 Naomi Deacon-Diaz
Naomi Deacon-Diaz Atul Malhotra
Atul Malhotra- Department of Medicine, Pulmonary and Critical Care Medicine, University of California, San Diego, San Diego, CA, United States
Unstable ventilatory chemoreflex control, quantified as loop gain, is recognized as one of four key pathophysiological traits that contribute to cause obstructive sleep apnea (OSA). Novel treatments aimed at reducing loop gain are being investigated, with the intention that future OSA treatment may be tailored to the individual's specific cause of apnea. However, few studies have evaluated loop gain in OSA and non-OSA controls and those that have provide little evidence to support an inherent abnormality in either overall chemical loop gain in OSA patients vs. non-OSA controls, or its components (controller and plant gain). However, intermittent hypoxia may induce high controller gain through neuroplastic changes to chemoreflex control, and may also decrease plant gain via oxidative stress induced inflammation and reduced lung function. The inherent difficulties and limitations with loop gain measurements are discussed and areas where further research are required are highlighted, as only by understanding the mechanisms underlying OSA are new therapeutic approaches likely to emerge in OSA.
Introduction
Obstructive sleep apnea (OSA) is a condition in which the upper airway either partially or completely obstructs during sleep. The repeated bouts of concomitant hypercapnia and hypoxia, surges in sympathetic neural activity and frequent arousals lead to a wide range of life-threatening comorbidities, including cardiovascular disease, metabolic disorder, depression, neurocognitive damage and increased mortality. OSA affects 24% of men and 9% of women in the US aged 30–60 years. As obesity is one of the key risk factors for developing OSA, the increasing obesity epidemic is estimated to increase OSA prevalence (1). Although several treatment options for OSA exist, issues of high cost, discomfort, invasiveness and poor efficacy cause many patients to discontinue long-term treatment (2). Thus, there is a need for new OSA treatments to reduce the growing public health burden.
In recent years it has been recognized that four quantifiable traits of upper airway anatomy and neuromuscular control contribute to variable degrees in each patient to cause OSA (3). New research has focused on treatments targeting each trait, with the intention that future OSA treatments may be tailored to the individual's pathology to increase both efficacy and adherence (4). One of the four traits is the stability of ventilatory chemoreflex control, called loop gain. Therefore, there is growing interest in treatments that may alter ventilatory chemoreflex control as a potential treatment for OSA (5, 6). However, it is currently uncertain whether ventilatory chemoreflex control, and therefore loop gain, is in fact consistently abnormal in OSA patients. It is also uncertain what physiological mechanisms may be responsible for increased loop gain in OSA, whether any abnormalities are inherent and therefore part of OSA pathogenesis, or whether abnormalities are induced due to OSA and are possibly reversible. This article aims to summarize the current knowledge on loop gain in OSA which may help guide the development of treatments intended to modulate ventilatory chemoreflex control in OSA.
Ventilatory Loop Gain
Obstructive sleep apnea is a fairly unique condition in that it only occurs during sleep. The state dependent nature of OSA is due to the fact that during wakefulness ventilation is not only governed by metabolic chemoreflex control, but also by a conscious drive originating from supra-pontine brain centers which allow, to a degree, conscious override of the chemical control system. This conscious control is essential to be able to coordinate breathing with emotion and locomotion and breath hold while performing functions such as swallowing and swimming (7, 8). However, it also affords protection as conscious drive will maintain breathing even in the event where chemical drive to breathe may be very low (9). At sleep onset the wakefulness drive is lost and ventilation is primarily governed by the metabolic chemoreflex control system (8). During non-REM sleep, if arterial CO2 (PaCO2) decreases only 3-6 mmHg below eupnic levels the central chemoreceptor drive to breathe ceases and central apnea ensues, until PaCO2 rises high enough to reinitiate central ventilatory drive (10). Ventilatory drive not only determines the level of activity of the thoracic pump muscles, but also the upper airway dilator muscles (11). Therefore, although central apnea is not characteristic of OSA, instability in ventilatory chemoreflex control may promote OSA as the upper airway is susceptible to collapse when PaCO2, and therefore neural drive to the upper airway muscles, is low (12).
Loop gain is an engineering method used to measure the stability of the negative feedback chemoreflex control system. The overall loop gain of the ventilatory system reflects the ratio of the ventilatory response to the disturbance that elicited the response (LG = ventilatory response/ventilatory disturbance). Therefore, when breathing deviates from eupnea (the point where ventilation matches metabolic demand), such as during a hypopnea, if the ventilatory response that is elicited is equal to the disturbance (LG = 1), ventilation will correct blood gases to re-establish eupneic levels (Figure 1). If the ventilatory response is disproportionately larger than the disturbance (LG > 1), ventilation will not only correct the disturbance to blood gases, but will overshoot such that PaCO2 will be reduced below eupnic levels. The resulting hypocapnia will then induce hypoventilation, upper airway muscle hypotonia and a secondary airway obstruction (apnea or hypopnea depending on prevailing upper airway mechanics), such that respiratory events become self-perpetuating. Thus higher loop gain reflects less stable ventilatory chemoreflex control (13, 14).
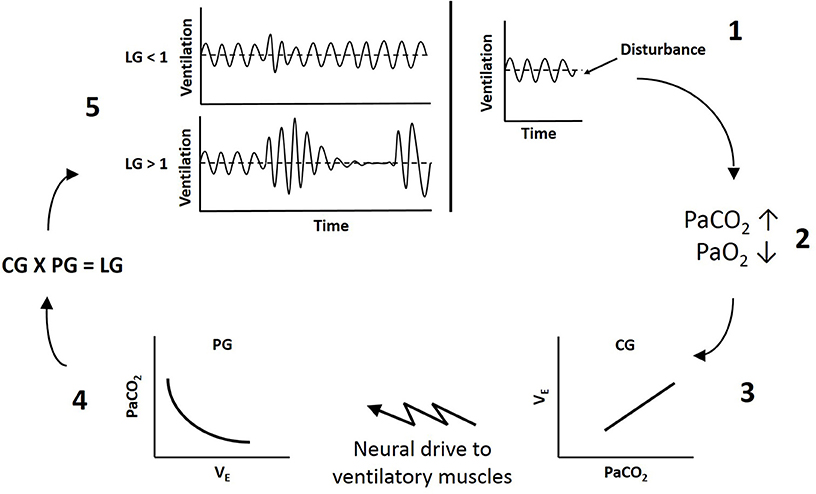
Figure 1. Schematic of ventilatory loop gain. 1, A disturbance to breathing causes a reduction in ventilation below eupnea. 2, Reduced ventilation increases arterial CO2 (PaCO2) and reduces arterial O2 (PaO2). 3, Controller gain (CG) reflects the sensitivity of the peripheral and central chemoreceptors to blood gases and dictates the magnitude of neural drive to ventilatory muscles (ΔVE/ΔPaCO2). 4, Plant gain (PG) represents the effectiveness of the lungs to change blood gases (ΔPaCO2/ΔVE). 5, The product of controller and plant gain determines overall loop gain (LG). If loop gain is less than 1 (LG < 1), the fluctuations in ventilation will dampen out and breathing will stabilize. If loop gain is greater than 1 (LG > 1), the fluctuations in ventilation will increase in amplitude and instability will be self-perpetuating.
