 Thomas Fleetwood
Thomas Fleetwood Roberto Cantello1
Roberto Cantello1 Cristoforo Comi
Cristoforo Comi- 1Section of Neurology, Department of Translational Medicine, University of Eastern Piedmont, Novara, Italy
- 2Interdisciplinary Research Centre of Autoimmune Diseases, University of Eastern Piedmont, Novara, Italy
Antiphospholipid syndrome (APS) is an autoimmune antibody-mediated condition characterized by thrombotic events and/or pregnancy morbidity in association with persistent positivity to antiphospholipid antibodies (aPL). The nervous system is frequently affected, as intracranial vessels are the most frequent site of arterial pathology. Over the course of years, many other neurological conditions not included in the diagnostic criteria, have been associated with APS. The pathogenic mechanisms behind the syndrome are complex and not fully elucidated. aPL enhance thrombosis, interfering with different pathways. Nevertheless, ischemic injury is not always sufficient to explain clinical features of the syndrome and immune-mediated damage has been advocated. This may be particularly relevant in the context of neurological complications. The reason why only a subgroup of patients develop non-criteria nervous system disorders and what determines the clinical phenotype are questions that remain open. The double nature, thrombotic and immunologic, of APS is also reflected by therapeutic strategies. In this review we summarize known neurological manifestations of APS, revisiting pathogenesis and current treatment options.
Introduction
Antiphospholipid syndrome (APS) is an autoimmune antibody-mediated disorder defined by the occurrence of thrombosis and/or pregnancy morbidity in presence of persistent antiphospholipid antibodies (aPL) (1). The estimated incidence of APS is approximately 5 cases every 100,000 subjects/year with a prevalence of 40–50 every 100,000 subjects (2). The diseases can occur alone (primary APS) or in the context of other autoimmune conditions, in particular systemic lupus erythematosus (SLE), Sjögren's syndrome and rheumatoid arthritis (secondary APS). Veins, arteries and small vessels can all be affected by thrombosis, with deep veins of the legs and intracranial arteries being the most common sites of venous and arterial thrombosis, respectively. Pregnancy morbidity includes embryonic losses, fetal death, and premature birth. The diagnosis is made according to the updated international Sydney consensus criteria (Table 1) (1). However, patients with persistent aPL may present with clinical manifestations not included in the criteria (the so called “non-criteria” symptoms), among which thrombocytopenia, hemolytic anemia, cardiac valve disease, renal microangiopathy, livedo reticularis, and neurologic disturbances other than ischemic cerebrovascular accidents (CVA) (3).
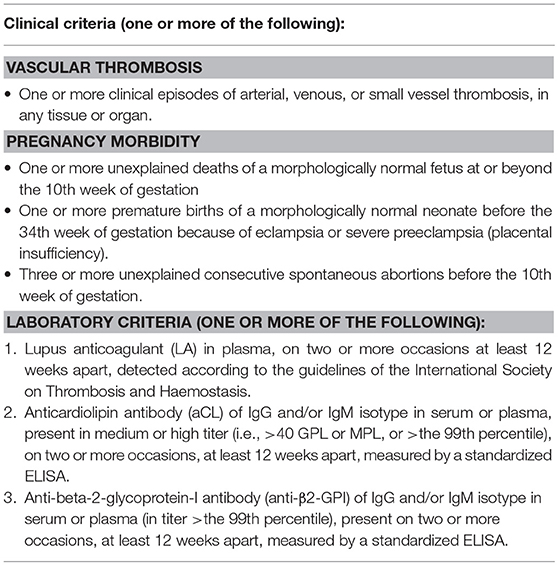
Table 1. Revised classification criteria for APS [adapted from Miyakis et al. (1)].
In this paper we summarize current concepts regarding the pathogenesis of APS, reviewing neurological clinical features and related therapeutic implications.
Pathogenesis of APS: Thrombosis and Other Mechanisms
Clinical manifestations in APS are associated with aPL presence. aPL, namely lupus anticoagulant (LA), anti-cardiolipin (aCL), anti-β2-glycoprotein-I (anti-β2-GPI), are an heterogeneous group of auto-antibodies directed against phospholipid binding proteins (4). The current hypothesis is that susceptible individuals develop aPL upon exposure to an external trigger, such as an infective agent, through a mechanism of molecular mimicry (2). β2-GPI, one of the main antigens recognized by aPL, is a plasma protein composed of five domains (I–V) (5). Circulating β2-GPI presents a circular form, in which epitopes present in domain I and recognized by B-cells are hidden. Upon binding to an anionic phospholipid surface, β2-GPI undergoes a conformational change exposing the cryptic epitopes (6). Oxidative stress seems to enhance the immune reaction, leading to the formation of disulfide bonds in domain I that further increase its immunogenic potential (7). Prevalence of aPL in the general population is around 1–5% (8) but only a minority of subjects develops APS, suggesting that the presence of autoantibodies alone is not sufficient to cause pathology. Following aPL formation, a “second hit” thus is necessary. It has been demonstrated that β2-GPI only binds its ApoE receptor 2 (ApoER2) when vessel endothelial cells become activated (9), inducing dimerization of the receptor and activation of intracellular signaling. A number of conditions that increase oxidative stress, including infection, malignancy, pregnancy and smoking, may act as triggers for endothelium priming (10, 11). The mechanisms through which aPL enhance thrombosis is complex and in part still to be understood. Different mediators are likely to be involved, among which endothelium, platelets, complement, and innate immune systems (9) (Figure 1). First, aPL stimulate the expression of proadhesive, procoagulant, and proinflammatory molecules. Tissue factor (TF) is a key component of the extrinsic coagulation pathway, implicated in the activation of thrombin, that becomes exposed upon vessel injury. aPL enhance surface expression of TF on endothelial cells by binding to annexin A2 and toll-like receptor 4 (TLR4) and activating the nuclear factor κB (NF-κB) signaling pathway (12–14). aPL also seem to interact directly with monocytes and neutrophils, inducing mitochondrial dysfunction and subsequent expression of TF and proinflammatory tumor necrosis factor α (TNF-α) (15). Annexin A5 is a protein involved in many biological processes. On endothelial cells it binds to phosphatidylserine molecules, forming a shield that inhibits the activation of procoagulant complexes. In vitro studies have shown that aPL binding to annexin A5 disrupts the shield leading to thrombosis (16). Endothelial cells also contribute to modulate the activity of vessel wall muscular cells through the