 Atsuko Katsumoto
Atsuko Katsumoto Hideyuki Takeuchi
Hideyuki Takeuchi Keita Takahashi
Keita Takahashi- Department of Neurology and Stroke Medicine, Yokohama City University Graduate School of Medicine, Yokohama, Japan
Microglia are resident immune cells in the central nervous system (CNS) that originate from myeloid progenitor cells in the embryonic yolk sac and are maintained independently of circulating monocytes throughout life. In the healthy state, microglia are highly dynamic and control the environment by rapidly extending and retracting their processes. When the CNS is inflamed, microglia can give rise to macrophages, but the regulatory mechanisms underlying this process have not been fully elucidated. Recent genetic studies have suggested that microglial function is compromised in Alzheimer's disease (AD), and that environmental factors such as diet and brain injury also affect microglial activation. In addition, studies of triggering receptor expressed on myeloid cells 2-deficiency in AD mice revealed heterogeneous microglial reactions at different disease stages, complicating the therapeutic strategy for AD. In this paper, we describe the relationship between genetic and environmental risk factors and the roles of microglia in AD pathogenesis, based on studies performed in human patients and animal models. We also discuss the mechanisms of inflammasomes and neurotransmitters in microglia, which accelerate the development of amyloid-β and tau pathology.
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disease. AD brains are characterized by the combined presence of two structures: extracellular amyloid-β (Aβ) plaques and intraneuronal neurofibrillary tangles. Aβ plaques create an environment that facilitates the rapid amplification and spread of pathological tau into large aggregates, initially appearing as the neuritic, phosphorylated, microtubule-associated protein tau. This is followed by the formation and spread of neurofibrillary tangles and neuropil threads to other neurons (1).
Recent genetic studies have identified variants in immune-related genes that increase the risk of developing AD (2), implicating the neuroinflammatory response in AD pathogenesis. Notably in this regard, coding variants in the triggering receptor expressed on myeloid cells 2 (TREM2) gene confer the highest AD risk, indicating that microglial neuroinflammation plays a critical role in AD progression (3, 4). In accordance with these findings, a single-nucleotide polymorphism in the gene encoding the microglial surface receptor CD33 reduces Aβ phagocytosis by peripheral macrophages isolated from carriers of heterozygous and homozygous mutations (5–7) supporting the hypothesis that microglial function is compromised.
The microglial phenotype may change drastically over the course of neurodegeneration, as demonstrated by studies of TREM2 deficiency in a mouse model of AD (8). A recent comprehensive survey of the transcriptome of hippocampal microglia over the course of progression from the healthy to neurodegenerative state, performed at a single-cell resolution, revealed the remarkable
phenotypic heterogeneity of microglia: the early response state is characterized by marked proliferation, whereas the late response state is associated with mounting immune responses (9). In the latter state, two functionally distinct reactive microglial phenotypes, typified by modules of co-regulated type 1 and type 2 interferon response genes, have been identified (9). These functional changes in microglia are also influenced by environmental factors such as diet, brain injury, or smoking.
Here, we review how genetics and environmental factors influence microglial functions, and then illustrate the therapeutic targets in AD, with special emphasis on microglial inflammasomes and neurotransmitters.
AD Risk Factors and Microglia
Genetic Variants in TREM2
TREM2 is a type I transmembrane receptor expressed in a subset of myeloid lineage cells including microglia, dendritic cells, osteoclasts, monocytes, and tissue macrophages (10, 11). Homozygous mutations in TREM2 cause Nasu-Hakola disease, and rare heterozygous variants are associated with other neurodegenerative diseases such as late-onset AD (3, 4), frontotemporal dementia (12), and Parkinson's disease (13). Although the exact molecular mechanisms underlying the development of neurodegeneration in the brain remain unknown, abnormalities in TREM2 and its interacting partner DNAX activating protein of 12 kDa (DAP12) appear to cause dysregulation of microglial inflammatory responses and neuronal debris clearance (14, 15). In addition, TREM2 affects microglial survival in an AD mouse model, as TREM2-deficient microglia are not able to sustain microgliosis and undergo apoptosis rather than becoming activated (15). Transcriptome analysis also revealed the role of TREM2 in chemotaxis, migration, and mobility (16), as TREM2 deficiency results in ineffective plaque encapsulation of Aβ and reduced plaque compaction, which is associated with worsened axonal pathology. Data from TREM2 knockout mice revealed that CCL2, IL-1β, TNF-α, and secreted phosphoprotein 1 (SPP1) are the direct targets of TREM2 signaling (16). Furthermore, TREM2 deficiency influences the microglial metabolic state through the mammalian target of rapamycin pathway (17). Microglia lacking TREM2 undergo global changes in their metabolism, resulting in reduced ATP levels and signs of stress and death. These observations imply that TREM2 is a critical regulator of microglial phenotypes.