Loop gain theory dictates there is a controller and a plant component, with a delay between the two (13). The controller senses a stimulus and dictates the output of the plant, which responds to decrease the stimulus. In ventilatory control, chemoreceptor sensitivity to blood gases reflects controller gain, and the effectiveness of the lungs to alter blood gases reflects plant gain. The product of controller and plant gain provides the overall loop gain of the system (LG = CG × PG) (14). Because there is a circulation delay between when ventilation begins to alter blood gases and when the chemoreceptors sense the change, if the gain of either the controller or the plant is too high, there is the potential for ventilatory overshoot producing instability in the system (13).
Carbon dioxide is considered the primary stimulus to chemoreflex control, because if PaCO2 is below a threshold level, hypoxic sensitivity is depressed and decreasing PaO2 will not alter ventilation (15). Therefore, controller gain reflects the slope of the ventilatory response to increasing CO2 (CG = ΔVE/ΔPaCO2) (12). Under hyperoxic conditions (albeit never naturally occurring) the peripheral chemoreceptors of the carotid bodies have diminished response to PaCO2. Only the central chemoreceptors in the brainstem will respond to increase ventilation, the gain of which describes central controller gain. During normoxia both the central and peripheral chemoreceptors contribute to the drive to breathe and the gain of the ventilatory response to CO2 will increase. Central chemoreceptors in the brainstem do not alter ventilation in response to changing PaO2. Only the peripheral chemoreceptors of the carotid bodies are sensitive to PaO2, increasing neural drive to ventilation in response to decreasing PaO2. If the additional stimuli of hypoxia is combined with hypercapnia, the contribution of the peripheral chemoreceptor drive to ventilation will increase and the gain of the ventilatory response to CO2 will increase further; a relationship described as hypoxic sensitization of the peripheral chemoreflex response to CO2 (16). Therefore, changes to either the central or peripheral chemoreceptor responses to CO2, or the peripheral chemoreceptor response to hypoxia, will alter controller gain. Chemoreflex sensitivity to both hypercapnia and hypoxia decrease progressively from wake to light NREM sleep to slow wave NREM sleep and are lowest in REM sleep (17, 18). Therefore, controller gain decreases with progressively deeper NREM sleep. REM sleep is characterized by tonic motor inhibition with bursts of phasic activity (19, 20). During the tonic phase ventilatory control is largely governed by the metabolic chemoreflex control system. However, the phasic phase provides additional stimulation to both inspiratory and expiratory muscles which somewhat overrides chemoreflex control and disorganizes breathing (19). In agreement with lower chemosensitivity in REM sleep than NREM sleep and additional neural drive to ventilatory muscles during REM sleep, loop gain in OSA patients is lowest in REM sleep and highest in lighter NREM sleep (21). Consequently, although breathing in REM sleep is characterized by an increase in respiratory events, breathing is typically more erratic without the underlying Cheyne-Stokes pattern characteristic of events driven by instability in chemical control (19, 20).
Plant gain quantifies the effectiveness of the lungs to alter blood gases, represented by the function of the isometabolic hyperbola (ΔPaCO2/ΔVA) (12). At rest ventilation is regulated to meet metabolic demand such that alveolar ventilation and PaCO2 are maintained relatively stable (i.e., eupnea). Due to the decreasing slope of the metabolic hyperbola during hypercapnia, plant gain increases as every unit increase in ventilation would produce a greater change in blood gases. Conversely, during hypocapnia the increasing slope of the metabolic hyperbola results in a reduction in plant gain, as every unit increase in ventilation would produce progressively smaller changes in blood gases (12). In people free of major cardiac or lung disease, it would be expected there would be no abnormalities in blood gas diffusion rates or circulation to affect plant gain. Therefore, in most OSA patients the main factor affecting plant gain is functional residual capacity. Reductions in lung stores of either CO2 or O2 will increase plant gain, because smaller tidal volumes will produce greater fluctuations in alveolar gas tensions. Thus reduced functional residual capacity increases plant gain (12, 13). Functional residual capacity decreases in the supine position (22), and further during sleep (23). Central obesity also decreases functional residual capacity due to increased abdominal compression (24). Therefore, obesity, particularly during supine sleep, would be expected to increase plant gain.
It is important to note that in OSA, the delay in the closed loop system is not just a function of lung to chemoreceptor circulatory delay. When the airway is obstructed, the delay is the duration of the event, as chemical drive continues to build and the ventilatory response cannot be expressed until the airway re-opens (25). Loop gain is also not a static value, rather it changes constantly. During an apneic event, as there is no ventilatory response, loop gain is actually zero (12). However, as chemical drive accumulates, because hypoxia sensitizes the carotid body hypercapnic response, progressive hypoxia and hypercapnia increases controller gain. As PaO2 is depleted and CO2 accumulates in the lungs, plant gain progressively increases such that when the airway opens, every unit increase in minute ventilation would produce greater fluctuations in alveolar gas tensions (12, 13). Thus, in OSA, the loop gain that is most relevant to apnea propagation is the loop gain at airway opening, when both controller and plant gains are at their peak. However, if the airway does not fully open the ventilatory response will be restricted, such that the net loop gain, being the loop gain that is actually expressed, will be lower than the chemical loop gain, being the actual drive to breathe (25). Most methods to quantify loop gain are performed during sleep with therapeutic continuous positive airway pressure (CPAP) to ensure the airway is fully dilated and there is no restriction to ventilation, thereby allowing quantification of chemical loop gain (26, 27). Upper airway mechanics are quantified within two other traits of OSA pathophysiology. The critical closing pressure is the airway pressure at which the passive airway collapses, and the upper airway recruitment threshold is the level of chemical drive required to activate the upper airway dilator muscles sufficiently to enable it to re-open after obstruction (28). Therefore, although upper airway mechanics will greatly influence net loop gain and therefore the loop gain contributing to event propagation in OSA, in patients free of cardiac and lung disease, the main factors that will affect chemical loop gain are factors that affect controller and plant gains.
Evidence for Pathophysiological Role of Unstable Chemoreflex Control in OSA
The pathophysiological role of unstable ventilatory chemoreflex control in promoting airway collapse in OSA due to hypocapnic hypotonia of the upper airways is well established. Obstructive apneas are followed by hyperventilation producing hypocapnia and respiratory depression (29). During spontaneous apneas, it is during the nadir of ventilation and upper airway dilator muscle activity, such as the genioglossus and alae nasi, that the airway collapses (30, 31). Imaging of the airway during spontaneous apneas has also shown the airway to collapse passively at end expiration (32). Even in healthy participants not normally susceptible to OSA, if hyperventilation and hypocapnia are induced during sleep, either with hypoxia or mechanical hyperventilation, participants develop periodic breathing and exhibit increased upper airway resistance, pharyngeal narrowing and obstruction during the nadir of ventilation (33–35). Additionally, in OSA patients, both hyperoxia to blunt the peripheral chemoreceptor response (i.e., reduce controller gain) and supplemental CO2 to prevent hyperventilation induced hypocapnic hypotonia of the upper airways, has been shown to stabilize ventilation and reduce the severity and frequency of apneic events (5, 29, 36).