production of nitric oxide by endothelial nitric oxide synthase (eNOS). In murine models, aPL inhibit the activity of eNOS (17). This can lead to impaired regulation of vascular tone, increase in superoxide and peroxynitrite, and cell adhesion (18, 19). Confirming this hypothesis, APS patients have reduced plasma levels of nitric oxide compared to controls (20). Platelets play a pivotal role in thrombus formation. β2-GPI binds to the von Willebrand factor (vWF) receptor glycoprotein Ibα and to the ApoER2 on the platelet surface inducing the release of thromboxane A2 and enhancing aggregation and adhesiveness (21). Interestingly, binding of aPL to platelet membrane phospholipids leads to activation and possible dysregulation of serotonin metabolism, which could be involved in the pathogenesis of aPL-mediated migraine (22). Some authors have also identified anti-platelet antibodies in the setting of APS (23). aPL can activate the classical complement pathway, inducing production of C5a, which in turn can bind to neutrophils and stimulate the expression of TF (24). Previous studies have also suggested that aPL impair thrombolysis by interfering with tissue plasminogen activator (tPA) and plasmin (25). Despite the amount of evidence gathered over the course of years, thrombosis alone is perhaps not sufficient to explain all of the clinical effects of aPL. Direct binding of aPL to nervous system antigens has been proposed to explain some neurologic manifestations of APS, which may be mediated by inflammation and neurodegeneration (26, 27). An experimental model of APS (eAPS) can be obtained by immunizing normal mice with β2GPI, therefore inducing aPL and typical clinical features of APS (28). Interestingly, in eAPS mice, long-term exposition to aPL also leads to behavioral hyperactivity and decline in cognitive performances (29, 30), which resolve after elimination of aPL through ultraviolet irradiation (26). A proposed mechanism of neuronal injury focuses on the disruption of the blood-brain barrier (BBB), secondary to diffuse endothelial dysfunction caused by aPL binding (31). Katzav et al. have demonstrated impaired integrity of the BBB and accumulation of aPL in cortical and inhibitory hippocampal neurons of immunized mice (32), which may be linked to behavioral changes and cognitive impairment. Furthermore, in a previous study eAPS mice displayed higher brain levels of proinflammatory TNF-α and prostaglandin E (PGE) and lower levels of thrombin inhibitors compared to controls (33). Treatment with aspirin and enoxaparin ameliorated concentrations of TNF-α, PGE and thrombin inhibitors, as well as behavioral patterns. An interesting correlation between coagulation and autoimmunity has been investigated in eAPS mice carrying the factor V Leiden mutation (FVL), either in homozygosis (FVLQ/Q) or in heterozygosis (FVLQ/−). Induced aPL levels were higher in FVLQ/Q mutated mice compared to FVLQ/− and controls, as well as the burden of behavioral and cognitive impairment and of neurodegenerative changes on histological examination (34). It is known from previous studies that aPL have the potential to bind to myelin, brain ependyma and choroid epithelium epitopes in the animal model (35). A direct interaction between aPL and specific, yet still unidentified, basal ganglia epitopes, may lead to the development of movement disorders in APS patients. Supporting this hypothesis Dale et al. in 2011 demonstrated binding of IgG from the serum of pediatric APS patients with chorea to neuronal cell-surface antigens of cultured neuronal cells with dopaminergic characteristics (36). Furthermore it has been shown that aPL can impair GABA receptor activity and induce depolarization of synaptoneurosomes, disrupting neuronal function by acting on nerve terminals (37, 38), with possible implications in APS-associated epileptogenesis. Energetic dysfunction in neuronal cells and altered neurotransmission may also play a role in APS pathology, since it has been demonstrated that aPL from patients with neurological involvement also bind to adenosine triphosphate (ATP) (39).
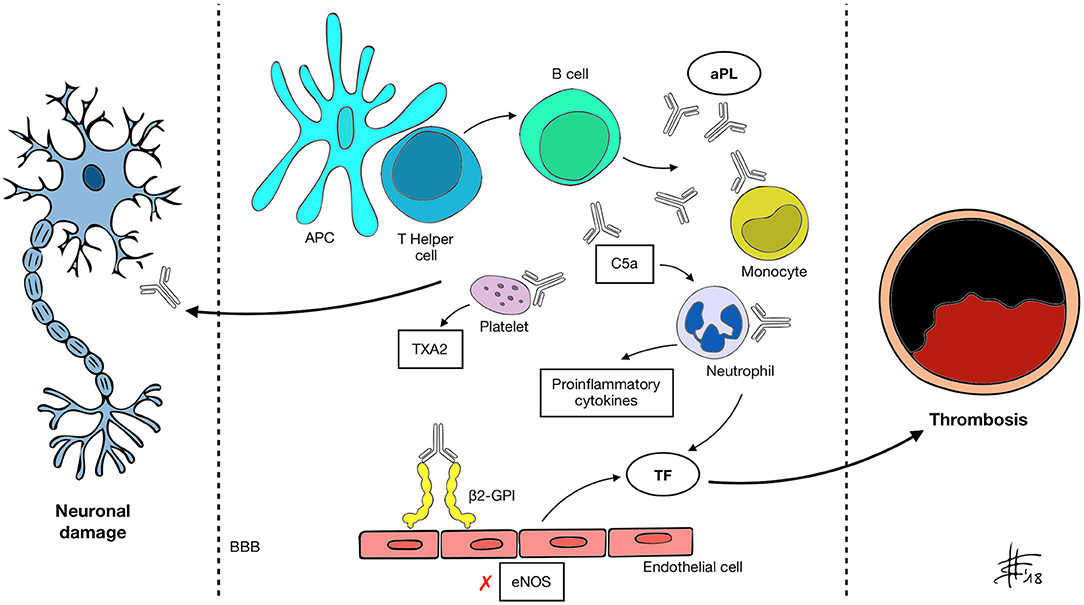
Figure 1. APC, antigen presenting cell; aPL, antiphospholipid antibody; β2-GPI, β2 glycoprotein-I; BBB, blood-brain barrier; C5a, activated complement 5 fraction; eNOS, endothelial nitric oxide synthase; TXA2, thromboxane A2; TF, tissue factor. A mechanism of molecular mimicry may underlie B cell activation by the APC-T helper system and subsequent aPL production in susceptible individuals. Through recognized antigens (mainly β2-GPI) aPL interact with different cell types and activate the classical complement pathway, inducing expression of pro-inflammatory cytokines, enhancing platelet adhesiveness and aggregation, impairing vascular tone by inhibiting eNOS and increasing the expression of TF. These changes determine a prothrombotic state and increase the risk of vascular events. aPL also appear to be able to recognize and target nervous system antigens, possibly through the disruption of the BBB. Neurological manifestations of APS may be determined by both mechanisms.
Neurological Manifestations of APS
The nervous system is a major target of APS. This appeared clear since the first description of the syndrome by Hughes in 1983 in which he described CVA and transverse myelitis (40). Although thrombotic damage has been advocated to explain many neurologic manifestations, direct immune-mediated processes may also be involved (41). Neurologic symptoms have been therefore classified in thrombotic and non-thrombotic according to the supposed primary pathogenic mechanism (Table 2) (42). The reason why some patients develop neurological symptoms is unknown as well as what determines the site of lesion (e.g., central vs. peripheral nervous system) and the clinical phenotype. Possible explanations may be related to antibody subtypes, individual susceptibility or even genetic predisposition.
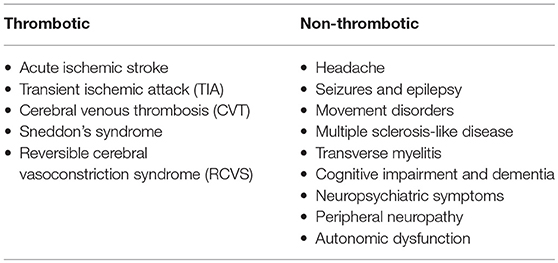
Table 2. Neurological manifestations of APS.
