulbar muscular atrophy. J Neurol. (2001) 248:856–60. doi: 10.1007/s004150170069
10. Guidetti D, Sabadini R, Ferlini A, Torrente I. Epidemiological survey of X-linked bulbar and spinal muscular atrophy, or Kennedy disease, in the province of Reggio Emilia, Italy. Eur J Epidemiol. (2001) 17:587–91. doi: 10.1023/A:1014580219761
11. Lund A, Udd B, Juvonen V, Andersen PM, Cederquist K, Davis M, et al. Multiple founder effects in spinal and bulbar muscular atrophy (SBMA, Kennedy disease) around the world. Eur J Hum Genet. (2001) 9:431–6. doi: 10.1038/sj.ejhg.5200656
12. Lund A, Udd B, Juvonen V, Andersen PM, Cederquist K, Ronnevi LO, et al. Founder effect in spinal and bulbar muscular atrophy (SBMA) in Scandinavia. Eur J Hum Genet. (2000) 8:631–6. doi: 10.1038/sj.ejhg.5200517
13. Tanaka F, Doyu M, Ito Y, Matsumoto M, Mitsuma T, Abe K, et al. Founder effect in spinal and bulbar muscular atrophy (SBMA). Hum Mol Genet. (1996) 5:1253–7. doi: 10.1093/hmg/5.9.1253
14. Weydt P, Sagnelli A, Rosenbohm A, Fratta P, Pradat PF, Ludolph AC, et al. Clinical trials in spinal and bulbar muscular atrophy-past, present, and future. J Mol Neurosci. (2016) 58:394–400. doi: 10.1007/s12031-015-0682-7
15. El Mendili M-M, Lenglet T, Stojkovic T, Behin A, Guimarães-Costa R, Salachas F, et al. Cervical spinal cord atrophy profile in adult SMN1-linked SMA. PLoS ONE (2016) 11:e0152439. doi: 10.1371/journal.pone.0152439
16. Querin G, Lenglet T, Debs R, Stojkovic T, Behin A, Salachas F, et al. The motor neuron number index (MUNIX) profile of patients with adult spinal muscular atrophy (SMA). Clin Neurophysiol. (2018) 129:2333–40. doi: 10.1016/j.clinph.2018.08.025
17. Fratta P, Nirmalananthan N, Masset L, Skorupinska I, Collins T, Cortese A, et al. Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology (2014) 82:2077–84. doi: 10.1212/WNL.0000000000000507
18. Finsterer J, Soraru G. Onset manifestations of spinal and bulbar muscular atrophy (Kennedy's Disease). J Mol Neurosci. (2016) 58:321–9. doi: 10.1007/s12031-015-0663-x
19. Hashizume A, Katsuno M, Banno H, Suzuki K, Suga N, Mano T, et al. Longitudinal changes of outcome measures in spinal and bulbar muscular atrophy. Brain (2012) 135:2838–48. doi: 10.1093/brain/aws170
20. Nishiyama A, Sugeno N, Tateyama M, Nishiyama S, Kato M, Aoki M. Postural leg tremor in X-linked spinal and bulbar muscular atrophy. J Clin Neurosci. (2014) 21:799–802. doi: 10.1016/j.jocn.2013.07.026
21. Warnecke T, Oelenberg S, Teismann I, Suntrup S, Hamacher C, Young P, et al. Dysphagia in X-linked bulbospinal muscular atrophy (Kennedy disease). Neuromuscul Disord. (2009) 19:704–8. doi: 10.1016/j.nmd.2009.06.371
22. Sumner CJ, Fischbeck KH. Jaw drop in Kennedy's disease. Neurology (2002) 59:1471–2. doi: 10.1212/01.WNL.0000033325.01878.13
23. Finsterer J. Perspectives of Kennedy's disease. J Neurol Sci. (2010) 298:1–10. doi: 10.1016/j.jns.2010.08.025
24. Sperfeld A-D, Hanemann CO, Ludolph AC, Kassubek J. Laryngospasm: an underdiagnosed symptom of X-linked spinobulbar muscular atrophy. Neurology (2005) 64:753–4. doi: 10.1212/01.WNL.0000151978.74467.E7
25. Antonini G, Gragnani F, Romaniello A, Pennisi EM, Morino S, Ceschin V, et al. Sensory involvement in spinal-bulbar muscular atrophy (Kennedy's disease). Muscle Nerve (2000) 23:252–8. doi: 10.1002/(SICI)1097-4598(200002)23:2<252::AID-MUS17>3.0.CO;2-P
26. Nagashima T, Seko K, Hirose K, Mannen T, Yoshimura S, Arima R, et al. Familial bulbo-spinal muscular atrophy associated with testicular atrophy and sensory neuropathy (Kennedy-Alter-Sung syndrome): autopsy case report of two brothers. J Neurol Sci. (1988) 87:141–52. doi: 10.1016/0022-510X(88)90240-7
27. Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy: a clinicopathological study. Brain (1989) 112:209–32. doi: 10.1093/brain/112.1.209
28. Ferrante MA, Wilbourn AJ. The characteristic electrodiagnostic features of Kennedy's disease. Muscle Nerve (1997) 20:323–9.