Is Loop Gain Inherently High in OSA?
Although much evidence supports that unstable chemoreflex control contributes to propagating apneas in OSA, whether loop gain is elevated in OSA, and whether high controller or plant gain contribute to high loop gain, is currently unclear. Younes et al. found loop gain correlated with AHI (apnea-hypopnea index) in OSA patients (27), which some had extrapolated out to mean loop gain was higher in OSA patients vs. non-OSA participants. However subsequent studies have shown that the different traits of OSA pathogenesis contribute to varying degrees in different patients, such that it is possible for patients to exhibit a high AHI despite normal loop gain, and loop gain only correlated with AHI in patients with airways that collapse near atmospheric pressures (37, 38). Although these later findings highlight that loop gain is highly variable between patients and that loop gain is not the sole determinant of whether a patient has severe, mild or no OSA, as these studies did not include non-OSA control participants, they do not help to determine whether loop gain is elevated in OSA patients compared to people without OSA.
While several methods have been developed to quantify loop gain, each method yields vastly different results. For example, in two studies in OSA patients, loop gain has been reported as ranging from 0.33 to 0.42 when quantified using proportional assist ventilation to induce ventilatory instability (37), while a newer method employing CPAP drops has produced loop gain values ranging from 0.7 to 10.6 (28). Consequently it is not possible to make meaningful comparison of results from studies employing different techniques, and both OSA and control participants must be compared in the same study to determine if loop gain is elevated in OSA. However, likely due to limitations of currently available techniques to quantify loop gain (discussed below under Methodological limitations), few studies have actually compared loop gain between OSA patients and non-OSA controls.
We are aware of only nine published studies that have quantified loop gain in both OSA and controls. One study was conducted in elderly, in which OSA pathophysiology is known to be different from younger adults (39). Three were methods papers, two of which made no direct comparison between groups (3, 28). Of the six published studies that have compared loop gain between adult OSA and non-OSA controls (26, 38, 40–43), three found no difference in loop gain between groups (38, 41, 42). Hudgel et al. found loop gain was higher in OSA and concluded this finding was driven by higher plant gain in the OSA participants, with no difference in controller gain between groups (26). Gederi et al. found loop gain was higher in OSA due solely to higher controller gain, with no difference between groups in plant gain (40). Whereas Deacon et al. found loop gain was higher in OSA patients, but found no difference between groups in either controller or plant gains (43).
Although Gederi and colleagues reported loop gain was higher in OSA patients due to higher controller gain, the age, gender, height, weight and BMI of the participants was not reported (40). All of these factors influence both controller and plant gain. For example, obesity alone is known to increase sensitivity to CO2 (44). Thus, it is not possible to determine whether the higher controller gain exhibited in the OSA patients was associated with having OSA, or whether it was due to some other difference between the patients and control participants. Hudgel et al. used a pseudorandom binary CO2 stimulation test in which the ventilatory response to rapid perturbations in FICO2 were used to compare ventilatory control in obese OSA patients and lean non-OSA control participants (26). By analyzing breath-by-breath variations in ventilation and CO2, this method allows quantification of both the controller gain component without plant gain feedback effects, called the open-loop response, and also the closed-loop response which incorporates both the controller and plant gain feedback. The authors did not report summary values of loop gain or its components, rather they compared the rate and magnitude of the ventilatory response to CO2. They found that the OSA patients had a greater peak and faster recovery in the closed-loop response, indicative of higher instability. However, there was no difference between patients and controls in the open-loop response (26). Therefore, loop gain was higher in the OSA patients due to higher plant gain. However, as the OSA patients were obese and the control participants were lean, and obesity is known to reduce functional residual volume which would be expected to increase plant gain, the authors concluded that less stable control in the closed-loop response in OSA patients may have been due to obesity, rather than being associated with OSA (26). Similarly, a more recent study by Sands et al. compared loop gain quantified using the CPAP drop method between obese OSA and both obese non-OSA and lean non-OSA (41). The authors reported that while there was no difference in loop gain between obese OSA patients and obese non-OSA control participants, loop gain was higher in both obese OSA patients and obese non-OSA controls compared to lean non-OSA controls. Although this method only quantifies the overall loop gain and controller and plant gain cannot be separately quantified, these findings also suggest high loop gain was associated with obesity dependent increased plant gain, rather than OSA (41). On the contrary, Deacon and colleagues rigorously matched control and OSA participants for gender, age, height and weight and found loop gain was higher in the OSA patients. Thus the findings of Deacon and colleagues are the first to support loop gain is higher in OSA patients independent of confounding variables such as obesity (43).
Of the published studies that have quantified loop gain in both OSA and controls, most have been conducted in patients that had been treated for several months to years with continuous positive airway pressure therapy (CPAP) (3, 38, 40, 41). Although two studies did not report treatment status, it is possible that patients in these studies were also treated with CPAP (26, 42). Therefore, the lack of strong evidence for high loop gain in OSA patients in these studies only suggests that loop gain is not inherently elevated in OSA patients. To determine whether loop gain is affected by treatment, Deacon and colleagues assessed newly diagnosed patients prior to commencing treatment and again following 2 and 6 weeks of CPAP treatment, and found no change in overall loop gain, or controller or plant gains, across the course of treatment. As controller and plant gains were not different between groups or affected by treatment, it is not possible to determine what mechanism contributed to the higher loop gain in the OSA patients in this study. However, the lack of treatment effect suggests loop gain may be inherently higher in OSA patients, or possibly that OSA induces increased loop gain through mechanisms which are not amenable to short-term treatment (43). As very few studies have compared loop gain between both OSA and controls, and the published data are inconclusive, it is necessary to evaluate the literature regarding the two major components of loop gain in OSA, being controller and plant gain.
Intermittent Hypoxia-Induced High Controller Gain
Although data from loop gain studies are conflicting, there is strong evidence that when compared to well-matched controls, untreated OSA patients' exhibit abnormalities in chemoreflex control that increase controller gain. These abnormalities are treatment-reversible, suggesting high controller gain is not an inherent trait in OSA, but is induced by OSA itself. Importantly, the abnormalities seen in chemoreflex control of untreated OSA patients reflect neuroplastic changes that can be induced by intermittent hypoxia and which normalize following return to stable normoxia, strongly suggesting that high controller gain in OSA patients is induced by intermittent hypoxia. Supporting publications and key findings are summarized in Table 1 and discussed in detail below.

Table 1. Intermittent hypoxia-induced treatment-reversible high controller gain in OSA.