Interestingly, TREM2 plays distinct functional roles at different stages: in a mouse model of AD, TREM2 deficiency ameliorates amyloid pathology in the early disease stage, but exacerbates the pathology as the disease progresses (8). One possible explanation might be that TREM2 deficiency affects different myeloid cell subsets at different stages of AD pathology. TREM2 deficiency first affects CD45hi myeloid cells, where it is primarily expressed, but subsequent loss of these CD45hi cells also affects the function of CD45lo myeloid cells, decreasing their proliferation and potentially altering other AD-related phenotypes. Regarding the change in microglial phenotypes, immune memory in microglia has been shown to modify Aβ pathology in AD mice (18), in which repeated stimulation shift from inflammatory to phagocytic microglia by differential epigenetic reprogramming. The blocking of epigenetic factors enhanced immune training in microglia, decreases Aβ levels and improves memory in AD mice (19).
Recent studies reported that binding of apolipoproteins including apolipoprotein E (APOE) with TREM2 facilitates microglial uptake of Aβ (20) and that the TREM2-APOE pathway was identified as the mechanism responsible for switching from a homeostatic to a neurodegenerative microglial phenotype after phagocytosis of apoptotic neurons (21) (Figure 1). Targeting the TREM2-APOE pathway restored the homeostatic signature of microglia in AD mouse models and prevented neuronal loss in an acute model of neurodegeneration (21). Moreover, the APOE-mediated neurodegenerative microglia lost their tolerogenic function. These findings imply that the TREM2-APOE pathway is a major regulator of the microglial functional phenotype in neurodegenerative diseases. On the contrary, the transition from homeostatic microglia expressing Cx3cr1, P2ry12, and Tmem119 to the disease-associated microglia (DAM) state with induction of ApoE was independent of TREM2 (22). Following loss of homeostatic signature, microglia increase phagocytic and lipid metabolism activity including upregulation of TREM2 and lipoprotein lipase to be the full DAM, which depends on TREM2. Moreover, loss of microglial CX3CR1 has opposing effects Aβ and tau pathologies (23, 24). Further studies are needed to uncover the precise mechanism of TREM2-APOE pathway in AD pathology.
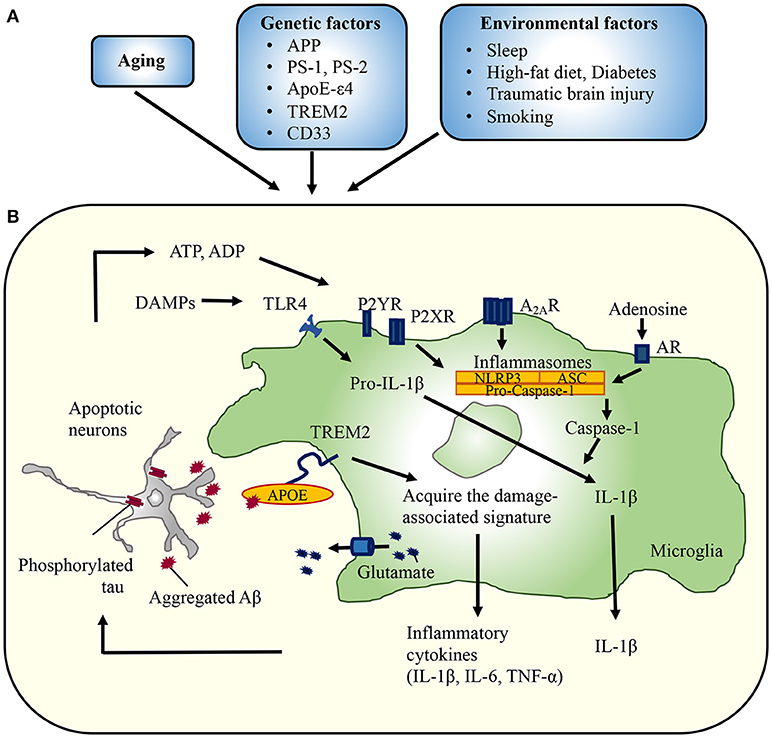
Figure 1. Implication of microglia in the development of Alzheimer's disease. (A) Several conditions are associated with an increased risk of developing AD. For example, variants in TREM2 ameliorate amyloid pathology in the early disease stage, but exacerbate pathology as the disease progresses. The TREM2-APOE pathway is responsible for switching from a homeostatic to a neurodegenerative microglial phenotype after phagocytosis of apoptotic neurons. Environmental factors also affect the microglial reaction to aggregated Aβ or phosphorylated tau. Head trauma also leads to a local increase in the levels of inflammatory mediators, which may stimulate Aβ generation and restrict phagocytic clearance. Likewise, microbiota influenced by diabetes or diet may regulate microglial phenotypes. (B) Aggregated Aβ or phosphorylated tau impairs synaptic functions, triggering the release of neurotoxic mediators from microglia. ATP, ADP, and adenosine activate NLRP3 inflammasomes, followed by the release of IL-1β. Similarly, glutamate released from gap junction hemichannels lead to massive neuronal damage. APP, amyloid precursor protein; PS, presenilin; ApoEε4, apolipoprotein Eε4; TREM2, triggering receptor expressed on myeloid cells 2; P2X(Y)R, purinergic receptor; A2ARs, adenosine A2A receptors; AR, adenosine receptor; DAMPs, damage-associated molecular patterns; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain.