29. Suzuki K, Katsuno M, Banno H, Takeuchi Y, Atsuta N, Ito M, et al. CAG repeat size correlates to electrophysiological motor and sensory phenotypes in SBMA. Brain (2008) 131:229–39. doi: 10.1093/brain/awm289
30. Manganelli F, Iodice V, Provitera V, Pisciotta C, Nolano M, Perretti A, et al. Small-fiber involvement in spinobulbar muscular atrophy (Kennedy's disease). Muscle Nerve (2007) 36:816–20. doi: 10.1002/mus.20872
31. Dejager S, Bry-Gauillard H, Bruckert E, Eymard B, Salachas F, LeGuern E, et al. A comprehensive endocrine description of Kennedy's disease revealing androgen insensitivity linked to CAG repeat length. J Clin Endocrinol Metab. (2002) 87:3893–901. doi: 10.1210/jcem.87.8.8780
32. Rosenbohm A, Hirsch S, Volk AE, Grehl T, Grosskreutz J, Hanisch F, et al. The metabolic and endocrine characteristics in spinal and bulbar muscular atrophy. J Neurol. (2018) 265:1026–36. doi: 10.1007/s00415-018-8790-2
33. Guber RD, Takyar V, Kokkinis A, Fox DA, Alao H, Kats I, et al. Nonalcoholic fatty liver disease in spinal and bulbar muscular atrophy. Neurology (2017) 89:2481–90. doi: 10.1212/WNL.0000000000004748
34. Nakatsuji H, Araki A, Hashizume A, Hijikata Y, Yamada S, Inagaki T, et al. Correlation of insulin resistance and motor function in spinal and bulbar muscular atrophy. J Neurol. (2017) 264:839–47. doi: 10.1007/s00415-017-8405-3
35. Querin G, Melacini P, D'Ascenzo C, Morandi L, Mazzini L, Silani V, et al. No evidence of cardiomyopathy in spinal and bulbar muscular atrophy. Acta Neurol Scand. (2013) 128:e30–2. doi: 10.1111/ane.12140
36. Araki A, Katsuno M, Suzuki K, Banno H, Suga N, Hashizume A, et al. Brugada syndrome in spinal and bulbar muscular atrophy. Neurology (2014) 82:1813–21. doi: 10.1212/WNL.0000000000000434
37. Romigi A, Liguori C, Placidi F, Albanese M, Izzi F, Uasone E, et al. Sleep disorders in spinal and bulbar muscular atrophy (Kennedy's disease): a controlled polysomnographic and self-reported questionnaires study. J Neurol. (2014) 261:889–93. doi: 10.1007/s00415-014-7293-z
38. Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS (2010) 5:463–6. doi: 10.1097/COH.0b013e32833ed177
39. Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med. (2012) 31:2973–84. doi: 10.1002/sim.5403
40. Bede P, Querin G, Pradat PF. The changing landscape of motor neuron disease imaging: the transition from descriptive studies to precision clinical tools. Curr Opin Neurol. (2018) 31:431–8. doi: 10.1097/WCO.0000000000000569
41. Scotton C, Passarelli C, Neri M, Ferlini A. Biomarkers in rare neuromuscular diseases. Exp Cell Res. (2014) 325:44–9. doi: 10.1016/j.yexcr.2013.12.020
