 Sean C. Dougherty
Sean C. Dougherty Amy Harper
Amy Harper Hind Al Saif
Hind Al Saif Gregory Vorona3
Gregory Vorona3 Scott R. Haines
Scott R. Haines- 1Department of Neurology, Virginia Commonwealth University, Richmond, VA, United States
- 2Department of Human and Molecular Genetics, Virginia Commonwealth University, Richmond, VA, United States
- 3Department of Radiology, Virginia Commonwealth University, Richmond, VA, United States
Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) is a rare, progressive, neurodegenerative disease characterized by ataxia, spasticity and polyneuropathy. First described in the French-Canadian population of Quebec in 1978, ARSACS has since been identified in multiple patients worldwide. In this clinical case report, we describe the evaluation of an 11-years-old African-American male who presented to neuromuscular clinic for assessment of a gait abnormality. He had a history of gross motor delay since early childhood, frequent falls and a below average IQ. Chromosomal microarray revealed a 1.422 megabase loss in the 13q12.12 region, which includes the SACS gene. Next Generation Sequencing then showed a novel, predicted to be pathogenic missense mutation (c.11824dup) of this gene. His clinical presentation and neurological imaging further confirmed the diagnosis of ARSACS. To our knowledge, this is the first reported case of this disease in the African-American population of the United States. This case report further highlights the growing trend of identifying genetic diseases previously restricted to single, ethnically isolated regions in many different ethnic groups worldwide.
Background
Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) is a rare neurodegenerative condition initially identified in the French-Canadian population in 1978 (1). Secondary to a mutation of the SACS gene on chromosome 13q, this disease is characterized by cerebellar ataxia, spasticity, polyneuropathy, nystagmus and retinal changes (2). It is inherited in an autosomal recessive manner and typically manifests in early childhood as difficulty with ambulation and gait unsteadiness. ARSACS is slowly progressive, eventually leading to loss of ambulation around age 40 (range 17–58) and death around age 50 (range 21–72) (3).
Originally characterized in two regions of northeastern Quebec previously settled by individuals of French descent (4), ARSACS has since been identified in multiple patients worldwide. Whereas, most of the affected patients in French-Canada had the same single nucleotide deletion in the exon of the SACS gene, new missense mutations have been identified in individuals from Tunisia, Turkey, Italy, Japan and multiple countries in Europe (5–11).
The worldwide incidence of ARSACS is unknown, though it is thought to be underdiagnosed. Within the Saguenay-Lac-St-Jean region of Quebec, however, the estimated incidence is between 1 in 1,500 to 2,000 individuals. The estimated carrier frequency of SACS pathogenic variants is 1 in 21 individuals based on population data accumulated between 1941 and 1985 (4, 12, 13).
In this clinical case report, we detail the first published case of ARSACS in an African-American patient in the United States, highlighting the clinical presentation, neurological imaging and genetic analysis involved in diagnosis. Furthermore, we identified a novel, predicted to be pathogenic missense mutation in the SACS gene (c.11824dup) resulting in the ARSACS phenotype.
Case Presentation
An 11 years-old African-American male presented to pediatric neuromuscular clinic for evaluation of gait abnormality. He had a history of gross motor delay since age 3 and was enrolled in both physical and occupational therapy. His mother noted frequent falls, particularly when running or getting onto the school bus, as well as chronic headaches and blurry vision. Previously performed neuropsychological testing showed a below average IQ and an MRI of the brain at age 3 was normal (Figure 1A). An MRI of the lumbosacral spine from age 5 was also reportedly normal. Prior laboratory evaluation, including CPK, ANA and Acetylcholine receptor antibodies, was normal. He had never had a formal eye exam. Family history was significant for a maternal great grandfather who required leg braces starting at age 6 (with removal around age 10), who also reportedly had a similar posture as the patient. There was also a family history of non-specific visual symptoms in multiple family members on his mother's side as well as an older sibling who previously had seizures. He was not of French-Canadian descent, was the child of a non-consanguineous conception and had two half-brothers and one half-sister, all of whom were alive and well aside from the aforementioned seizures. Both of his parents and all of his grandparents were African-American as well.