These studies were performed on animal models of Aβ-related pathologies, but little is known regarding the role of TREM2 in regulating intracellular tau pathology. Elevated levels of soluble TREM2 in the cerebrospinal fluid (CSF) of AD patients, as determined by mass spectrometry, are correlated with levels of CSF total tau and phosphorylated-tau, but not the level of CSF Aβ42 (25). Notably in this regard, CSF analysis revealed that a recently reported rare variant in TREM2 (p.R47H, rs75932628) is significantly associated with the risk of AD (26). In addition, carriers of the risk allele exhibited similar phenotypes (significantly elevated levels of CSF total tau, but not Aβ42, in AD patients). In addition, our group has recently reported that TREM2 deficiency leads to heightened tau pathology coupled with widespread activation of neuronal stress kinases, including ERK1/2 and JNK, in a mouse model of tauopathy (27). These observations support the hypothesis that CSF TREM2 is a marker for tau dysfunction in AD.
Traumatic Brain Injury (TBI)
TBI is associated with the development of neurodegenerative conditions such as AD and chronic traumatic encephalopathy. A prominent feature of TBI is the development of an inflammatory reaction within minutes of the injury event. Damage-associated molecular patterns (DAMPs) (e.g., ATP, reactive oxygen species, damaged mitochondria, and necrotic cells) activate microglia and resident mononuclear phagocytes in the CNS, which promote neuroprotection and repair through the clearance of tissue debris and subsequent resolution of the inflammatory response (28, 29). Unless properly controlled, microglial activity leads to further neuronal damage through secretion of pro-inflammatory cytokines and reactive species, as well as, other mechanisms (29). Analysis of mRNA expression in microglia/macrophages revealed a rapid rise and fall in the protective phenotype (CD206, Arg1, Ym1/2, and TGF-β) and a sustained rise in the inflammatory phenotype (iNOS, CD11b, CD16, and CD86) after TBI (30). On the other hand, blocking neural/microglial interaction via CX3CR1 deficiency conferred neurological protection at early time points after TBI, but caused appreciable impairments accompanied by persistent neuronal death at later times (31, 32). In vivo imaging with positron emission tomography for activated microglia in patients revealed elevated microglial activation for several years after TBI (33).
How, then, can microglia activated by TBI trigger rapid and insidiously progressive AD-like pathological changes? Elevation of the Aβ burden and phosphorylated tau has been observed in patients within hours after TBI (34, 35). TBI-induced axonal injury is among the first perturbations of tau that results in dissociation from microtubules. Cis phosphorylated-tau (p-tau) appears within hours after closed head injury and long before other known pathogenic p-tau conformations, including oligomers, pre-fibrillary tangles, and NFTs (36). In particular, cis p-tau contributes to functional impairment in an animal model of TBI, as well as, in humans (37). Murine microglia rapidly internalize and degrade hyperphosphorylated tau (38), and expression of tau by microglia themselves also promotes their activation (39). Thus, robust and persistent inflammation may be sufficient to promote tauopathy.