Cerebrovascular Disease
Acute ischemic stroke and transient ischemic attack (TIA) are the most common manifestations of arterial pathology in APS (2). According to the Euro-Phospholipid Project Study Group the cumulative prevalence of stroke and TIA in APS patients are 19.8 and 11.1%, respectively, (43). On the other hand, aPL may be detected in up to 13.5% of stroke patients (44), with higher rates in young subjects. Moreover, age at onset in aPL-positive patients is anticipated. A previous study on 128 patients with CVA and aPL showed a mean age was 46 years (45). It has been suggested that APS accounts for over 20% of cases in the young population (46). A 2015 systematic review of 43 studies showed that the presence of aPL in subjects under the age of 50 increased the risk for thrombotic cerebrovascular events by 5.48-fold (47). The association between aPL and stroke incidence in older patients is less clear, due to the higher prevalence of other vascular risk factors (48). Clinical manifestations depend on the site and entity of the lesion. In a sequence of 110 SLE and non-SLE aPL-positive patients undergoing neuroradiological evaluation, the most common finding was large infarcts (22%), followed by white matter changes (17%), small cortical infarcts (10%), and lacunar infarcts (9%) (49). In a cohort of 55 APS patients, 25 of which suffered an ischemic stroke, the most common site of occlusion was the middle cerebral artery (31%) (50). In situ thrombosis is thought to be the most frequent pathogenic mechanism, whereas in other cases, cardiac embolisms can arise from involvement of the cardiac valvular apparatus, with thickening of valve leaflets by deposition of immune complexes (Liebman-Sacks endocarditis) (51). Besides stem or branch occlusion of intracranial arteries, a vasculitis-like pattern, with multiple sites of narrowing and dilation has also been described through arteriography (52). Alterations of extracranial arteries appear to be less frequent (52), though early atherosclerosis seems to affect APS patients (53), and some authors have proposed a direct role of the autoantibodies through oxidative damage (54). Although rare, cerebral venous thrombosis (CVT) can complicate APS or, in some cases, be the presenting symptom (55). Presence of aPL in non-SLE patients with CVT has been reported in previous studies (56). aCL positivity may be found in 7–22% of patients (51). APS accounts for ~6–17% of all CVT cases, being one of the most frequent prothrombotic conditions associated (57). Other vascular conditions have also been reported in association with APS, including Sneddon's syndrome and reversible cerebral vasoconstriction syndrome (RCVS). Sneddon's syndrome is a rare non-inflammatory thrombotic vasculopathy, characterized by livedo reticularis and recurrent cerebral infarctions (53). Other clinical manifestations include headache, seizures and cognitive decline (58). Details of the pathogenetic mechanisms are still to be clarified but non-inflammatory thrombotic vasculopathy is seen in medium- and small-sized arteries in the brain and skin (59). Indeed, neuroradiological findings show that leukoaraiosis and small lacunar infarcts are more common than infarcts in the territory of large cerebral arteries (60). Reversible cerebral vasoconstriction syndrome (RCVS) is a neurological disorder marked by severe headaches variably associated with seizures, ischemic stroke, and subarachnoid hemorrhage (61). The cause has been related to a possible disturbance in the regulation of cerebrovascular tone (62).
Headache
Headache, specifically in the form of migraine, is the most common neurologic symptom of APS patients, with an estimated prevalence of 20.2% (43). Indeed, aPL antibodies are more frequently found in migraineurs than in age-matched controls (63). However, given its high prevalence in the general population, it is difficult to establish whether APS is a risk factor for developing migraine or if this represents a comorbid condition. Previous CVA represent a strong risk factor for developing migraine. However, not all patients have a positive history for stroke or TIA, which suggests non-ischemic mechanisms (64). On the contrary some authors have found long-standing migraine to be a risk factor for stroke in APS patients (65). Further studies are necessary to elucidate such interplay. Notably, APS-associated migraine may be difficult to control with classic analgesic regimens (66).
Seizures and Epilepsy
APS seems to confer a higher risk of developing seizures, with an estimated prevalence of ~8% (67), which may further increase in APS secondary to SLE (68). All forms of epilepsy may be seen, including subclinical forms, determined by the presence of abnormal electroencephalography findings alone (46). It has been suggested that 20% of idiopathic juvenile epilepsy cases may be associated with aPL (69). According to existing studies, previous CVA have been identified has the most solid risk factor for developing seizures (68). It is therefore easy to speculate that the most plausible pathogenic mechanism is ischemic damage to brain tissue, leading to the formation of cortical epileptogenic foci (64). However, seizures may also develop in structurally normal brains, suggesting an antibody-mediated mechanism (70).
Movement Disorders
Movement disorders are a possible, though rare, neurological manifestations in the setting of APS. Chorea in particular occurs in 1.3–4.5% of patients (71), and may be the first symptom of the syndrome (72, 73). Other less frequently reported conditions include parkinsonism (corticobasal-like syndrome and progressive supranuclear palsy phenotype), dystonia, ballismus, paroxysmal dyskinesias, tremor, tic, myoclonus, cerebellar ataxia (41). Mixed clinical presentations have also been described (74). The pathogenesis of movement disorders in the setting of APS is matter of debate. On the one hand, cerebral infarctions and white matter changes on MRI suggest a thrombo-occlusive mechanism, which could account for the majority of cases (51, 75). On the other, immune-mediated attack against basal ganglia epitopes has been suggested and reported in some cases (41). No specific correlations between the type of antibody and clinical features have been reported (41). Nevertheless, it is likely that genetic predisposition might partially explain phenotypic variability, in analogy to what was reported in Parkinson's disease (76).
Multiple Sclerosis-Like Disease
Overlap of clinical and laboratory findings of multiple sclerosis (MS) and SLE have been described long ago, leading to the proposal of a hybrid condition named “lupoid sclerosis” (77, 78). Diagnostic criteria, as well as the role of anti-nuclear antibodies (ANA) and aPL are still matter of debate (79). Several studies have provided different estimates of aPL prevalence in definite MS patients, ranging from 2 to 88% (80), with higher titers observed during exacerbations of the disease (81). aCL and anti-β2GPI appear to be more prevalent compared to LA, although the latter have been studied to a lesser extent and the real prevalence remains to be clarified (80). Some authors have suggested that aPL may alter the integrity of the BBB and facilitate the access of immune cells to the central nervous system (CNS) compartment (81, 82). APS can also present with symptoms resembling MS, including visual, sensitive or motor deficits with a relapsing remitting course, with similar MRI T2 lesions (83–87). This condition is referred to as MS-like disease (64). Indeed, the differential diagnosis between these two conditions may be challenging. Acute onset of atypical MS symptoms, coexistence of other typically APS-related neurological manifestations (for example headache or epilepsy), connective tissue-like features or a history of thrombosis, pregnancy morbidity should orientate toward APS (88, 89). Neuroimaging studies may also help in the differential diagnosis. APS lesions on MRI are smaller, frequently localized in the subcortical area, are stable over time and may also improve with anticoagulation therapy (80, 90). Normal cell count and absence of oligoclonal bands on CSF analysis also suggests APS (91). Some authors have suggested that aPL may alter the integrity of the BBB and facilitate the access of immune cells to the central nervous system (CNS) compartment (81, 82).