Chronic intermittent hypoxia in rats, designed to mimic OSA with 8 h daily exposures for several days to weeks, induces neuroplasticity at the carotid bodies which increases both basal neural discharge and hypoxic sensitivity (58, 59). In humans even acute intermittent hypoxia increases hypoxic sensitivity, while chronic exposure to intermittent hypoxia enhances this response (51, 52). In both humans and animals, hypoxic sensitivity returns to baseline following several days of re-exposure to stable normoxia (54, 56, 60). When compared to age and BMI matched non-OSA controls, OSA patients' exhibit elevated hypoxic sensitivity (45) and 1 month of continuous positive airway pressure treatment significantly reduces hypoxic sensitivity in previously treatment naïve patients (49). These findings support that carotid body hypoxic sensitivity is increased in untreated OSA patients due to intermittent hypoxia-induced neuroplasticity.
Acute intermittent hypoxia consisting of several short bursts can induce neuroplastic changes in cell bodies of ventilatory motor neurons such as those of the phrenic or hypoglossal nerves, which results in a persistent increase in neural activity for several hours after the last hypoxic stimulus, called long-term facilitation (LTF) (61). In intact animals, phrenic LTF increases ventilation for the same level of chemical stimulation (62). However, the increased ventilation is not due to increased sensitivity to CO2. Following intermittent hypoxia and the induction of ventilatory LTF in both animals and humans, chemoreflex tests have consistently shown there is no change in the sensitivity to hypercapnia (53, 54, 60, 63). Rather, when rats or humans are exposed to intermittent hypoxia to induce ventilatory LTF and then returned to a poikilocapnic environment, ventilatory feedback reduces PETCO2 which then reduces minute ventilation, thereby constraining the expression of LTF (51, 53, 64). Thus, ventilatory LTF increases the magnitude of the ventilatory response to CO2 by causing a leftward shift along the metabolic hyperbola, without changing the sensitivity to CO2 above eupnea. As the apneic threshold in humans is not altered by intermittent hypoxia, ventilatory LTF reduces the CO2 reserve below eupnea, resulting in an increase in controller gain below eupnea (55). Like carotid body neuroplasticity, in both animals and humans, ventilatory LTF has also been shown to be reversible, with breathing and PETCO2 returning to baseline levels several days after returning to stable normoxic breathing (54, 56, 57).
Data regarding hypercapnic ventilatory sensitivity in OSA patients have been conflicting, possibly due to lack of matching for obesity between groups, as obesity increases sensitivity to hypercapnia (44, 65), and inclusion of chronically hypercapnic patient in which CO2 sensitivity is depressed (66). Most studies report that untreated normocapnic OSA patients exhibit no difference in hypercapnic ventilatory sensitivity to well matched non-OSA controls (45, 46, 67, 68). This finding is in agreement with the lack of change in the sensitivity to CO2 following experimental exposure to intermittent hypoxia in both animals and humans (53, 54, 60, 63). However, whether OSA patients exhibit ventilatory LTF is uncertain. This may be because ventilatory LTF, as defined by an elevated minute ventilation, is usually experimentally determined with supplemental CO2 to maintain PETCO2, thus preventing ventilatory feedback constraint of minute ventilation (51, 64). Although ventilatory LTF has not been reported as such in OSA patients, when compared to age, sex and BMI matched non-OSA participants, untreated OSA patients exhibit a reduced PETCO2, reduced CO2 reserve and increased CO2 sensitivity below eupnea, which reflects the expression of ventilatory LTF and ventilatory feedback (47, 55). Following CPAP treatment PETCO2 and the CO2 reserve increased in the OSA patients, further supporting the possible presence of ventilatory LTF in untreated OSA patients (47).
Intermittent hypoxia has also been shown to induce an increase in the ventilatory response of humans to combined hypercapnic hypoxia (53). In this study the authors reported LTF had not been induced, as minute ventilation was only elevated for the first 60–90s after the last hypoxic episode, then returned to baseline levels. However, PETCO2 was reduced below baseline levels for the next 15 min of the room air breathing recovery period. This finding reflects the expression of LTF and ventilatory feedback constraint of ventilation observed in several other studies (51, 55, 64). During hyperoxia, there was no difference in the ventilatory sensitivity to hypercapnia post-intermittent hypoxia. As hypocapnia inhibits the peripheral chemoreceptor response to hypoxia, and intermittent hypoxia had induced a reduction in room air breathing PETCO2 (due to LTF), the authors also found that there was no difference in the hypoxic ventilatory response when PETCO2 was maintained. However, when CO2 was supplemented to raise PETCO2 above the peripheral chemoreceptor threshold, hypoxic sensitivity was increased. Therefore, the findings of Mateika and colleagues reflects the combined effects of intermittent hypoxia-induced LTF and increased hypoxic sensitivity (53). Untreated OSA patients also exhibit an elevated ventilatory response to combined hypercapnic hypoxia, a finding which significantly attenuates following several months of CPAP treatment (48, 50). By definition, as the ventilatory response to chemostimulation (which incorporates both controller and plant gain) decreased following CPAP, these data show that loop gain decreased in OSA patients following CPAP treatment. Although non-OSA control participants were not included in these studies to determine whether the ventilatory response to hypercapnic hypoxia was higher in untreated OSA patients than non-OSA controls, a reduction in loop gain post-treatment supports that loop gain is increased in untreated OSA patients. Also, that elevated loop gain is not an inherent trait, rather it is induced by OSA itself and can be normalized with adequate treatment to prevent obstructive apneas.
Collectively, these data suggest that controller gain, and overall loop gain, could be elevated in untreated OSA patients due to the combined effects of intermittent hypoxia-induced ventilatory LTF and increased hypoxic sensitivity. As intermittent hypoxia-induced neuroplasticity is reversible, studies that have failed to find elevated loop gain in OSA patients may be because most [some authors did not provide treatment status (26, 42)] studies have evaluated OSA patients that had been treatment compliant for several months to years prior to evaluation (3, 38, 40, 41). Thus, any intermittent hypoxia-induced elevation of controller gain would have likely already been ameliorated. Although Deacon et al. found 6 weeks of CPAP treatment did not reduce elevated loop gain in previously treatment naïve patients, the lack of treatment effect may be due to limitations of the method used to quantify loop gain (43). The pseudorandom binary stimulation of CO2 method employed in this study only evaluates the chemoreflex loop gain to hypercapnia. Therefore, if patients did exhibit increased hypoxic sensitivity and increased sensitivity to CO2 below eupnea prior to treatment, and CPAP normalized these abnormalities, the method employed to quantify loop gain would not have evaluated these changes. Unfortunately, there are no loop gain methods currently available that separately quantify controller gain, which incorporate hypoxic sensitivity or CO2 sensitivity below eupnea.