42. Pennuto M, Greensmith L, Pradat P-F, Sorarù G. 210th ENMC International Workshop: Research and clinical management of patients with spinal and bulbar muscular atrophy, 27-29 March, 2015, Naarden, The Netherlands. Neuromuscul Disord. (2015) 25:802–12. doi: 10.1016/j.nmd.2015.06.462
43. Pareyson D, Fratta P, Pradat P, Sorarù G, Finsterer J, Vissing J, et al. Towards a European registry and biorepository for patients with spinal and bulbar muscular atrophy. J Mol Sci. (2016) 58:394–400. doi: 10.1007/s12031-015-0704-5
44. Takeuchi Y, Katsuno M, Banno H, Suzuki K, Kawashima M, Atsuta N, et al. Walking capacity evaluated by the 6-minute walk test in spinal and bulbar muscular atrophy. Muscle Nerve (2008) 38:964–71. doi: 10.1002/mus.21077
45. Querin G, D'Ascenzo C, Peterle E, Ermani M, Bello L, Melacini P, et al. Pilot trial of clenbuterol in spinal and bulbar muscular atrophy. Neurology (2013) 80:2095–8. doi: 10.1212/WNL.0b013e318295d766
46. Harris-Love MO, Fernandez-Rhodes L, Joe G, Shrader JA, Kokkinis A, La Pean Kirschner A, et al. Assessing function and endurance in adults with spinal and bulbar muscular atrophy: validity of the adult myopathy assessment tool. Rehabil Res Pract. (2014) 2014:873872. doi: 10.1155/2014/873872
47. Shrader JA, Kats I, Kokkinis A, Zampieri C, Levy E, Joe GO, et al. A randomized controlled trial of exercise in spinal and bulbar muscular atrophy. Ann Clin Transl Neurol. (2015) 2:739–47. doi: 10.1002/acn3.208
48. Hashizume A, Katsuno M, Suzuki K, Banno H, Suga N, Mano T, et al. A functional scale for spinal and bulbar muscular atrophy: cross-sectional and longitudinal study. Neuromuscul Disord. (2015) 25:554–62. doi: 10.1016/j.nmd.2015.03.008
49. Querin G, DaRe E, Martinelli I, Bello L, Bertolin C, Pareyson D, et al. Validation of the Italian version of the SBMA functional rating scale as outcome measure. Neurol Sci. (2016) 37:1815–21. doi: 10.1007/s10072-016-2666-y
50. Lu M, Guo H, Fan D. Kennedy's disease 1234 scale: preliminary design and test. J Clin Neurosci. (2017) 40:185–9. doi: 10.1016/j.jocn.2017.02.007
51. Banno H, Katsuno M, Suzuki K, Takeuchi Y, Kawashima M, Suga N, et al. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. (2009) 65:140–50. doi: 10.1002/ana.21540
52. Fernández-Rhodes LE, Kokkinis AD, White MJ, Watts CA, Auh S, Jeffries NO, et al. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. (2011) 10:140–7. doi: 10.1016/S1474-4422(10)70321-5
53. Hijikata Y, Katsuno M, Suzuki K, Hashizume A, Araki A, Yamada S, et al. Treatment with creatine monohydrate in spinal and bulbar muscular atrophy: protocol for a randomized, double-blind, placebo-controlled trial. JMIR Res Protoc. (2018) 7:e69. doi: 10.2196/resprot.8655