Transverse Myelitis
Transverse myelitis (TM) is an inflammatory condition affecting the gray and white matter of the spinal cord (92). Symptoms include motor and sensory level deficits and sphincter abnormalities. Estimated prevalence in APS is around 0.4–4% (71, 93). Although the exact pathogenesis is unsure, vasculitis and arterial thrombosis resulting in ischemic cord necrosis have been suggested (89). Furthermore, APS has been described in overlap with neuromyelitis optica spectrum disorder (NMOSD) (94, 95), another autoimmune condition characterized by recurrent episodes of optic neuritis and longitudinally extensive transverse myelitis (that is, extended over 3 or more spinal cord segments) and positivity for anti-aquaporin-4 (anti-AQP4) or anti-myelin oligodendrocyte glycoprotein (anti-MOG) (96). Therefore, some authors suggest screening APS patients presenting with optic neuritis or myelitis for NMO-associated autoantibodies (95).
Cognitive Impairment and Dementia
A high percentage of patients with primary APS (which may reach 42–80%) develop some degree of cognitive impairment, usually with a subcortical pattern (64, 97). In some cases, deficits may even precede the diagnosis of APS, as demonstrated in a study by Jacobson et al. on aPL-positive non-elderly subjects who displayed differences in executive functioning, verbal learning and memory, and visuospatial ability compared to age- and education-matched controls (98). Dementia frequency has been estimated around 2.5% in APS patients (71). MRI studies have shown a high burden of white matter lesions in APS patients with cognitive impairment (99) resembling multi-infarct dementia. However, vascular damage may not be the only mechanism. Findings of degenerative rather than multi-infarct dementia have also been described in aPL-positive elderly subjects (100). Other studies and a meta-analysis have underlined a strong association with aCL antibodies (101). Furthermore, some animal models showed that cognitive dysfunction can be induced by intraventricular injection of neuronal-binding antibodies from APS patients (26), while other failed to demonstrate an association with ischemic lesions (102). Such findings support the idea of a direct effect of aPL on congnition. aPL-mediated dysregulation of the dopaminergic system has also been proposed (103). Given the clinical overlap, MS-like disease should also be considered in the differential diagnosis.
Neuropsychiatric Symptoms
Psychiatric symptoms, including psychosis, mania, depression, bipolar disorders, obsessive–compulsive disorders, and schizophrenia have been described in APS patients (64). Older age, cerebral lesions, and triple aPL positivity (i.e., anti-β2-GPI, aCL, and LA) are considered risk factors (104). A high prevalence of aPL has been described in patients with psychosis, though such finding must be interpreted carefully as antipsychotic medications are though to induce aPL and aCL in particular (66).
Peripheral Neuropathy
A small study by Santos et al. in 2010 has investigated the involvement of the peripheral nervous system in APS (105). The most frequent findings were sensori-motor neuropathy and isolated carpal tunnel syndrome. Most patients were asymptomatic and showed no sign of pathology upon physical examination. The underlying pathogenic mechanism is not clear, and may be linked to thrombosis of the vasa nervorum, vasculitis or even targeting of lipidic components of myelin by aPL. Reports of Guillain-Barré syndrome in APS patients support this hypothesis (106).
Autonomic Dysfunction
Several reports exist of autonomic dysfunction in the context of APS. A 2017 work by Schofield describes the clinical findings in 22 patients with autonomic dysfunction as the initial symptom of APS (107). Manifestations included postural tachycardia syndrome, neurocardiogenic syncope, inappropriate sinus tachycardia, labile hypertension, complex regional pain syndrome, severe gastrointestinal dysmotility, and neurogenic bladder, with 45% of subjects presenting more than one disorder. Reduction in autonomic and sensory small fibers, assessed by skin punch biopsy was widespread among subjects. Ten patients (45%) subsequently developed arterial thrombosis, of which 8 (36%) presented with stroke, TIA, or amaurosis fugax. Pathogenesis of small fiber dysfunction may derive either from microthrombosis or from direct antibody binding to neuronal epitopes leading to nerve dysfunction. The latter hypothesis is supported by reported improvement of symptoms with immune modulatory therapy (107).
Treatment: Current Concepts
To date, antithrombotic therapy represents the cornerstone of APS management. This is primarily based on vitamin K antagonists (VKA), generally warfarin (108). The high risk of thrombosis recurrence, which can be seen in 5–16% of subjects (109), warrants long-term anticoagulation. In the acute phase, ischemic cerebrovascular events are managed according to clinical standards as for patients without APS. Reports of intravenous thrombolysis in APS patients who develop acute ischemic stroke date back to 1997 (110). aPL-induced thrombocytopenia or prolongation of prothrombin time are issues to be considered before starting treatment (111). Primary endovascular thrombectomy may be considered as an alternative in these patients (112). Anticoagulation with warfarin and INR 2–3 is the most common strategy for secondary thromboprophylaxis (113). Risk stratification combining antibody profile and comorbidities could help to identify patients needing a more aggressive treatment but to date no model has been validated (3). Although no strong evidence exists in this sense, long-term antiplatelet therapy with low-doses aspirin may be beneficial in addition to warfarin (114). Recurrence of thrombosis frequently depends from inadequate anticoagulation (115), sometimes linked to aPL artifactual prolongation of prothrombin time. Nevertheless, in a series of 66 APS patients treated to a target INR of 3.5 a recurrence rate of 9.1/100 patient-years was detected, with recurrences often affecting the same vascular bed as the original thrombosis (116). Due to the lack of solid evidence, management of recurrences is not standardized and may include higher-intensity warfarin therapy (INR 3–4), the addition of low-dose aspirin or the use of low-molecular-weight heparin (3). Prophylactic treatment in asymptomatic aPL carriers is matter of debate. The risk of a first thrombotic event is likely liked to the presence of other concomitant factors (117–120). The use of low-dose antiplatelet therapy for primary prophylaxis is controversial, due to the lack of solid evidence of efficacy and some authors suggest management following guidelines for prevention of cardiovascular disease in the general population (3). Under specific conditions, immunosuppressive treatment may be an option in addition to antithrombotic therapy. The paradigm of this is represented by catastrophic APS (CAPS), a rare condition characterized by widespread small vessel thrombosis and subsequent multiorgan failure in the context of APS (2) with a mortality rate up to 37% (121). In the case of CAPS, aggressive treatment is recommended and consists of a “triple therapy” composed by anticoagulants plus glucocorticoids plus plasma exchange (PE) and/or intravenous immunoglobulins (IVIG) (122). Rituximab (RTX) is an anti-CD20 monoclonal antibody that depletes B-cells, currently used to treat several autoimmune diseases and hematologic malignancies. B-cells seem to play a pivotal role in the pathogenesis of APS (123). Several case reports describe the use of RTX in APS patients, suggesting a beneficial role in the management of hemolytic anemia (124), thrombocytopenia (125) (126), thrombotic microangiopathy (127). Despite no strong evidence, RTX may be used in patients with thrombotic relapses while on AVK and adequate INR, especially in the setting of APS secondary to SLE (128–130). No standard treatment exists for non-thrombotic neurological manifestations of APS and available evidence mostly derives from retrospective non-randomized trials or case reports (64). Besides cerebrovascular events, anticoagulation has proven effective in the treatment of conditions that are not primarily thrombotic, including migraine, transverse myelitis, and neuropsychiatric disturbances (131–133). Improvement of cognitive performance with anticoagulation therapy has also been described (46). Notably, response to anticoagulation may help distinguish APS from atypical presentations of CNS inflammatory disorders (80). The combined use of neuroleptics and antiplatelet or anticoagulant therapy, with or without steroids, can help control aPL-associated chorea (74, 134, 135). On the other hand the potential role of aPL-mediated damage provides the rational for the use of immunosuppressive therapy (136). Reports describe clinical remission of pediatric aPL-associated chorea achieved with mycophenolate mofetil, an immunosuppressant agent primarily used to prevent rejection in organ transplant induced (137), and IVIG (138). Steroids have been successfully used to treat psychotic symptoms in the context of APS (139). Furthermore, a 2013 non-randomized pilot study (RITAPS) demonstrated partial or complete remission of cognitive dysfunction in a small sample of patients treated with RTX (140).