Obesity, Lung Volume and Plant Gain
Although loop gain studies support a role for obesity dependent reduced lung volume and increased plant gain in contributing to high loop gain in OSA, there are very little data comparing lung volumes or plant gain measurements between OSA and non-OSA controls matched for factors known to alter these variables. However, Deacon and colleagues found functional residual capacity and plant gain were not different between OSA patients and matched controls and CPAP treatment did not alter either measure. Although forced expiratory volume and forced vital capacity were only assessed at baseline prior to patients commencing CPAP treatment, so treatment effects could not be evaluated, both measures were significantly lower in the OSA patients (43). In a much larger study of 170 participants, Zerah-Lancner and colleagues compared spirometry, forced oscillation mechanics and gas exchange between moderate and severe OSA patients and non-OSA snorers, in which groups were matched for age, gender, BMI and smoking history. The authors also found no difference in total lung capacity, vital capacity, functional residual capacity or expiratory reserve volume between groups. However, lung function was impaired in OSA patients, as forced expiratory volumes and respiratory conductance decreased and respiratory resistance increased with increasing OSA severity (69). Gas exchange was also compromised in the OSA patients and correlated with OSA severity as PaO2 and O2 saturation negatively correlated with AHI and PaCO2 positively correlated with AHI (69).
These findings are in agreement with a growing body of work which supports that intermittent hypoxia experienced in OSA not only induces oxidative stress and inflammation systemically, but also within the lower airways, which may induce obstructive airways disease (70). This notion is supported by evidence that OSA commonly coexists with both asthma and COPD and has been shown to exacerbate disease progression in both conditions (70–72). CPAP treatment prevents the OSA associated decline in FEV1 in patients with comorbid asthma (72). In patients with comorbid COPD, CPAP improves FEV1 and reduces risk of exacerbations leading to hospitalization and also risk of increased mortality (71, 73). Exhaled nitric oxide (eNO) is a marker of respiratory inflammation and OSA patients exhibit elevated levels of eNO compared to non-OSA control participants. Also, eNO significantly increases in the morning after sleep in OSA patients but not in controls, the post-sleep increase in eNO levels correlates with AHI, nadir SaO2 and arousal index, and long-term CPAP treatment reduces eNO and markers of systemic oxidative stress (74, 75). Non-smoker OSA patients have also been shown to exhibit elevated levels of bronchial neutrophilia and IL-8 concentration in sputum compared to non-OSA participants. IL-8 concentration correlated with AHI and negatively correlated with nadir SaO2. One month of CPAP treatment did not significantly alter either marker of inflammation. However after 1 week of CPAP treatment airway hyperresponsiveness was increased and remained elevated at 4 weeks, which indicates that short duration CPAP treatment may aggravate airway inflammation, and that longer treatment duration may be required to observe improvements (76). Intermittent hypoxia has also been shown to induce oxidative stress, inflammation, altered immune responses, airway hyperresponsiveness and airflow limitation in rodents (77–80).
Although elevated plant gain may be acquired due to obesity dependent reduced lung volume, which may contribute to OSA pathogenesis and disease progression, the current data support that there is no inherent abnormality in lung volumes or plant gain in OSA. If OSA does reduce lung function, it would be expected that by compromising gas exchange, reduced lung function would decrease plant gain, not increase it. Therefore, OSA may induce a reduction in plant gain. However, due to the limited studies comparing plant gain, lung volumes and gas exchange between OSA and non-OSA participants matched for factors known to alter these variables, further research is needed to clarify the effects of obesity vs. OSA on plant gain.
Methodological Limitations
Discrepancies between the currently published studies comparing loop gain between OSA patients and non-OSA participants is likely due to the inherent difficulties and limitations with conducting this type of research. As obesity is one of the major causes of OSA, it is difficult to find control participants matched for BMI that do not have some OSA. It is also difficult to recruit OSA participants without commonly associated comorbidities that may affect loop gain, such as respiratory and cardiac disease. Similarly, while several methods have been developed to quantify loop gain, all have some limitations. Methods such as proportional assist ventilation, CPAP drops and cycle duty ratio of spontaneously occurring respiratory events essentially quantify the ratio of the ventilatory response to the disturbance, and therefore only provide the overall loop gain and cannot separate controller and plant components (27, 28, 81). Proportional assist ventilation produces hyperventilation to induce periodic breathing. However periodic breathing can only reliably be induced in severe OSA, therefore this method is not suitable for comparing loop gain between OSA and controls (27). Similarly, methods requiring spontaneous respiratory events cannot be used to calculate loop gain in non-OSA participants (82). To quantify controller and plant gains, it is necessary to analyze breath-to-breath variations in inspiratory and expiratory gases as well as ventilation. The pseudorandom binary stimulation of CO2 method does separately quantify controller and plant gains and can be conducted in both OSA and non-OSA participants (26). However, chemosensitivity, lung volumes and many other factors that affect loop gain change with sleep onset, and due to difficulty with maintaining sleep during chemostimulation and the fact that OSA patients would require CPAP during sleep to stabilize the airway, pseudorandom binary stimulation of CO2 is practically difficult to do during sleep. Methods have been developed that use spontaneous variations in ventilation and expiratory gases which allow for quantification of both controller and plant gain in both OSA and controls during sleep (40). However, to our knowledge there are no models in humans that have been published that incorporate O2 or oxyhemoglobin measurements. Therefore, the carotid body hypoxic contribution to controller gain is not evaluated. As intermittent hypoxia is known to increase hypoxic sensitivity and OSA patients exhibit treatment-reversible elevated hypoxic sensitivity, inability to quantify hypoxic sensitivity contribution to controller gain with currently available methods likely dramatically underestimates loop gain in untreated OSA patients.
Summary
As several factors of upper airway anatomy and neuromuscular control interact to promote airway collapse, even a low/normal loop gain may be associated with pharyngeal airway collapse in participants with highly susceptible airways (38). Therefore, the lack of strong evidence for inherently high loop gain in OSA patients does not imply that loop gain does not contribute to OSA pathogenesis. Few studies have evaluated loop gain in both OSA and non-OSA controls, and data from studies that have provide conflicting results. Discrepancies between studies may be due to lack of matching for morphological traits known to alter both controller and plant gains, because most studies have not evaluated OSA patients both pre and post treatment, and due to limitations in all currently available methods to quantify loop gain.
Although loop gain studies are inconclusive, there is no evidence to suggest that lung volumes or plant gain are inherently different in OSA patients independent of other factors known to alter lung volume, such as obesity. There is also no evidence to suggest controller gain is inherently abnormal in OSA patients. However, intermittent hypoxia experienced in OSA may increase controller gain via neuroplasticity, and decrease plant gain via inflammation-induced compromised lung function. Although CPAP appears to ameliorate both abnormalities in chemoreflex control and lung function, whether total recovery is possible, or whether abnormalities persist with treatment, is unknown. Despite the inherent difficulties in conducting such research, future studies should aim to compare OSA patients both pre and post-treatment to well matched non-OSA participants, to properly assess treatment effects on controller, plant and overall loop gain. However, to thoroughly assess these aspects in both OSA and controls, development of new techniques that can separately quantify controller and plant gain contributions to overall loop gain, which can be conducted during sleep in both OSA and non-OSA participants, and which incorporate contributions of hypoxic sensitivity and CO2 sensitivity below eupnea to controller gain, will likely be necessary. Only by understanding the mechanism by which OSA appears to modulate both controller and plant gains, can treatments be developed which treat the cause of loop gain abnormalities in OSA.