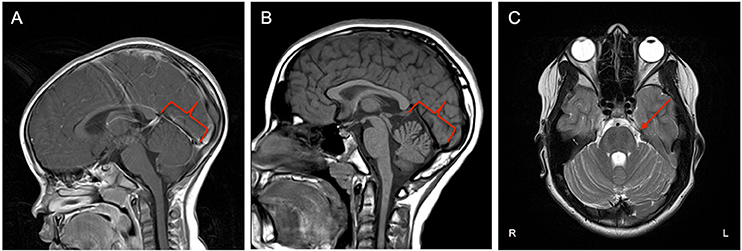
Figure 1. (A) Midsagittal MRI at age 3 showing no cerebellar changes at that time. (B) Midsagittal MRI at age 11 showing evidence of asymmetric volume loss within the superior and middle components of the cerebellar vermis with preserved volume of the inferior component (red outline). (C) Axial plane MRI exhibiting faint linear/T2 signal hypointensities within the Pons (red arrow).
On examination, he had gaze-evoked nystagmus and saccadic dysmetria with undershoot saccades in all directions, prominent white striations emanating adjacent to the optic nerve withdistinct optic disc margins on fundoscopy (Figure 2A), and symmetric pupils with no afferent defect. Subtle choreiform movements were noted in the fingers with outstretched hands and end point tremor on finger to nose testing was present. In the lower extremities, there was increased tone of spastic nature. Patellar reflexes were brisk and ankle jerks were decreased with associated weakness. Cross adduction was present. His gait was wide based and staggering; he was unable to perform tandem gait testing. Spasticity with mild scissoring was apparent with casual gait testing.
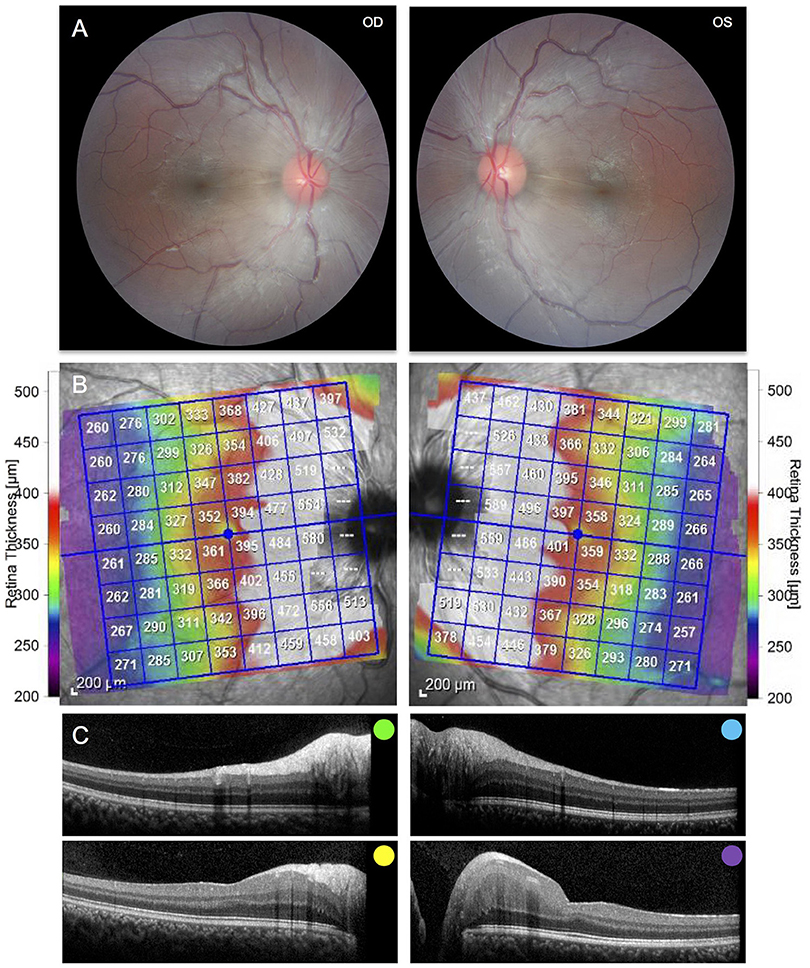
Figure 2. (A) Fundus photography of the right and left eyes demonstrating peripapillary retinal nerve fiber hypertrophy and white striations emanating adjacent to the optic nerve. (B) Posterior pole Optical Coherence Tomography demonstrating hypertrophy of the retinal nerve fiber layer and other inner retinal layers. (C) Selected cross sections of the right and left eye demonstrating hypertrophy of the retinal nerve fiber layer and other inner retinal layers. Colors correspond to the retinal layers depicted above.