Future Directions in the Treatment of APS
Antithrombotic Agents
Direct oral anticoagulants (DOACs) represent in interesting possible alternative to warfarin, especially considering the young age of many patients and the need to monitor INR with VKA. Furthermore, it has been suggested that the therapeutic effect of DOACs may go beyond anticoagulation and exert an anti-inflammatory and anti-angiogenic effect (141). Many case reports and case series have been published on the use of DOACs (mainly rivaroxaban) in APS, with heterogeneous results (142). However, only a few solid randomized controlled trials are available. In 2016 the RAPS trial by Cohen et al. failed to demonstrate the non-inferiority of rivaroxaban over warfarin in APS patients (143). Indeed, questions have been raised regarding thrombotic risk and safety issues related to DOACs, especially in triple-positive patients (144). Given the lack of solid evidence, the 15th International Congress on Antiphospholipid Antibodies Task Force on Antiphospholipid Syndrome Treatment Trends in 2017 did not support the use of DOACs instead of warfarin (142). Another trial by Pengo et al. on high-risk APS patients treated with rivaroxaban or warfarin was prematurely terminated because of the higher rate of thromboembolic events and major bleeding in the rivaroxaban group (145). Although other studies are currently ongoing investigating the effect of rivaroxaban and other DOACs, warfarin represents at the moment the only approved anticoagulant agent. Platelet activation and aggregation represents a further target for therapeutic agents. Glycoprotein IIb/IIIa (GPIIb/IIIa) receptor inhibitors are a relatively new class of antiplatelet drugs compared to aspirin, mainly used in the field of coronary disease. In murine models aPL-enhanced thrombosis can be inhibited by infusion of a GPIIb/IIIa antagonist monoclonal antibody and GPIIb/IIIa-deficient mice are resistant to aPL-mediated thrombosis (146, 147). Management of acute myocardial infarction in APS can represent a challenge due to the high risk of restenosis after angioplasty (148). Abciximab, a monoclonal antibody targeting GPIIb/IIIa on platelets, has been proposed as a promising alternative to stenting (149, 150). However, Abciximab is not approved for the management of cerebrovascular disease due to the negative results of previous studies (151) limiting its possible use in aPL-ischemic stroke.
Agents Targeting Inflammation
Several agents with anti-inflammatory and anti-oxidative properties may also prove beneficial in APS. Among them, one of the most studied is hydroxichloroquine (HCQ), which has been used for the treatment of SLE for many years. Available data show a potential role in the inhibition of toll-like receptors (TLRs) (152), and reduction of proinflammatory IL-1, TNF-α, and IL-2 (153). HCQ may also target the coagulation pathway by inhibiting platelet aggregation (154) and preventing the disruption of the annexin A5 shield by anti-β2-GPI antibodies (155). Interestingly, a 2013 non-randomized study by Schmidt-Tanguy et al. demonstrated a lower rate of recurrence of thrombosis in APS patients treated with oral anticoagulation plus HCQ vs. oral anticoagulation alone (156). Other potential agents include N-Acetylcysteine (NAC) and mitochondrial cofactor coenzyme Q10 (9). Statins, a class of HMG-CoA reductase inhibitors commonly used to treat dyslipidemia and atherosclerosis, have shown a potential to inhibit aPL-mediated thrombogenesis and modulate the proinflammatory milieu in APS, and both in vitro and in vivo, through the down-regulation of the expression of intracellular adhesion molecule 1 (ICAM1), vascular endothelial growth factor (VEGF) and of pro-inflammatory cytokines, including IL-1β, TNF-α, and interferon (IFN)-α (157, 158).
Monoclonal Antibodies
Belimumab (BEL) is a monoclonal antibody directed against the B-cell activating factor (BAFF or BLyS) which promotes B-cell survival and differentiation, approved for unresponsive SLE. A report by Yazici et al. described its use in two primary APS patients with pulmonary and skin manifestations, respectively. One of the patients showed partial benefit while the other complete remission of symptoms, while aPL profile remained unchanged (159). On the contrary, a report by Sciascia et al. demonstrated disappearance of aPL in three patients with APS secondary to SLE treated with belimumab (160). Eculizumab is a monoclonal antibody directed against the C5 fraction of complement approved for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. To date a series of case reports have been published regarding its use in APS, specifically in the setting of CAPS and post-transplantation renal thrombotic microangiopathy, with encouraging results (161–170).
Other Agents
Sirolimus is a macrolide with inhibiting properties on the mammalian target of rapamycin (mTOR), a kinase involved in many signaling pathways related to cellular growth, proliferation, and survival (112). Previous studies have highlighted the possibility of the involvement of mTOR in the genesis of vascular stenosis in the context of endothelial injury (171, 172). Vascular cellular infiltrates and changes in the vessels intima and media layers have been observed in APS patients, especially in the context of CAPS (110, 111), suggesting a vasculopathic pathogenic mechanism. In a 2014 publication Canaud et al. described a series of patients with APS nephropathy undergoing transplantation. Interestingly, those receiving sirolimus had no recurrence of vascular lesions and showed decreased vascular proliferation on biopsy (173). Ongoing researches aim at identifying other therapeutic targets, including TLR 4 inhibition, TF inhibition, protease-activated receptor (PAR) antagonists, intracellular signaling blockers, and tolerogenic dendritic cells (142).