Microglia may play a dual role in Aβ accumulation and clearance. Increased expression of the gamma secretase complex proteins on microglia and astrocytes have been observed in a closed head injury model (40). On the other hand, microglia containing Aβ have been found in association with plaques after TBI (41), suggesting phagocytic clearance of Aβ by proteases such as neprilysin and insulin-degrading enzyme (42). Suppression of microglial activation is associated with decreases in TBI-induced Aβ and restores depressed neurogenesis (43). It should be noted, however, that no studies have conclusively determined whether Aβ is the cause of microglial activation and inflammation following TBI.
Given that recent clinicopathologic and biomarker studies have failed to confirm the relationship between TBI and development of AD dementia or pathologic changes (44–46), it is possible that TBI exposure is a risk for late-life neurodegeneration but not AD. Therefore, further investigation is clearly needed to determine the relationship between TBI and cognitive decline.
Gut-Brain Axis
Recent studies have revealed the relationship between the gastrointestinal tract and the brain. Germ-free mice exhibit global defects in microglia with altered cell proportions and an immature phenotype, leading to impaired innate immune responses. Limited microbiota complexity also resulted in dramatic alterations in microglial properties (47). In addition, short-chain fatty acids and microbiota-derived bacterial fermentation products, have been demonstrated to regulate microglia maturation and function (47). In AD mice, perturbations in microbial diversity following antibiotic exposure diminish amyloid pathology (48). Microglia, which lie at the interface between environmental signals and brain circuitry throughout embryonic and adult life, are prime candidates as mediators of these effects.
Sleep
Lack of sleep is suggested as a risk for AD. Chronic lack of sleep increases Aβ plaque deposition (49), and sleep promotes efficient soluble Aβ clearance (50). Lack of sleep affects microglial morphology, phagocytosis, and Aβ clearance (51, 52). A recent study revealed that upregulation of complement C1q and C3 promotes synapse loss by microglial phagocytosis in AD (53). Even a short period of sleep loss enhances the mouse cerebral cortex expression level of complement C3 which activates synapse loss by microglia, and impaired sleep-wake cycle reduces microglial Aβ clearance (51). Moreover, chronic sleep restriction, but not acute sleep deprivation, promotes microglial phagocytosis without neuroinflammation (52). More detailed studies are needed to clarify how sleep affects microglial function and AD pathogenesis.
Inflammatory Cues in AD
Inflammasomes
Inflammasomes are a group of cytosolic protein complexes that form to mediate host immune responses to microbial infection and cellular damage (54). Assembly of an inflammasome triggers proteolytic cleavage of dormant procaspase-1 into active caspase-1, which converts IL-1 family cytokine precursors, pro-IL-1β, and pro-IL-18, into mature and biologically active IL-1β and IL-18, respectively (55). IL-1β and IL-18, in turn, initiate multiple signaling pathways and drive inflammatory responses, which results in neuronal injury or death (Figure 1).
Because IL-1β and IL-18 are key contributors to the progression of chronic inflammation—associated neurodegenerative diseases, including AD, inflammasomes are considered to be major players in chronic neuroinflammation (56, 57). The Aβ oligomer promotes the processing of pro-IL-1β into mature IL-1β in microglia, which in turn enhances microglial neurotoxicity (57). Levels of nucleotide-binding oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing 3 (NLRP3) inflammasomes and caspase-1 are substantially elevated in the brains of AD patients (56, 58), and elevated expression of IL-1β and IL-18 initiates inflammatory processes in the brain of AD patients. Elevated expression of these cytokines has also been detected in microglia and astrocytes, as well as, in neurons, co-localized with both Aβ plaques and tau deposition. Chronic inflammation may be responsible for increases in Aβ accumulation and tau phosphorylation in the brain (59). Halle et al. identified the NLRP3 inflammasome as a sensor of Aβ in a process involving phagocytosis of Aβ and subsequent lysosomal damage and release of cathepsin B (60).