Brain MRI revealed asymmetric volume loss within the superior and middle aspects of the cerebellar vermis relative to the inferior aspect (Figure 1B) and faint linear T2 signal hypointensities within the pons (Figure 1C), findings that have been shown to be associated with ARSACS (14, 15). Fundoscopic photography and Optical Coherence Tomography both demonstrated hypertrophy of the retinal nerve fiber layer and other retinal layers (Figures 2B,C), findings also observed in ARSACS (16). Electromyography performed was abnormal for both sensory and motor components in multiple nerves tested, providing electrophysiological evidence for a sensory greater than motor axonal polyneuropathy.
Initial chromosome microarray then showed a 1.422 megabase loss in the 13q12.12 region (Figure 3), of which there are 14 genes present. Three of these genes have autosomal recessive genetic conditions associated with them, including Limb-girdle muscular dystrophy type 2C (SGCG gene), ARSACS (SACS gene), and Combined oxidative phosphorylation deficiency 31 (MIPEP gene). Subsequent Next Generation Sequencing of these genes revealed that the patient had hemizygous, likely pathogenic variant in the SACS gene (c.11824dup), which was predicted to result in a frameshift mutation with premature protein termination (p.Met3942Asnfs*4). Taken together, these clinical and genetic findings are consistent with the diagnosis of ARSACS.
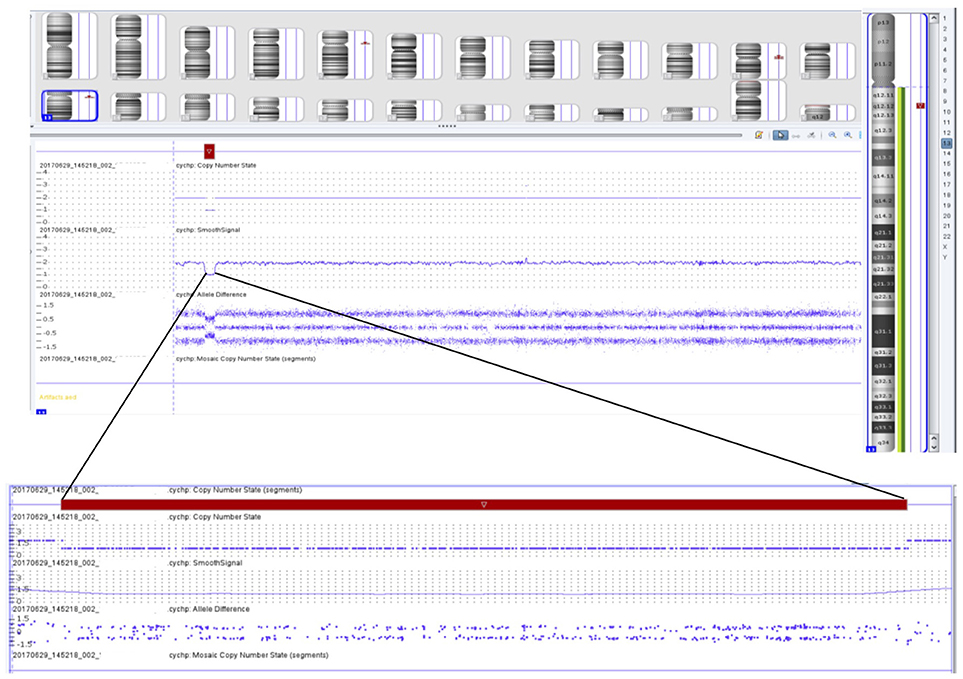
Figure 3. Chromosomal microarray showing 1.422 megabase loss in the 13q12.12 region containing 14 different genes, including the SACS gene.
Current treatment is focused on symptomatic management and improvement of daily life. Gait stabilization with the wearing of ankle foot orthoses has been observed and he has been receiving scheduled botox injections to the bilateral hamstrings every 90 days with improvement in flexibility.
Discussion
With the implementation of targeted high throughput sequencing and identification of novel genetic mutations, ARSACS, a disease previously reported primarily in the French-Canadian population of Quebec, is now being identified worldwide (17). Here, we have described the clinical presentation, diagnostic evaluation and management of the first reported case of ARSACS in the African-American population. Furthermore, we identified a previously unreported mutation in the SACS gene, designated c. 11824dup.