Conclusions
APS is a rare and heterogeneous condition the neurologist may have to deal with. The diagnosis is not easy and a high level of suspicion must be held. APS should always be considered in young patients with CVA, especially in the absence of other vascular risk factors. Moreover, publications from the past decades have shown that many neurologic disorders not comprised in the original description by Hughes may harbor the disease. Elements such as autoimmune and connective tissue comorbidities, a positive history for obstetrical complications and refractoriness to standard therapy may guide the neurologist toward the correct diagnosis. A great amount of research aims at clarifying the complex etiopathogenesis of APS but many aspects remain obscure. The mechanism behind aPL-mediated pathology appears to be strongly, but not exclusively, thrombotic. Immune-mediated damage may be the key to some aPL-related neurologic manifestations. What favors the attack against nervous structures and what lies beneath the great phenotypic variability even among neurological presentations are open questions for researchers in the near future. On the one hand, mechanisms of immunological surveillance might be involved, as it was demonstrated in other more common forms of nervous system autoimmunity (174). On the other, genetic variability in neurobiological pathways may be worth careful investigation (76, 175). The double nature, thrombotic and immune-mediated, of APS is exemplified by current therapeutic strategies. Anticoagulation with warfarin represents at the moment the most effective therapy in the setting of thrombotic events. Anticoagulation has also been reported to reverse non-criteria neurologic manifestations that are not primarily thrombotic in their origin, further supporting the role of thrombosis in the pathogenesis of APS. On the other hand targeting the immune response through steroids, rituximab, IVIG or PE, can improve many non-criteria symptoms. A great number of other treatments are under investigation. DOACs represent an appealing alternative to warfarin, especially considering the young age of APS patients and the necessity of INR monitoring. Nevertheless, available evidence does not support their use. Other interesting options include antioxidant agents, monoclonal antibodies, and several agents targeting specific cells or molecules in the complex pathogenetic pathway of APS. One last interesting issues concerns the relationship between APS and SLE. A 2010 report by Veres et al. on a group of 165 patients with primary APS showed that 23% of cases converted to definite SLE within 10 years (176). The correlation between these two entities is not fully understood. The term neurolupus indicates the involvement of the nervous system in the setting of SLE. Pathogenic mechanisms, similarly to APS, include thrombosis, vascular proliferative changes and, possibly, anti-neuronal and anti-glial antibody activity (177). Further studies are needed to understand the relation between these two entities, shared mechanisms of nervous system injury and possible therapeutic options.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x
2. Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers (2018) 4:18005. doi: 10.1038/nrdp.2018.5
3. Garcia D, Erkan D. Diagnosis and management of the antiphospholipid syndrome. N Eng J Med. (2018) 378:2010–21. doi: 10.1056/NEJMra1705454
4. Shoenfeld Y, Twig G, Katz U, Sherer Y. Autoantibody explosion in antiphospholipid syndrome. J Autoimmun. (2008) 30:74–83. doi: 10.1016/j.jaut.2007.11.011
5. de Laat B, Mertens K, de Groot PG. Mechanisms of disease: antiphospholipid antibodies-from clinical association to pathologic mechanism. Nat Clin Pract Rheumatol. (2008) 4:192–9. doi: 10.1038/ncprheum0740
6. Agar C, van Os GM, Morgelin M, Sprenger RR, Marquart JA, Urbanus RT, et al. Beta2-glycoprotein I can exist in 2 conformations: implications for our understanding of the antiphospholipid syndrome. Blood (2010) 116:1336–43. doi: 10.1182/blood-2009-12-260976
7. Ioannou Y, Zhang JY, Qi M, Gao L, Qi JC, Yu DM, et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen beta2-glycoprotein I. Arthritis Rheum. (2011) 63:2774–82. doi: 10.1002/art.30383
8. Cervera R. Antiphospholipid syndrome. Thromb Res. (2017) 151(Suppl. 1):S43–7. doi: 10.1016/S0049-3848(17)30066-X
9. Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. (2013) 368:1033–44. doi: 10.1056/NEJMra1112830
10. Erkan D, Lockshin MD. What is antiphospholipid syndrome? Curr Rheumatol Rep. (2004) 6:451–7. doi: 10.1007/s11926-004-0024-1
11. Pengo V, Ruffatti A, Legnani C, Testa S, Fierro T, Marongiu F, et al. Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: a multicenter prospective study. Blood (2011) 118:4714–8. doi: 10.1182/blood-2011-03-340232
12. Ma K, Simantov R, Zhang JC, Silverstein R, Hajjar KA, McCrae KR. High affinity binding of beta 2-glycoprotein I to human endothelial cells is mediated by annexin II. J Biol Chem. (2000) 275:15541–8. doi: 10.1074/jbc.275.20.15541
13. Allen KL, Fonseca FV, Betapudi V, Willard B, Zhang J, McCrae KR. A novel pathway for human endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood (2012) 119:884–93. doi: 10.1182/blood-2011-03-344671
14. Raschi E, Testoni C, Bosisio D, Borghi MO, Koike T, Mantovani A, et al. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood (2003) 101:3495–500. doi: 10.1182/blood-2002-08-2349
15. Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C, et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum. (2007) 56:2687–97. doi: 10.1002/art.22802
16. de Laat B, Wu XX, van Lummel M, Derksen RH, de Groot PG, Rand JH. Correlation between antiphospholipid antibodies that recognize domain I of beta2-glycoprotein I and a reduction in the anticoagulant activity of annexin A5. Blood (2007) 109:1490–4. doi: 10.1182/blood-2006-07-030148
17. Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, Girardi G, et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via beta2GPI and apoER2. J Clin Invest. (2011) 121:120–31. doi: 10.1172/JCI39828
18. Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. (1990) 87:1620–4. doi: 10.1073/pnas.87.4.1620
19. Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. (2012) 90:467–77. doi: 10.1016/j.ajhg.2012.01.017
20. Ames PR, Batuca JR, Ciampa A, Iannaccone L, Delgado Alves J. Clinical relevance of nitric oxide metabolites and nitrative stress in thrombotic primary antiphospholipid syndrome. J Rheumatol. (2010) 37:2523–30. doi: 10.3899/jrheum.100494
21. Urbanus RT, Pennings MT, Derksen RH, de Groot PG. Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2'. J Thrombos Haemost. (2008) 6:1405–12. doi: 10.1111/j.1538-7836.2008.03021.x
22. Noureldine MHA, Haydar AA, Berjawi A, Elnawar R, Sweid A, Khamashta MA, et al. Antiphospholipid syndrome (APS) revisited: would migraine headaches be included in future classification criteria? Immunol Res. (2017) 65:230–41. doi: 10.1007/s12026-016-8831-9
23. Bidot CJ, Jy W, Bidot C, Horstman LL, Fontana V, Yaniz M, et al. Antibodies against platelet glycoprotein target antigens in Antiphospholipid Syndrome (APS). Blood (2006) 108:3973.
24. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. (2006) 177:4794–802. doi: 10.4049/jimmunol.177.7.4794
25. Forastiero R, Martinuzzo M. Prothrombotic mechanisms based on the impairment of fibrinolysis in the antiphospholipid syndrome. Lupus (2008) 17:872–7. doi: 10.1177/0961203308091633
26. Shoenfeld Y, Nahum A, Korczyn AD, Dano M, Rabinowitz R, Beilin O, et al. Neuronal-binding antibodies from patients with antiphospholipid syndrome induce cognitive deficits following intrathecal passive transfer. Lupus (2003) 12:436–42. doi: 10.1191/0961203303lu409oa
27. Gris JC, Nobile B, Bouvier S. Neuropsychiatric presentations of antiphospholipid antibodies. Thromb Res. (2015) 135(Suppl. 1):S56–9. doi: 10.1016/S0049-3848(15)50445-3
28. Garcia CO, Kanbour-Shakir A, Tang H, Molina JF, Espinoza LR, Gharavi AE. Induction of experimental antiphospholipid antibody syndrome in PL/J mice following immunization with beta 2 GPI. Am J Reprod Immunol. (1997) 37:118–24. doi: 10.1111/j.1600-0897.1997.tb00200.x
29. Katzav A, Pick CG, Korczyn AD, Oest E, Blank M, Shoenfeld Y, et al. Hyperactivity in a mouse model of the antiphospholipid syndrome. Lupus (2001) 10:496–9. doi: 10.1191/096120301678416060
30. Shrot S, Katzav A, Korczyn AD, Litvinju Y, Hershenson R, Pick CG, et al. Behavioral and cognitive deficits occur only after prolonged exposure of mice to antiphospholipid antibodies. Lupus (2002) 11:736–43. doi: 10.1191/0961203302lu255oa
31. Katzav A, Shoenfeld Y, Chapman J. The pathogenesis of neural injury in animal models of the antiphospholipid syndrome. Clin Rev Allergy Immunol. (2010) 38:196–200. doi: 10.1007/s12016-009-8154-x
32. Katzav A, Menachem A, Maggio N, Pollak L, Pick CG, Chapman J. IgG accumulates in inhibitory hippocampal neurons of experimental antiphospholipid syndrome. J Autoimmun. (2014) 55:86–93. doi: 10.1016/j.jaut.2014.07.006
33. Tanne D, Katzav A, Beilin O, Grigoriadis NC, Blank M, Pick CG, et al. Interaction of inflammation, thrombosis, aspirin and enoxaparin in CNS experimental antiphospholipid syndrome. Neurobiol Dis. (2008) 30:56–64. doi: 10.1016/j.nbd.2007.12.004
34. Katzav A, Grigoriadis NC, Ebert T, Touloumi O, Blank M, Pick CG, et al. Coagulopathy triggered autoimmunity: experimental antiphospholipid syndrome in factor V Leiden mice. BMC Med. (2013) 11:92. doi: 10.1186/1741-7015-11-92
35. Kent M, Alvarez F, Vogt E, Fyffe R, Ng AK, Rote N. Monoclonal antiphosphatidylserine antibodies react directly with feline and murine central nervous system. J Rheumatol. (1997) 24:1725–33.
36. Dale RC, Yin K, Ding A, Merheb V, Varadkhar S, McKay D, et al. Antibody binding to neuronal surface in movement disorders associated with lupus and antiphospholipid antibodies. Dev Med Child Neurol. (2011) 53:522–8. doi: 10.1111/j.1469-8749.2011.03922.x
37. Liou HH, Wang CR, Chou HC, Arvanov VL, Chen RC, Chang YC, et al. Anticardiolipin antisera from lupus patients with seizures reduce a GABA receptor-mediated chloride current in snail neurons. Life Sci. (1994) 54:1119–25. doi: 10.1016/0024-3205(94)00422-6
38. Chapman J, Cohen-Armon M, Shoenfeld Y, Korczyn AD. Antiphospholipid antibodies permeabilize and depolarize brain synaptoneurosomes. Lupus (1999) 8:127–33. doi: 10.1191/096120399678847524
39. Chapman J, Soloveichick L, Shavit S, Shoenfeld Y, Korczyn AD. Antiphospholipid antibodies bind ATP: a putative mechanism for the pathogenesis of neuronal dysfunction. Clin Dev Immunol. (2005) 12:175–80. doi: 10.1080/17402520500217844
40. Hughes GR. Thrombosis, abortion, cerebral disease, and the lupus anticoagulant. Br Med J. (1983) 287:1088–9. doi: 10.1136/bmj.287.6399.1088
41. Carecchio M, Cantello R, Comi C. Revisiting the molecular mechanism of neurological manifestations in antiphospholipid syndrome: beyond vascular damage. J Immunol Res. (2014) 2014:239398. doi: 10.1155/2014/239398
42. Sanna G, D'Cruz D, Cuadrado MJ. Cerebral manifestations in the antiphospholipid (Hughes) syndrome. Rheumat Dis Clin North Am. (2006) 32:465–90. doi: 10.1016/j.rdc.2006.05.010
43. Cervera R, Boffa MC, Khamashta MA, Hughes GR. The Euro-Phospholipid project: epidemiology of the antiphospholipid syndrome in Europe. Lupus (2009) 18:889–93. doi: 10.1177/0961203309106832
44. Andreoli L, Chighizola CB, Banzato A, Pons-Estel GJ, Ramire de Jesus G, Erkan D. Estimated frequency of antiphospholipid antibodies in patients with pregnancy morbidity, stroke, myocardial infarction, and deep vein thrombosis: a critical review of the literature. Arthritis Care Res. (2013) 65:1869–73. doi: 10.1002/acr.22066
45. Clinical and laboratory findings in patients with antiphospholipid antibodies and cerebral ischemia. The antiphospholipid antibodies in stroke study group. Stroke (1990) 21:1268–73. doi: 10.1161/01.STR.21.9.1268
46. Hughes GR. Migraine, memory loss, and “multiple sclerosis ”. Neurological features of the antiphospholipid (Hughes') syndrome. Postgraduate Med J. (2003) 79:81–3. doi: 10.1136/pmj.79.928.81
47. Sciascia S, Sanna G, Khamashta MA, Cuadrado MJ, Erkan D, Andreoli L, et al. The estimated frequency of antiphospholipid antibodies in young adults with cerebrovascular events: a systematic review. Ann Rheumat Dis. (2015) 74:2028–33. doi: 10.1136/annrheumdis-2014-205663
48. Roldan JF, Brey RL. Neurologic manifestations of the antiphospholipid syndrome. Curr Rheumatol Rep. (2007) 9:109–15. doi: 10.1007/s11926-007-0004-3
49. Provenzale JM, Barboriak DP, Allen NB, Ortel TL. Patients with antiphospholipid antibodies: CT and MR findings of the brain. Am J Roentgenol. (1996) 167:1573–8. doi: 10.2214/ajr.167.6.8956600
50. Miesbach W, Gilzinger A, Gokpinar B, Claus D, Scharrer I. Prevalence of antiphospholipid antibodies in patients with neurological symptoms. Clin Neurol Neurosurg. (2006) 108:135–42. doi: 10.1016/j.clineuro.2005.03.005
51. Ricarte IF, Dutra LA, Abrantes FF, Toso FF, Barsottini OGP, Silva GS, et al. Neurologic manifestations of antiphospholipid syndrome. Lupus (2018) 2018:961203318776110. doi: 10.1177/0961203318776110
52. Provenzale JM, Barboriak DP, Allen NB, Ortel TL. Antiphospholipid antibodies: findings at arteriography. AJNR Am J Neuroradiol. (1998) 19:611–6.