Damaged neurons injured by insoluble Aβ oligomers and fibrils release DAMPs, which are sensed by NLRP3 inflammasomes, initiating a chain of events that leads to the maturation of pro-IL-1β and pro-IL-18 and release of their active forms (Figure 1) (60, 61). In addition, NLRP3 inflammasomes sense disease-associated extracellular amyloid and unique protein aggregates caused by inappropriate oligomerization or misfolding (62), likely as DAMPs within the resident microglia/macrophages after engulfment in the brain. Deficiency of NLRP3 or caspase-1 substantially attenuates spatial memory impairment and enhances Aβ clearance in AD model mice, indicating the importance of inflammasome-mediated neuroinflammation in AD pathogenesis (56). Furthermore, upon activation, microglia release ASC specks (63). These bodies have a direct molecular link to classical hallmarks of neurodegeneration: ASC specks bind to Aβ in the extracellular space and promote its aggregation, thereby directly activating innate immunity in association with the progression of AD pathology. Lysates derived from APP/PS1;Asc−/− brains had a reduced capacity to increase the Aβ load. Furthermore, a specific anti-ASC antibody prevented Aβ aggregation (63). Given that tau oligomers are known to spread to neighboring cells, their relationship to inflammasome activation should be examined further. Of interest, recent data emphasize that pathological tau promotes IL-1β secretion by activating inflammasomes.
Neurotransmitters
Microglia are closely associated with astrocytes and neurons, particularly at synapses, and recent data indicate that neurotransmitters play an important role in regulating the morphology and function of surveying/resting microglia, which express receptors for most known neurotransmitters (64, 65). In particular, microglia express receptors for ATP and glutamate, which regulate their motility. When Aβ induces ATP secretion by neurons (66) and microglia (67), effector functions such as phagocytosis and cytokine secretion are triggered.
Glutamate clearance and regulation at synaptic clefts is primarily mediated by glial transporter 1, and that expression is reduced in human AD hippocampal tissue (68). Consequently, glutamate overload triggers synaptic and neuronal loss influenced by AMPA receptors, which potentially contributes to AD. Levels of AMPA receptor subunit GluA2 are reduced in accordance with the Braak stages of AD (69). Lack of GluA2 in microglia leads to Ca2+ permeability in response to glutamate and may cause excess release of inflammatory cytokines, thereby increasing glutamate toxicity to neurons. Inhibition of glutamate receptor signaling has been proposed as a therapeutic approach for several neurodegenerative diseases. Because gap junctions/hemichannels are the main avenues for release of excessive glutamate from neurotoxin-activated microglia (70), their blockade by glycyrrhetinic acid derivatives significantly prevents activated microglia/macrophage-mediated neuronal death in rodent models of AD (71, 72). Moreover, because gap junctions/hemichannels are the main source of ATP, UTP, and glutamate, their blockade can halt the vicious cycle of transmission and amplification of neuroinflammation, and this also represents a promising therapeutic strategy for CNS diseases (65).
Adenosine A2A receptors (A2ARs) expressed by astrocytes and microglia are at the center of a neuromodulatory network that interacts with and integrates several neurotransmitter pathways. A2ARs modulate both glial activation and the ability of glia to release inflammatory factors or take up glutamate (73), and also mediates microglial process retraction (74). Expression of A2AR in microglial cells is elevated in the hippocampus and cerebral cortex of AD patients (75). Interestingly, consumption of caffeine, a non-selective adenosine A2ARs antagonist, reduces the risk of developing AD (76) and mitigates both amyloid and tau burden in transgenic mouse models (77, 78). Blockade of adenosine A2ARs decreases both hippocampal tau phosphorylation and neuroinflammatory response in a tauopathy mouse model (79), and also decreases amyloid burden in the brain and improves cognitive performance in an Aβ-injection model (80, 81). Therefore, regulation of inflammatory responses by microglial transmitters may have effects on AD.
Conclusions
Here, we briefly discussed the role of microglial functions in the development of AD. Microglial reactions in neurological disorders are complex and vary among disease stages; indeed, pro-inflammatory and anti-inflammatory microglia co-exist in some contexts. Newly emerging data reveal that microglia are a unique cell-population, to which the simple M1/M2 classification does not fit. Further investigation focusing on the microglial regulation will be required to develop new therapeutic interventions targeting CNS neuroinflammatory pathways.