54. Mano T, Katsuno M, Banno H, Suzuki K, Suga N, Hashizume A, et al. Tongue pressure as a novel biomarker of spinal and bulbar muscular atrophy. Neurology (2014) 82:255–62. doi: 10.1212/WNL.0000000000000041
55. Mano T, Katsuno M, Banno H, Suzuki K, Suga N, Hashizume A, et al. Head lift exercise improves swallowing dysfunction in spinal and bulbar muscular atrophy. Eur Neurol. (2015) 74:251–8. doi: 10.1159/000431088
56. Hashizume A, Banno H, Katsuno M, Hijikata Y, Yamada S, Inagaki T, et al. Quantitative assessment of swallowing dysfunction in patients with spinal and bulbar muscular atrophy. Intern Med. (2017) 56:3159–65. doi: 10.2169/internalmedicine.8799-16
57. Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2010) 9:875–84. doi: 10.1016/S1474-4422(10)70182-4
58. Suzuki K, Katsuno M, Banno H, Takeuchi Y, Kawashima M, Suga N, et al. The profile of motor unit number estimation (MUNE) in spinal and bulbar muscular atrophy. J Neurol Neurosurg Psychiatry (2010) 81:567–71. doi: 10.1136/jnnp.2009.190462
59. Lehky TJ, Chen CJ, di Prospero NA, Rhodes LE, Fischbeck K, Floeter MK. Standard and modified statistical MUNE evaluations in spinal-bulbar muscular atrophy. Muscle Nerve (2009) 40:809–14. doi: 10.1002/mus.21399
60. Hamano T, Mutoh T, Hirayama M, Kawamura Y, Nagata M, Fujiyama J, et al. Muscle MRI findings of X-linked spinal and bulbar muscular atrophy. J Neurol Sci. (2004) 222:93–7. doi: 10.1016/j.jns.2004.04.028
61. ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. (2002) 166:111–7. doi: 10.1164/ajrccm.166.1.at1102
62. Dunaway Young S, Montes J, Kramer SS, Marra J, Salazar R, Cruz R, et al. Six-minute walk test is reliable and valid in spinal muscular atrophy. Muscle Nerve (2016) 54:836–42. doi: 10.1002/mus.25120
63. Andersen LK, Knak KL, Witting N, Vissing J. Two- and 6-minute walk tests assess walking capability equally in neuromuscular diseases. Neurology (2016) 86:442–5. doi: 10.1212/WNL.0000000000002332
64. Meriggioli MN, Rowin J. Fatigue and abnormal neuromuscular transmission in Kennedy's disease. Muscle Nerve (2003) 27:249–51. doi: 10.1002/mus.10295
65. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. (1999) 169:13–21.
66. Bohannon RW. Manual muscle testing: does it meet the standards of an adequate screening test? Clin Rehabil. (2005) 19:662–7. doi: 10.1191/0269215505cr873oa
67. Hogrel JY, Ollivier G, Desnuelle C. Manual and quantitative muscle testing in neuromuscular disorders. How to assess the consistency of strength measurements in clinical trials? Rev Neurol. (2006) 162:427–36.