Author Contributions
ND-D developed the topic and wrote the manuscript. AM helped structure the topic, edited the manuscript and approved the final draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AM is PI on NIH RO1 HL085188, K24 HL132105, T32 HL134632 and co-investigator on R21 HL121794, RO1 HL 119201, RO1 HL081823. As an Officer of the American Thoracic Society, AM has relinquished all outside personal income since 2012. ResMed, Inc. provided a philanthropic donation to the UC San Diego in support of a sleep center.
References
1. Young T, Palta M, Dempsey J, Peppard PE, Nieto FJ, Hla KM. Burden of sleep apnea: rationale, design, and major findings of the Wisconsin Sleep Cohort study. WMJ (2009) 108:246–9.
2. Weaver TE, Grunstein RR. Adherence to continuous positive airway pressure therapy: the challenge to effective treatment. Proc Am Thoracic Soc. (2008) 5:173–8. doi: 10.1513/pats.200708-119MG
3. Wellman A, Eckert DJ, Jordan AS, Edwards BA, Passaglia CL, Jackson AC, et al. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol. (2011) 110:1627–37. doi: 10.1152/japplphysiol.00972.2010
4. Owens RL, Edwards BA, Eckert DJ, Jordan AS, Sands SA, Malhotra A, et al. An integrative model of physiological traits can be used to predict obstructive sleep apnea and response to non positive airway pressure therapy. Sleep (2015) 38:961–70. doi: 10.5665/sleep.4750
5. Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol. (2008) 162:144–51. doi: 10.1016/j.resp.2008.05.019
6. Edwards BA, Sands SA, Eckert DJ, White DP, Butler JP, Owens RL, et al. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. (2012) 590:1199–211. doi: 10.1113/jphysiol.2011.223925
7. Horn EM, Waldrop TG. Suprapontine control of respiration. Respir Physiol. (1998) 114:201–11. doi: 10.1016/S0034-5687(98)00087-5
8. Horner RL. Emerging principles and neural substrates underlying tonic sleep-state-dependent influences on respiratory motor activity. Philos Trans R Soc Lond B Biol Sci. (2009) 364:2553–64. doi: 10.1098/rstb.2009.0065
9. Fink BR. Influence of cerebral activity in wakefulness on regulation of breathing. J Appl Physiol. (1961) 16:15–20. doi: 10.1152/jappl.1961.16.1.15
10. Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol. (1983) 55:813–22. doi: 10.1152/jappl.1983.55.3.813
11. Haxhiu MA, Mitra J, van Lunteren E, Prabhakar N, Bruce EN, Cherniack NS. Responses of hypoglossal and phrenic nerves to decreased respiratory drive in cats. Respiration (1986) 50:130–8. doi: 10.1159/000194919
12. Dempsey JA, Smith CA, Przybylowski T, Chenuel B, Xie A, Nakayama H, et al. The ventilatory responsiveness to CO2 below eupnoea as a determinant of ventilatory stability in sleep. J Physiol. (2004) 560:1–11. doi: 10.1113/jphysiol.2004.072371
13. Khoo MC. Determinants of ventilatory instability and variability. Respir Physiol. (2000) 122:167–82. doi: 10.1016/S0034-5687(00)00157-2
14. Khoo MCK. (ed.). Physiological Control Systems: Anlaysis, Simulation, and Estimation. New York, NY: The Institute of Electrical and Electronics Engineers, Inc. Press (2000).
15. Corne S, Webster K, Younes M. Hypoxic respiratory response during acute stable hypocapnia. Am J Respir Crit Care Med. (2003) 167:1193–9. doi: 10.1164/rccm.2203019
16. Duffin J. Measuring the ventilatory response to hypoxia. J Physiol. (2007) 584:285–93. doi: 10.1113/jphysiol.2007.138883
17. Douglas NJ, White DP, Weil JV, Pickett CK, Martin RJ, Hudgel DW, et al. Hypoxic ventilatory response decreases during sleep in normal men. Am Rev Respir Dis. (1982) 125:286–9.
18. Douglas NJ, White DP, Weil JV, Pickett CK, Zwillich CW. Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis. (1982) 126:758–62.
19. Fraigne JJ, Orem JM. Phasic motor activity of respiratory and non-respiratory muscles in REM sleep. Sleep (2011) 34:425–34. doi: 10.1093/sleep/34.4.425
20. Xie A. Effect of sleep on breathing—Why recurrent apneas are only seen during sleep. J Thorac Dis. (2012) 4:194–7. doi: 10.3978/j.issn.2072-1439.2011.04.04
21. Landry SA, Andara C, Terrill PI, Joosten SA, Leong P, Mann DL, et al. Ventilatory control sensitivity in patients with obstructive sleep apnea is sleep stage dependent. Sleep (2018) 41:zsy040. doi: 10.1093/sleep/zsy040
22. Joosten SA, Edwards BA, Wellman A, Turton A, Skuza EM, Berger PJ, et al. The effect of body position on physiological factors that contribute to obstructive sleep apnea. Sleep (2015) 38:1469–78. doi: 10.5665/sleep.4992
23. Hudgel DW, Devadatta P. Decrease in functional residual capacity during sleep in normal humans. J Appl Physiol Respir Environ Exerc Physiol. (1984) 57:1319–22. doi: 10.1152/jappl.1984.57.5.1319
24. Parameswaran K, Todd DC, Soth M. Altered respiratory physiology in obesity. Can Respir J. (2006) 13:203–10. doi: 10.1155/2006/834786
25. Younes M. CrossTalk proposal: elevated loop gain is a consequence of obstructive sleep apnoea. J Physiol. (2014) 592:2899–901. doi: 10.1113/jphysiol.2014.271833
26. Hudgel DW, Gordon EA, Thanakitcharu S, Bruce EN. Instability of ventilatory control in patients with obstructive sleep apnea. Am J Respir Crit Care Med. (1998) 158:1142–9. doi: 10.1164/ajrccm.158.4.9712105
27. Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. (2001) 163:1181–90. doi: 10.1164/ajrccm.163.5.2007013
28. Wellman A, Edwards BA, Sands SA, Owens RL, Nemati S, Butler J, et al. A simplified method for determining phenotypic traits in patients with obstructive sleep apnea. J Appl Physiol. (2013) 114:911–22. doi: 10.1152/japplphysiol.00747.2012
29. Iber C, Davies SF, Chapman RC, Mahowald MM. A possible mechanism for mixed apnea in obstructive sleep apnea. Chest (1986) 89:800–5. doi: 10.1378/chest.89.6.800
30. Remmers JE, deGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol. (1978) 44:931–8. doi: 10.1152/jappl.1978.44.6.931