Author Contributions
AK wrote the manuscript. HT, KT, and FT edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and grants from the Ministry of Health, Labor, and Welfare of Japan.
Abbreviations
A2ARs, adenosine A2A receptors; ASC, apoptosis-associated speck–like protein containing a caspase-recruitment domain; DAMPs, damage-associated molecular patterns; NLRP3, nucleotide-binding oligomerization domain-, leucine-rich repeat–and pyrin domain–containing 3; TBI, traumatic brain injury; TREM2, triggering receptor expressed on myeloid cells 2.
References
1. He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, et al. Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. (2018) 24:29–38. doi: 10.1038/nm.4443
2. Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry (2015) 77:43–51. doi: 10.1016/j.biopsych.2014.05.006
3. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
4. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
5. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. (2011) 43:429–35. doi: 10.1038/ng.803
6. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. (2011) 43:436–41. doi: 10.1038/ng.801
7. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. (2013) 16:848–50. doi: 10.1038/nn.3435
8. Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J Neurosci. (2017) 37:637–47. doi: 10.1523/JNEUROSCI.2110-16.2016
9. Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. (2017) 21:366–80. doi: 10.1016/j.celrep.2017.09.039
10. Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. (2003) 3:445–53. doi: 10.1038/nri1106
11. Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol. (2014) 88:495–8. doi: 10.1016/j.bcp.2013.11.021
12. Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging (2014) 35:e937–10. doi: 10.1016/j.neurobiolaging.2013.09.017
13. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. (2013) 8:19. doi: 10.1186/1750-1326-8-19
14. Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. (2015) 125:2161–70. doi: 10.1172/JCI77983
15. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell (2015) 160:1061–71. doi: 10.1016/j.cell.2015.01.049
16. Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. (2017) 18:1186–98. doi: 10.15252/embr.201743922
17. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell (2017) 170: 649–63.e13. doi: 10.1016/j.cell.2017.07.023
18. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
19. Datta M, Staszewski O, Raschi E, Frosch M, Hagemeyer N, Tay TL, et al. Histone deacetylases 1 and 2 regulate microglia function during development, homeostasis, and neurodegeneration in a context-dependent manner. Immunity (2018) 48: 514–29.e516. doi: 10.1016/j.immuni.2018.02.016
20. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
21. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
22. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's Disease. Cell (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
23. Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron (2010) 68:19–31. doi: 10.1016/j.neuron.2010.08.023
24. Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. (2010) 177:2549–62. doi: 10.2353/ajpath.2010.100265
25. Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener. (2016) 11:3. doi: 10.1186/s13024-016-0071-x
26. Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement. (2015) 11:1407–16. doi: 10.1016/j.jalz.2014.12.009
27. Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. (2017) 12:74. doi: 10.1186/s13024-017-0216-6
28. Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. (2010) 41:242–7. doi: 10.1007/s12035-010-8105-9
29. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. (2014) 14:463–77. doi: 10.1038/nri3705
30. Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. (2013) 33:1864–74. doi: 10.1038/jcbfm.2013.146
31. Febinger HY, Thomasy HE, Pavlova MN, Ringgold KM, Barf PR, George AM, et al. Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J Neuroinflammation (2015) 12:154. doi: 10.1186/s12974-015-0386-5
32. Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T, et al. Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J Neurotrauma (2016) 33:1060–72. doi: 10.1089/neu.2015.4041
33. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. (2011) 70:374–83. doi: 10.1002/ana.22455
34. Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci. (2010) 11:361–70. doi: 10.1038/nrn2808
35. Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, et al. Amyloid pathology and axonal injury after brain trauma. Neurology (2016) 86:821–8. doi: 10.1212/WNL.0000000000002413
36. Albayram O, Herbert MK, Kondo A, Tsai CY, Baxley S, Lian X, et al. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci. (2016) 6:59. doi: 10.1186/s13578-016-0124-4
37. Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. (2017) 8:1000. doi: 10.1038/s41467-017-01068-4
38. Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep. (2015) 5:11161. doi: 10.1038/srep11161
39. Wang L, Jiang Q, Chu J, Lin L, Li XG, Chai GS, et al. Expression of Tau40 induces activation of cultured rat microglial cells. PLoS ONE (2013) 8:e76057. doi: 10.1371/journal.pone.0076057
40. Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KS, Shohami E, et al. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia (2008) 56:552–67. doi: 10.1002/glia.20638
41. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. (2009) 19:214–23. doi: 10.1111/j.1750-3639.2008.00176.x
42. Shimizu E, Kawahara K, Kajizono M, Sawada M, Nakayama H. IL-4-induced selective clearance of oligomeric beta-amyloid peptide(1-42) by rat primary type 2 microglia. J Immunol. (2008) 181:6503–13. doi: 10.4049/jimmunol.181.9.6503
43. Thau-Zuchman O, Gomes RN, Dyall SC, Davis M, Priestley JV, Groenendijk M, et al. Brain phospholipid precursors administered post-injury reduce tissue damage and improve neurological outcome in experimental traumatic brain injury. J Neurotrauma (2018). doi: 10.1089/neu.2017.5579. [Epub ahead of print].