A number of different mutations have been shown to cause the ARSACS phenotype (18–20), which is thought to result from loss of function of the encoded protein Sacsin. The exact role of this large, 520-kDa cytoplasmic protein remains unknown, though it has been proposed to have roles in mitochondrial function (21), protein chaperoning (22), and the ubiquitin-proteasome system (23). Further studies have shown that Sacsin knockout results in increased reactive oxygen species production (24) and alterations in synaptic input and intrinsic firing in cerebellar Purkinje cells (25).
The observed genetics in this patient are atypical in that he is a compound heterozygote, wherein full chromosomal deletion of one of the SACS genes combined with the observed frameshift mutation (hemizygous) resulted in the ARSACS phenotype. A hemizygous presentation of this disease has previously been described in the Belgian population (26). These two cases are unique in that they involve gross chromosomal deletion of the SACS gene, whereas all previously reported mutations in patients with ARSACS involved either point mutations or missense mutations in this gene. Moderate perceptive hearing loss was described as the only feature by which the Belgian patient with ARSACS differed from other cases of the disease, however this feature was not present in our patient.
Despite a family history of gait abnormalities on his mother's side, we cannot comment specifically on the nature of this patient's inheritance without formal genetic testing of his parents, which has not been performed at this time. De novo or inherited deletion of the SACS gene from one parent combined with another de novo or inherited likely pathogenic variant from the other parent would be a theory of inheritance compatible with the history and known genetics of this patient.
Concluding Remarks
In sum, a high index of clinical suspicion combined with appropriate genetic testing should allow for continued identification of the ARSACS phenotype, a disease whose worldwide prevalence will undoubtedly continue to rise as further novel mutations are characterized.
Ethics Statement
All clinical data in this case report was collected with the consent of the patient and his mother. A written informed consent was obtained from the patient and his parents for the participation in the study and the publication of this report. The case report is exempt from institutional review board approval.
Author Contributions
SD wrote the manuscript under the mentorship of SH. AH acquired and interpreted clinical data and aided in diagnosis. HA interpreted genetic data and provided commentary. GV interpreted radiologic images and provided commentary. SH acquired ophthalmologic photographs and clinical data, interpreted data and supervised case report formulation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the patient and his family members for entrusting them with his care.
Abbreviations
ARSACS, Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay.
References
1. Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci. (1978) 5:61–9.
2. Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. (2000) 24:120–5. doi: 10.1038/72769
3. Bouchard JP. Ressesive spastic ataxia of Charlevoix-Saguenay. In: de Jong MBV, editors. Hereditary Neuropathies and Spinocerebellar Atrophies. Handbook of Clinical Neurology. Amsterdam: Elsevier (1991). p. 451–9.
4. De Braekeleer M, Giasson F, Mathieu J, Roy M, Bouchard JP, Morgan K. Genetic epidemiology of autosomal recessive spastic ataxia of Charlevoix-Saguenay in northeastern Quebec. Genet Epidemiol. (1993) 10:17–25. doi: 10.1002/gepi.1370100103
5. El Euch-Fayache G, Lalani I, Amouri R, Turki I, Ouahchi K, Hung WY, et al. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol. (2003) 60:982–8. doi: 10.1001/archneur.60.7.982
6. Criscuolo C, Banfi S, Orio M, Gasparini P, Monticelli A, Scarano V, et al. A novel mutation in SACS gene in a family from southern Italy. Neurology (2004) 62:100–2. doi: 10.1212/WNL.62.1.100