31. Suratt PM, McTier R, Wilhoit SC. Alae nasi electromyographic activity and timing in obstructive sleep apnea. J Appl Physiol. (1985) 58:1252–6. doi: 10.1152/jappl.1985.58.4.1252
32. Morrell MJ, Arabi Y, Zahn B, Badr MS. Progressive retropalatal narrowing preceding obstructive apnea. Am J Respir Crit Care Med. (1998) 158:1974–81. doi: 10.1164/ajrccm.158.6.9712107
33. Onal E, Burrows DL, Hart RH, Lopata M. Induction of periodic breathing during sleep causes upper airway obstruction in humans. J Appl Physiol. (1986) 61:1438–43. doi: 10.1152/jappl.1986.61.4.1438
34. Warner G, Skatrud JB, Dempsey JA. Effect of hypoxia-induced periodic breathing on upper airway obstruction during sleep. J Appl Physiol. (1987) 62:2201–11. doi: 10.1152/jappl.1987.62.6.2201
35. Sankri-Tarbichi AG, Rowley JA, Badr MS. Expiratory pharyngeal narrowing during central hypocapnic hypopnea. Am J Respir Crit Care Med. (2009) 179:313–9. doi: 10.1164/rccm.200805-741OC
36. Hudgel DW, Hendricks C, Dadley A. Alteration in obstructive apnea pattern induced by changes in oxygen- and carbon-dioxide-inspired concentrations. Am Rev Respir Dis. (1988) 138:16–9. doi: 10.1164/ajrccm/138.1.16
37. Wellman A, Jordan AS, Malhotra A, Fogel RB, Katz ES, Schory K, et al. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. (2004) 170:1225–32. doi: 10.1164/rccm.200404-510OC
38. Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. (2013) 188:996–1004. doi: 10.1164/rccm.201303-0448OC
39. Wellman A, Malhotra A, Jordan AS, Schory K, Gautam S, White DP. Chemical control stability in the elderly. J Physiol. (2007) 581:291–8. doi: 10.1113/jphysiol.2006.126409
40. Gederi E, Nemati S, Edwards BA, Clifford GD, Malhotra A, Wellman A. Model-based estimation of loop gain using spontaneous breathing: a validation study. Respir Physiol Neurobiol. (2014) 201:84–92. doi: 10.1016/j.resp.2014.07.002
41. Sands SA, Eckert DJ, Jordan AS, Edwards BA, Owens RL, Butler JP, et al. Enhanced upper-airway muscle responsiveness is a distinct feature of overweight/obese individuals without sleep apnea. Am J Respir Crit Care Med. (2014) 190:930–7. doi: 10.1164/rccm.201404-0783OC
42. Bokov P, Essalhi M, Delclaux C. Loop gain in severely obese women with obstructive sleep apnoea. Respir Physiol Neurobiol. (2016) 221:49–53. doi: 10.1016/j.resp.2015.11.003
43. Deacon NL, Sands SA, McEvoy DR, Catcheside PG. Daytime loop gain is elevated in obstructive sleep apnea but not reduced by CPAP treatment. J Appl Physiol (2018). doi: 10.1152/japplphysiol.00175.2018. [Epub ahead of print].
44. Narkiewicz K, Kato M, Pesek CA, Somers VK. Human obesity is characterized by a selective potentiation of central chemoreflex sensitivity. Hypertension (1999) 33:1153–8. doi: 10.1161/01.HYP.33.5.1153
45. Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation (1999) 99:1183–9. doi: 10.1161/01.CIR.99.9.1183
46. Foster GE, Hanly PJ, Ostrowski M, Poulin MJ. Ventilatory and cerebrovascular responses to hypercapnia in patients with obstructive sleep apnoea: effect of CPAP therapy. Respir Physiol Neurobiol. (2009) 165:73–81. doi: 10.1016/j.resp.2008.10.011
47. Salloum A, Rowley JA, Mateika JH, Chowdhuri S, Omran Q, Badr MS. Increased propensity for central apnea in patients with obstructive sleep apnea: effect of nasal continuous positive airway pressure. Am J Respir Crit Care Med. (2010) 181:189–93. doi: 10.1164/rccm.200810-1658OC
48. Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A, Hanly P. Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol. (2007) 103:1929–41. doi: 10.1152/japplphysiol.00561.2007
49. Spicuzza L, Bernardi L, Balsamo R, Ciancio N, Polosa R, Di Maria G. Effect of treatment with nasal continuous positive airway pressure on ventilatory response to hypoxia and hypercapnia in patients with sleep apnea syndrome. Chest (2006) 130:774–9. doi: 10.1378/chest.130.3.774
50. Loewen A, Ostrowski M, Laprairie J, Atkar R, Gnitecki J, Hanly P, et al. Determinants of ventilatory instability in obstructive sleep apnea: inherent or acquired? Sleep (2009) 32:1355–65. doi: 10.1093/sleep/32.10.1355
51. Harris DP, Balasubramaniam A, Badr MS, Mateika JH. Long-term facilitation of ventilation and genioglossus muscle activity is evident in the presence of elevated levels of carbon dioxide in awake humans. Am J Physiol Regul Integr Comp Physiol. (2006) 291:R1111–9. doi: 10.1152/ajpregu.00896.2005
52. Lee DS, Badr MS, Mateika JH. Progressive augmentation and ventilatory long-term facilitation are enhanced in sleep apnoea patients and are mitigated by antioxidant administration. J Physiol. (2009) 587:5451–67. doi: 10.1113/jphysiol.2009.178053
53. Mateika JH, Mendello C, Obeid D, Badr MS. Peripheral chemoreflex responsiveness is increased at elevated levels of carbon dioxide after episodic hypoxia in awake humans. J Appl Physiol. (2004) 96:1197–205. doi: 10.1152/japplphysiol.00573.2003
54. Pialoux V, Hanly PJ, Foster GE, Brugniaux JV, Beaudin AE, Hartmann SE, et al. Effects of exposure to intermittent hypoxia on oxidative stress and acute hypoxic ventilatory response in humans. Am J Respir Crit Care Med. (2009) 180:1002–9. doi: 10.1164/rccm.200905-0671OC
55. Chowdhuri S, Shanidze I, Pierchala L, Belen D, Mateika JH, Badr MS. Effect of episodic hypoxia on the susceptibility to hypocapnic central apnea during NREM sleep. J Appl Physiol. (2010) 108:369–77. doi: 10.1152/japplphysiol.00308.2009
56. Brugniaux JV, Pialoux V, Foster GE, Duggan CT, Eliasziw M, Hanly PJ, et al. Effects of intermittent hypoxia on erythropoietin, soluble erythropoietin receptor and ventilation in humans. Eur Respir J. (2011) 37:880–7. doi: 10.1183/09031936.00156009
57. Reeves SR, Gozal E, Guo SZ, Sachleben LRJr, Brittian KR, Lipton AJ, et al. Effect of long-term intermittent and sustained hypoxia on hypoxic ventilatory and metabolic responses in the adult rat. J Appl Physiol. (2003) 95:1767–74. doi: 10.1152/japplphysiol.00759.2002