68. Banno H, Katsuno M, Suzuki K, Sobue G. Dutasteride for spinal and bulbar muscular atrophy. Lancet Neurol. (2011) 10:113–5. doi: 10.1016/S1474-4422(10)70324-0
69. Gooch C, Shefner J. MUNE. Amyotroph Lateral Scler Other Mot Neuron Disord. (2004) 5:104–7. doi: 10.1080/17434470410019889
70. Swoboda KJ, Prior TW, Scott CB, McNaught TP, Wride MC, Reyna SP, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. (2005) 57:704–12. doi: 10.1002/ana.20473
71. Neuwirth C, Barkhaus PE, Burkhardt C, Castro J, Czell D, De Carvalho M, et al. Tracking motor neuron loss in a set of six muscles in amyotrophic lateral sclerosis using the Motor Unit Number Index (MUNIX): a 15-month longitudinal multicentre trial. J Neurol Neurosurg Psychiatry (2015) 86:1172–9. doi: 10.1136/jnnp-2015-310509
72. Delmont E, Benvenutto A, Grimaldi S, Duprat L, Philibert M, Pouget J, et al. Motor unit number index (MUNIX): is it relevant in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP)? Clin Neurophysiol. (2016) 127:1891–4. doi: 10.1016/j.clinph.2015.12.002
73. El Mendili MM, Cohen-Adad J, Pelegrini-Issac M, Rossignol S, Morizot-Koutlidis R, Marchand-Pauvert V, et al. Multi-parametric spinal cord MRI as potential progression marker in amyotrophic lateral sclerosis. PLoS ONE (2014) 9:e95516. doi: 10.1371/journal.pone.0095516
74. Querin G, El Mendili MM, Lenglet T, Delphine S, Marchand-Pauvert V, Benali H, et al. Spinal cord multi-parametric magnetic resonance imaging for survival prediction in amyotrophic lateral sclerosis. Eur J Neurol. (2017) 24:1040–6. doi: 10.1111/ene.13329
75. Querin G, El Mendili MM, Bede P, Delphine S, Lenglet T, Marchand-Pauvert V, et al. Multimodal spinal cord MRI offers accurate diagnostic classification in ALS. J Neurol Neurosurg Psychiatry (2018) 20:jnnp-2017-317214. doi: 10.1136/jnnp-2017-317214
76. Paquin ME, El Mendili MM, Gros C, Dupont SM, Cohen-Adad J, Pradat PF. Spinal cord gray matter atrophy in amyotrophic lateral sclerosis. AJNR Am J Neuroradiol. (2018) 39:184–192. doi: 10.3174/ajnr.A5427
77. Agosta F, Rocca MA, Valsasina P, Sala S, Caputo D, Perini M, et al. A longitudinal diffusion tensor MRI study of the cervical cord and brain in amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry (2009) 80:53–5. doi: 10.1136/jnnp.2008.154252
78. Kassubek J, Juengling FD, Sperfeld AD. Widespread white matter changes in Kennedy disease: a voxel based morphometry study. J Neurol Neurosurg Psychiatry (2007) 78:1209–12. doi: 10.1136/jnnp.2006.112532
79. Unrath A, Müller HP, Riecker A, Ludolph AC, Sperfeld AD, Kassubek J. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum Brain Mapp. (2010) 31:1727–40. doi: 10.1002/hbm.20971
80. Pieper CC, Konrad C, Sommer J, Teismann I, Schiffbauer H. Structural changes of central white matter tracts in Kennedy's disease - a diffusion tensor imaging and voxel-based morphometry study. Acta Neurol Scand. (2013) 127:323–8. doi: 10.1111/ane.12018
81. Sperfeld AD, Bretschneider V, Flaith L, Unrath A, Hanemann CO, Ludolph AC, et al. MR-pathologic comparison of the upper spinal cord in different motor neuron diseases. Eurol Neurol. (2005) 53:74–7. doi: 10.1159/000084650
82. Lai T-H, Liu R-S, Yang B-H, Wang P-S, Lin K-P, Lee Y-C, et al. Cerebral involvement in spinal and bulbar muscular atrophy (Kennedy's disease): a pilot study of PET. J Neurol Sci. (2013) 335:139–44. doi: 10.1016/j.jns.2013.09.016
83. Kasper E, Wegrzyn M, Marx I, Korp C, Kress W, Benecke R, et al. Minor cognitive disturbances in X-linked spinal and bulbar muscular atrophy, Kennedy's disease. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:15–20. doi: 10.3109/21678421.2013.837927