7. Grieco GS, Malandrini A, Comanducci G, Leuzzi V, Valoppi M, Tessa A, et al. Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay type. Neurology (2004) 62:103–6. doi: 10.1212/01.WNL.0000104491.66816.77
8. Ogawa T, Takiyama Y, Sakoe K, Mori K, Namekawa M, Shimazaki H, et al. Identification of a SACS gene missense mutation in ARSACS. Neurology (2004) 62:107–9. doi: 10.1212/01.WNL.0000099371.14478.73
9. Richter AM, Ozgul RK, Poisson VC, Topaloglu H. Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics (2004) 5:165–70. doi: 10.1007/s10048-004-0179-y
10. Vermeer S, Meijer RP, Pijl BJ, Timmermans J, Cruysberg JR, Bos MM, et al. ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics (2008) 9:207–14. doi: 10.1007/s10048-008-0131-7
11. Baets J, Deconinck T, Smets K, Goossens D, Van den Bergh P, Dahan K, et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology (2010) 75:1181–8. doi: 10.1212/WNL.0b013e3181f4d86c
12. De Braekeleer M. Hereditary disorders in Saguenay-Lac-St-Jean (Quebec, Canada). Hum Hered. (1991) 41:141–6. doi: 10.1159/000153992
13. Dupre N, Bouchard JP, Brais B, Rouleau GA. Hereditary ataxia, spastic paraparesis and neuropathy in the French-Canadian population. Can J Neurol Sci. (2006) 33:149–57. doi: 10.1017/S031716710000490X
14. Bouchard JP, Richter A, Mathieu J, Brunet D, Hudson TJ, Morgan K, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuromuscul Disord. (1998) 8:474–9. doi: 10.1016/S0960-8966(98)00055-8
15. Martin MH, Bouchard JP, Sylvain M, St-Onge O, Truchon S. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of MR imaging in 5 patients. AJNR Am J Neuroradiol. (2007) 28:1606–8. doi: 10.3174/ajnr.A0603
16. Yu-Wai-Man P, Pyle A, Griffin H, Santibanez-Korev M, Horvath R, Chinnery PF. Abnormal retinal thickening is a common feature among patients with ARSACS-related phenotypes. Br J Ophthalmol. (2014) 98:711–3. doi: 10.1136/bjophthalmol-2013-304534
18. Vermeer S, van de Warrenburg BP, Kamsteeg EJ. Arsacs. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews((R)). Seattle, WA: University of Washington. (1993).
19. Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schule R, et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis. (2013) 8:41. doi: 10.1186/1750-1172-8-41
20. Thiffault I, Dicaire MJ, Tetreault M, Huang KN, Demers-Lamarche J, Bernard G, et al. Diversity of ARSACS mutations in French-Canadians. Can J Neurol Sci. (2013) 40:61–6. doi: 10.1017/S0317167100012968
21. Girard M, Lariviere R, Parfitt DA, Deane EC, Gaudet R, Nossova N, et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Natl Acad Sci USA. (2012) 109:1661–6. doi: 10.1073/pnas.1113166109
22. Duncan EJ, Lariviere R, Bradshaw TY, Longo F, Sgarioto N, Hayes MJ, et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum Mol Genet. (2017) 26:3130–43. doi: 10.1093/hmg/ddx197
23. Parfitt DA, Michael GJ, Vermeulen EG, Prodromou NV, Webb TR, Gallo JM, et al. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet. (2009) 18:1556–65. doi: 10.1093/hmg/ddp067
24. Criscuolo C, Procaccini C, Meschini MC, Cianflone A, Carbone R, Doccini S, et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J Neurol. (2015) 262:2755–63. doi: 10.1007/s00415-015-7911-4
25. Ady V, Toscano-Marquez B, Nath M, Chang PK, Hui J, Cook A, et al. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J Physiol. 596:4253–67. (2018). doi: 10.1113/JP275902
Keywords: ARSACS, ataxia, Charlevoix-Saguenay, cerebellum, neurogenetics, polyneuropathy, spasticity, retina
Citation: Dougherty SC, Harper A, Al Saif H, Vorona G and Haines SR (2018) A Chromosomal Deletion and New Frameshift Mutation Cause ARSACS in an African-American. Front. Neurol. 9:956. doi: 10.3389/fneur.2018.00956
Received: 07 September 2018; Accepted: 25 October 2018;
Published: 15 November 2018.
Edited by:
Kette D. Valente, Universidade de São Paulo, BrazilReviewed by:
Duccio Maria Cordelli, Università degli Studi di Bologna, ItalyAnna Lecticia Pinto, Boston Children's Hospital, United States
Rudá Alessi, Faculdade de Medicina do ABC, Brazil
Copyright © 2018 Dougherty, Harper, Al Saif, Vorona and Haines. This is an openaccess article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott R. Haines, c2NvdHQuaGFpbmVzQHZjdWhlYWx0aC5vcmc=