58. Peng Y, Kline DD, Dick TE, Prabhakar NR. Chronic intermittent hypoxia enhances carotid body chemoreceptor response to low oxygen. Adv Exp Med Biol. (2001) 499:33–8. doi: 10.1007/978-1-4615-1375-9_5
59. Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA. (2003) 100:10073–8. doi: 10.1073/pnas.1734109100
60. Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. (2004) 96:1236–42. doi: 10.1152/japplphysiol.00820.2003
61. Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. (1996) 104:251–60. doi: 10.1016/0034-5687(96)00017-5
62. Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, et al. Invited review: intermittent hypoxia and respiratory plasticity. J Appl Physiol. (2001) 90:2466–75. doi: 10.1152/jappl.2001.90.6.2466
63. Khodadadeh B, Badr MS, Mateika JH. The ventilatory response to carbon dioxide and sustained hypoxia is enhanced after episodic hypoxia in OSA patients. Respir Physiol Neurobiol. (2006) 150:122–34. doi: 10.1016/j.resp.2005.04.019
64. Olson EBJr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, et al. Ventilatory long-term facilitation in unanesthetized rats. J Appl Physiol. (2001) 91:709–16. doi: 10.1152/jappl.2001.91.2.709
65. Yuan H, Pinto SJ, Huang J, McDonough JM, Ward MB, Lee YN, et al. Ventilatory responses to hypercapnia during wakefulness and sleep in obese adolescents with and without obstructive sleep apnea syndrome. Sleep (2012) 35:1257–67. doi: 10.5665/sleep.2082
66. Verbraecken J, De Backer W, Willemen M, De Cock W, Wittesaele W, Van de H. Chronic CO2 drive in patients with obstructive sleep apnea and effect of CPAP. Respir Physiol. (1995) 101:279–87. doi: 10.1016/0034-5687(95)00037-E
67. Sin DD, Jones RL, Man GC. Hypercapnic ventilatory response in patients with and without obstructive sleep apnea: do age, gender, obesity, and daytime PaCO2 matter? Chest (2000) 117:454–9. doi: 10.1378/chest.117.2.454
68. Kara T, Narkiewicz K, Somers VK. Chemoreflexes–physiology and clinical implications. Acta Physiol Scand. (2003) 177:377–84. doi: 10.1046/j.1365-201X.2003.01083.x
69. Zerah-Lancner F, Lofaso F, Coste A, Ricolfi F, Goldenberg F, Harf A. Pulmonary function in obese snorers with or without sleep apnea syndrome. Am J Respir Crit Care Med. (1997) 156:522–7. doi: 10.1164/ajrccm.156.2.9609015
70. Alkhalil M, Schulman ES, Getsy J. Obstructive sleep apnea syndrome and asthma: the role of continuous positive airway pressure treatment. Ann Allergy Asthma Immunol. (2008) 101:350–7. doi: 10.1016/S1081-1206(10)60309-2
71. Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med. (2010) 182:325–31. doi: 10.1164/rccm.200912-1869OC
72. Wang TY, Lo YL, Lin SM, Huang CD, Chung FT, Lin HC, et al. Obstructive sleep apnoea accelerates FEV1 decline in asthmatic patients. BMC Pulm Med. (2017) 17:55. doi: 10.1186/s12890-017-0398-2
73. Sundar KM, Daly SE, Willis A. Is untreated obstructive sleep apnea (OSA) a cause of COPD in non-smokers? In: B42 COPD Pathogenesis: Cellular Expression and Protein Responses. American Thoracic Society (2011). A3023p.
74. Chua AP, Aboussouan LS, Minai OA, Paschke K, Laskowski D, Dweik RA. Long-term continuous positive airway pressure therapy normalizes high exhaled nitric oxide levels in obstructive sleep apnea. J Clin Sleep Med. (2013) 9:529–35. doi: 10.5664/jcsm.2740
75. Tichanon P, Wilaiwan K, Sopida S, Orapin P, Watchara B, Banjamas I. Effect of continuous positive airway pressure on airway inflammation and oxidative stress in patients with obstructive sleep apnea. Can Respir J. (2016) 2016:3107324. doi: 10.1155/2016/3107324
76. Devouassoux G, Levy P, Rossini E, Pin I, Fior-Gozlan M, Henry M, et al. Sleep apnea is associated with bronchial inflammation and continuous positive airway pressure-induced airway hyperresponsiveness. J Allergy Clin Immunol. (2007) 119:597–603. doi: 10.1016/j.jaci.2006.11.638
77. Broytman O, Braun RK, Morgan BJ, Pegelow DF, Hsu PN, Mei LS, et al. Effects of chronic intermittent hypoxia on allergen-induced airway inflammation in rats. Am J Respir Cell Mol Biol. (2015) 52:162–70. doi: 10.1165/rcmb.2014-0213OC
78. Tuleta I, Stockigt F, Juergens UR, Pizarro C, Schrickel JW, Kristiansen G, et al. Intermittent hypoxia contributes to the lung damage by increased oxidative stress, inflammation, and disbalance in protease/antiprotease system. Lung (2016) 194:1015–20. doi: 10.1007/s00408-016-9946-4
79. Lee EJ, Heo W, Kim JY, Kim H, Kang MJ, Kim BR, et al. Alteration of inflammatory mediators in the upper and lower airways under chronic intermittent hypoxia: preliminary animal study. Mediators Inflamm. (2017) 2017:4327237. doi: 10.1155/2017/4327237
80. Zhang X, Rui L, Wang M, Lian H, Cai L. Sinomenine attenuates chronic intermittent hypoxia-induced lung injury by inhibiting inflammation and oxidative stress. Med Sci Monit. (2018) 24:1574–80. doi: 10.12659/MSM.906577
81. Sands SA, Edwards BA, Kee K, Turton A, Skuza EM, Roebuck T, et al. Loop gain as a means to predict a positive airway pressure suppression of Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med. (2011) 184:1067–75. doi: 10.1164/rccm.201103-0577OC
Keywords: loop gain, obstructive sleep apnea, chemoreflex control, neuroplasticity, functional residual capacity
Citation: Deacon-Diaz N and Malhotra A (2018) Inherent vs. Induced Loop Gain Abnormalities in Obstructive Sleep Apnea. Front. Neurol. 9:896. doi: 10.3389/fneur.2018.00896
Received: 29 May 2018; Accepted: 02 October 2018;
Published: 02 November 2018.
Edited by:
Ahmed S. BaHammam, King Saud University, Saudi ArabiaReviewed by:
Andrea Romigi, Istituto Neurologico Mediterraneo (IRCCS), ItalyLeszek Kubin, University of Pennsylvania, United States
Copyright © 2018 Deacon-Diaz and Malhotra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naomi Deacon-Diaz, bmRlYWNvbkB1Y3NkLmVkdQ==