44. Crane PK, Gibbons LE, Dams-O'connor K, Trittschuh E, Leverenz JB, Keene CD, et al. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. (2016) 73:1062–9. doi: 10.1001/jamaneurol.2016.1948
45. Weiner MW, Crane PK, Montine TJ, Bennett DA, Veitch DP. Traumatic brain injury may not increase the risk of Alzheimer disease. Neurology (2017) 89:1923–5. doi: 10.1212/WNL.0000000000004608
46. Weiner MW, Harvey D, Hayes J, Landau SM, Aisen PS, Petersen RC, et al. Effects of traumatic brain injury and posttraumatic stress disorder on development of Alzheimer's disease in vietnam veterans using the alzheimer's disease neuroimaging initiative: preliminary report. Alzheimers Dement. (2017) 3:177–88. doi: 10.1016/j.trci.2017.02.005
47. Erny D, Hrabe De Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
48. Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of Alzheimer's disease. Sci Rep. (2017) 7:10411. doi: 10.1038/s41598-017-11047-w
49. Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science (2009) 326:1005–7. doi: 10.1126/science.1180962
50. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science (2013) 342:373–7. doi: 10.1126/science.1241224
51. An H, Cho MH, Kim DH, Chung S, Yoon SY. Orexin impairs the phagocytosis and degradation of amyloid-beta fibrils by microglial cells. J Alzheimers Dis. (2017) 58:253–61. doi: 10.3233/JAD-170108
52. Bellesi M, De Vivo L, Chini M, Gilli F, Tononi G, Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J Neurosci. (2017) 37:5263–73. doi: 10.1523/JNEUROSCI.3981-16.2017
53. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (2016) 352:712–6. doi: 10.1126/science.aad8373
54. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
55. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
56. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493:674–8. doi: 10.1038/nature11729
57. Parajuli B, Sonobe Y, Horiuchi H, Takeuchi H, Mizuno T, Suzumura A. Oligomeric amyloid β induces IL-1β processing via production of ROS: implication in Alzheimer's disease. Cell Death Dis. (2013) 4:e975. doi: 10.1038/cddis.2013.503
58. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol Neurodegener. (2016) 11:23. doi: 10.1186/s13024-016-0088-1
59. Lira-De Leon KI, Garcia-Gutierrez P, Serratos IN, Palomera-Cardenas M, Figueroa-Corona Mdel P, Campos-Pena V, et al. Molecular mechanism of tau aggregation induced by anionic and cationic dyes. J Alzheimers Dis. (2013) 35:319–34. doi: 10.3233/JAD-121765
60. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
61. Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer's pathology. J Cell Mol Med. (2008) 12:2255–62. doi: 10.1111/j.1582-4934.2008.00496.x
62. Masters SL, O'neill LA. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol Med. (2011) 17:276–82. doi: 10.1016/j.molmed.2011.01.005
63. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer's disease. Nature (2017) 552:355–61. doi: 10.1038/nature25158
64. Domercq M, Vazquez-Villoldo N, Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci. (2013) 7:49. doi: 10.3389/fncel.2013.00049
65. Takeuchi H, Suzumura A. Gap junctions and hemichannels composed of connexins: potential therapeutic targets for neurodegenerative diseases. Front Cell Neurosci. (2014) 8:189. doi: 10.3389/fncel.2014.00189
66. Saez-Orellana F, Godoy PA, Bastidas CY, Silva-Grecchi T, Guzman L, Aguayo LG, et al. ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of β-amyloid peptide in hippocampal neurons. Neuropharmacology (2016) 100:116–23. doi: 10.1016/j.neuropharm.2015.04.005