84. Soukup GR, Sperfeld AD, Uttner I, Karitzky J, Ludolph AC, Kassubek J, et al. Frontotemporal cognitive function in X-linked spinal and bulbar muscular atrophy (SBMA): a controlled neuropsychological study of 20 patients. J Neurol. (2009) 256:1869–75. doi: 10.1007/s00415-009-5212-5
85. Di Rosa E, Sorarù G, Kleinbub JR, Calvo V, Vallesi A, Querin G, et al. Theory of mind, empathy and neuropsychological functioning in X-linked Spinal and Bulbar Muscular Atrophy: a controlled study of 20 patients. J Neurol. (2015) 262:394–401. doi: 10.1007/s00415-014-7567-5
86. Marcato S, Querin G, Pick E, Kleinbub JR, Martinalli I, Bertolin C, et al. Not impaired neuropsychological performance and enhanced memory recall in patients with SBMA: a large sample comparative study. Sci Rep. (2018) 8:13627. doi: 10.1038/s41598-018-32062-5
87. Manzano R, Sorarú G, Grunseich C, Fratta P, Zuccaro E, Pennuto M, et al. Beyond motor neurons: expanding the clinical spectrum in Kennedy's disease. J Neurol Neurosurg Psychiatry (2018) 89:808–12. doi: 10.1136/jnnp-2017-316961
88. Rinaldi C, Bott LC, Fischbeck KH. Muscle matters in kennedy's disease. Neuron (2014) 82:251–3. doi: 10.1016/j.neuron.2014.04.005
89. Sorenson EJ, Klein CJ. Elevated creatine kinase and transaminases in asymptomatic SBMA. Amyotroph Lateral Scler. (2007) 8:62–4. doi: 10.1080/17482960600765040
90. Hijikata Y, Katsuno M, Suzuki K, Hashizume A, Araki A, Yamada S, et al. Impaired muscle uptake of creatine in spinal and bulbar muscular atrophy. Ann Clin Transl Neurol. (2016) 3:537–46. doi: 10.1002/acn3.324
91. Hijikata Y, Hashizume A, Yamada S, Inagaki T, Ito D, Hirakawa A, et al. Biomarker-based analysis of preclinical progression in spinal and bulbar muscular atrophy. Neurology (2018) 90:e1501–9. doi: 10.1212/WNL.0000000000005360
92. Hashizume A, Katsuno M, Suzuki K, Hirakawa A, Hijikata Y, Yamada S, et al. Long-term treatment with leuprorelin for spinal and bulbar muscular atrophy: natural history-controlled study. J Neurol Neurosurg Psychiatry (2017) 88:1026–32. doi: 10.1136/jnnp-2017-316015
93. Guber RD, Kokkinis AD, Schindler AB, Bendixen RM, Heatwole CR, Fischbeck KH, et al. Patient-identifed impact of symptoms in spinal and bulbar muscular atrophy. Muscle Nerve (2018) 57:40–4. doi: 10.1002/mus.25957
94. Rinaldi C, Malik B, Greensmith L. Targeted molecular therapies for SBMA. J Mol Sci. (2016) 58:335–42. doi: 10.1007/s12031-015-0676-5
95. Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. (2009) 8:94–109. doi: 10.1016/S1474-4422(08)70293-X
96. Chiò A, Montalcini RL, Traynor BJ. Motor neuron disease in 2014: biomarkers for ALS—in search of the Promised Land. Nat Rev Neurol. (2015) 11:72–4. doi: 10.1038/nrneurol.2014.250
Keywords: SBMA, biomarkers, clinical trials, multisystem involvement, outcome measures
Citation: Querin G, Bede P, Marchand-Pauvert V and Pradat P-F (2018) Biomarkers of Spinal and Bulbar Muscle Atrophy (SBMA): A Comprehensive Review. Front. Neurol. 9:844. doi: 10.3389/fneur.2018.00844
Received: 01 August 2018; Accepted: 20 September 2018;
Published: 10 October 2018.
Edited by:
Francesca Trojsi, Università degli Studi della Campania “Luigi Vanvitelli” Naples, ItalyReviewed by:
Fiore Manganelli, Università degli Studi di Napoli Federico II, ItalyGiuseppe Piscosquito, Fondazione Salvatore Maugeri, Telese (IRCCS), Italy
Patrick Weydt, Universität Bonn, Germany
Copyright © 2018 Querin, Bede, Marchand-Pauvert and Pradat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgia Querin, Z2lvcmdpYS5xdWVyaW5AZ21haWwuY29t