67. Sanz JM, Chiozzi P, Ferrari D, Colaianna M, Idzko M, Falzoni S, et al. Activation of microglia by amyloid {beta} requires P2X7 receptor expression. J Immunol. (2009) 182:4378–85. doi: 10.4049/jimmunol.0803612
68. Zumkehr J, Rodriguez-Ortiz CJ, Medeiros R, Kitazawa M. Inflammatory cytokine, IL-1β, regulates glial glutamate transporter via microRNA-181a in vitro. J Alzheimers Dis. (2018) 63:965–75. doi: 10.3233/JAD-170828
69. Carter TL, Rissman RA, Mishizen-Eberz AJ, Wolfe BB, Hamilton RL, Gandy S, et al. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer's disease patients according to Braak stage. Exp Neurol. (2004) 187:299–309. doi: 10.1016/j.expneurol.2003.12.010
70. Takeuchi H, Mizuno T, Zhang G, Wang J, Kawanokuchi J, Kuno R, et al. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem. (2005) 280:10444–54. doi: 10.1074/jbc.M413863200
71. Takeuchi H, Mizoguchi H, Doi Y, Jin S, Noda M, Liang J, et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer's disease. PLoS ONE (2011) 6:e21108. doi: 10.1371/journal.pone.0021108
72. Yi C, Ezan P, Fernandez P, Schmitt J, Saez JC, Giaume C, et al. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer's disease. Glia (2017) 65:1607–25. doi: 10.1002/glia.23182
73. Matos M, Augusto E, Agostinho P, Cunha RA, Chen JF. Antagonistic interaction between adenosine A2A receptors and Na+/K+-ATPase-α2 controlling glutamate uptake in astrocytes. J Neurosci. (2013) 33:18492–502. doi: 10.1523/JNEUROSCI.1828-13.2013
74. Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci. (2009) 12:872–8. doi: 10.1038/nn.2341
75. Angulo E, Casado V, Mallol J, Canela EI, Vinals F, Ferrer I, et al. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. (2003) 13:440–51. doi: 10.1111/j.1750-3639.2003.tb00475.x
76. Flaten V, Laurent C, Coelho JE, Sandau U, Batalha VL, Burnouf S, et al. From epidemiology to pathophysiology: what about caffeine in Alzheimer's disease? Biochem Soc Trans. (2014) 42:587–92. doi: 10.1042/BST20130229
77. Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, et al. Caffeine protects Alzheimer's mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience (2006) 142:941–52. doi: 10.1016/j.neuroscience.2006.07.021
78. Laurent C, Eddarkaoui S, Derisbourg M, Leboucher A, Demeyer D, Carrier S, et al. Beneficial effects of caffeine in a transgenic model of Alzheimer's disease-like tau pathology. Neurobiol Aging (2014) 35:2079–90. doi: 10.1016/j.neurobiolaging.2014.03.027
79. Laurent C, Burnouf S, Ferry B, Batalha VL, Coelho JE, Baqi Y, et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol Psychiatry (2016) 21:97–107. doi: 10.1038/mp.2014.151
80. Canas PM, Porciuncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci. (2009) 29:14741–51. doi: 10.1523/JNEUROSCI.3728-09.2009
Keywords: microglia, TBI, inflammasomes, NLRP3, TREM2, neuroinflammation, glutamate
Citation: Katsumoto A, Takeuchi H, Takahashi K and Tanaka F (2018) Microglia in Alzheimer's Disease: Risk Factors and Inflammation. Front. Neurol. 9:978. doi: 10.3389/fneur.2018.00978
Received: 18 September 2018; Accepted: 30 October 2018;
Published: 15 November 2018.
Edited by:
Jun-ichi Kira, Kyushu University, JapanReviewed by:
Katsuhisa Masaki, University of Chicago Medical Center, United StatesYasumasa Ohyagi, Ehime University, Japan
Copyright © 2018 Katsumoto, Takeuchi, Takahashi and Tanaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideyuki Takeuchi, aHRha2VAeW9rb2hhbWEtY3UuYWMuanA=