53. de Amorim LC, Maia FM, Rodrigues CE. Stroke in systemic lupus erythematosus and antiphospholipid syndrome: risk factors, clinical manifestations, neuroimaging, and treatment. Lupus (2017) 26:529–36. doi: 10.1177/0961203316688784
54. Jara LJ, Medina G, Vera-Lastra O, Shoenfeld Y. Atherosclerosis and antiphospholipid syndrome. Clin Rev Allergy Immunol. (2003) 25:79–88. doi: 10.1385/CRIAI:25:1:79
55. Tsai CL, Hueng DY, Tsao WL, Lin JC. Cerebral venous sinus thrombosis as an initial manifestation of primary antiphospholipid syndrome. Am J Emerge Med. (2013) 31:888.e1–3. doi: 10.1016/j.ajem.2012.12.020
56. Shlebak A. Antiphospholipid syndrome presenting as cerebral venous sinus thrombosis: a case series and a review. J Clin Pathol. (2016) 69:337–43. doi: 10.1136/jclinpath-2015-203077
57. Silvis SM, de Sousa DA, Ferro JM, Coutinho JM. Cerebral venous thrombosis. Nat Rev Neurol. (2017) 13:555–65. doi: 10.1038/nrneurol.2017.104
58. Boesch SM, Plorer AL, Auer AJ, Poewe W, Aichner FT, Felber SR, et al. The natural course of Sneddon syndrome: clinical and magnetic resonance imaging findings in a prospective six year observation study. J Neurol Neurosurg Psychiatry (2003) 74:542–4. doi: 10.1136/jnnp.74.4.542
59. Aladdin Y, Hamadeh M, Butcher K. The Sneddon syndrome. Arch Neurol. (2008) 65:834–5. doi: 10.1001/archneur.65.6.834
60. Fetoni V, Grisoli M, Salmaggi A, Carriero R, Girotti F. Clinical and neuroradiological aspects of Sneddon's syndrome and primary antiphospholipid antibody syndrome. A follow-up study. Neurol Sci. (2000) 21:157–64. doi: 10.1007/s100720070091
61. Ducros A. Reversible cerebral vasoconstriction syndrome. Lancet Neurol. (2012) 11:906–17. doi: 10.1016/S1474-4422(12)70135-7
62. Gupta S, Zivadinov R, Ramasamy D, Ambrus JLJr. Reversible cerebral vasoconstriction syndrome (RCVS) in antiphospholipid antibody syndrome (APLA): the role of centrally acting vasodilators. Case series and review of literature. Clin Rheumatol. (2014) 33:1829–33. doi: 10.1007/s10067-013-2434-9
63. Cavestro C, Micca G, Molinari F, Bazzan M, C DIP, Aloi R, et al. Migraineurs show a high prevalence of antiphospholipid antibodies. J Thromb Haemost. (2011) 9:1350–4. doi: 10.1111/j.1538-7836.2011.04348.x
64. Yelnik CM, Kozora E, Appenzeller S. Non-stroke central neurologic manifestations in antiphospholipid syndrome. Curr Rheumatol Rep. (2016) 18:11. doi: 10.1007/s11926-016-0568-x
65. Silvestrini M, Cupini LM, Matteis M, De Simone R, Bernardi G. Migraine in patients with stroke and antiphospholipid antibodies. Headache (1993) 33:421–6. doi: 10.1111/j.1526-4610.1993.hed3308421.x
66. Graf J. Central nervous system manifestations of antiphospholipid syndrome. Rheumat Dis Clin North Am. (2017) 43:547–60. doi: 10.1016/j.rdc.2017.06.004
67. Noureldine MH, Harifi G, Berjawi A, Haydar AA, Nader M, Elnawar R, et al. Hughes syndrome and epilepsy: when to test for antiphospholipid antibodies? Lupus (2016) 25:1397–411. doi: 10.1177/0961203316651747
68. Shoenfeld Y, Lev S, Blatt I, Blank M, Font J, von Landenberg P, et al. Features associated with epilepsy in the antiphospholipid syndrome. J Rheumatol. (2004) 31:1344–8.
69. Cimaz R, Meroni PL, Shoenfeld Y. Epilepsy as part of systemic lupus erythematosus and systemic antiphospholipid syndrome (Hughes syndrome). Lupus (2006) 15:191–7. doi: 10.1191/0961203306lu2272rr
70. de Carvalho JF, Pasoto SG, Appenzeller S. Seizures in primary antiphospholipid syndrome: the relevance of smoking to stroke. Clin Dev Immunol. (2012) 2012:981519. doi: 10.1155/2012/981519
71. Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. (2002) 46:1019–27. doi: 10.1002/art.10187
72. Safarpour D, Buckingham S, Jabbari B. Chorea associated with high titers of antiphospholipid antibodies in the absence of antiphospholipid antibody syndrome. Tremor Other Hyperkinet Move (New York, NY). (2015) 5:294. doi: 10.7916/D8DB80M9
73. Baizabal-Carvallo JF, Jankovic J. Autoimmune and paraneoplastic movement disorders: an update. J Neurol Sci. (2018) 385:175–84. doi: 10.1016/j.jns.2017.12.035
74. Carecchio M, Comi C, Varrasi C, Stecco A, Sainaghi PP, Bhatia K, et al. Complex movement disorders in primary antiphospholipid syndrome: a case report. J Neurol Sci. (2009) 281:101–3. doi: 10.1016/j.jns.2009.03.011
75. Tanne D, Hassin-Baer S. Neurologic manifestations of the antiphospholipid syndrome. Curr Rheumatol Rep. (2001) 3:286–92. doi: 10.1007/s11926-001-0032-3
76. Comi C, Ferrari M, Marino F, Magistrelli L, Cantello R, Riboldazzi G, et al. Polymorphisms of dopamine receptor genes and risk of L-dopa-induced dyskinesia in Parkinson's Disease. Int J Mol Sci. (2017) 18:2. doi: 10.3390/ijms18020242
77. Shepherd DI, Downie AW, Best PV. Systemic lupus erythematosis and multiple sclerosis. Trans Am Neurol Assoc. (1974) 99:173–6.
78. Stepien M, Cendrowski W. [Form of systemic lupus erythematosus (lupoid sclerosis) resembling multiple sclerosis]. Neurol Neurochir Polska (1974) 8:607–9.
79. Keiserman B, da Silva LF, Keiserman MW, von Muhlen CA, Staub HL. Lupoid sclerosis. Rheumatol Int. (2010) 30:431–4. doi: 10.1007/s00296-009-1175-1
80. Uthman I, Noureldine MH, Berjawi A, Skaf M, Haydar AA, Merashli M, et al. Hughes syndrome and multiple sclerosis. Lupus (2015) 24:115–21. doi: 10.1177/0961203314555539
81. Bidot CJ, Horstman LL, Jy W, Jimenez JJ, Bidot C Jr, Ahn YS, et al. Clinical and neuroimaging correlates of antiphospholipid antibodies in multiple sclerosis: a preliminary study. BMC Neurol. (2007) 7:36. doi: 10.1186/1471-2377-7-36
82. Roussel V, Yi F, Jauberteau MO, Couderq C, Lacombe C, Michelet V, et al. Prevalence and clinical significance of anti-phospholipid antibodies in multiple sclerosis: a study of 89 patients. J Autoimmun. (2000) 14:259–65. doi: 10.1006/jaut.2000.0367
83. Cuadrado MJ, Khamashta MA, Ballesteros A, Godfrey T, Simon MJ, Hughes GR. Can neurologic manifestations of Hughes (antiphospholipid) syndrome be distinguished from multiple sclerosis? Analysis of 27 patients and review of the literature. Medicine (2000) 79:57–68. doi: 10.1097/00005792-200001000-00006
84. Ahbeddou N, Ait Ben Haddou E, Hammi S, Slimani C, Regragui W, Benomar A, et al. [Multiple sclerosis associated with antiphospholipid syndrome: diagnostic and therapeutic difficulties]. Revue Neurol. (2012) 168:65–9. doi: 10.1016/j.neurol.2011.01.021
85. Fernandez-Fernandez FJ, Rivera-Gallego A, de la Fuente-Aguado J, Perez-Fernandez S, Munoz-Fernandez D. Antiphospholipid syndrome mimicking multiple sclerosis in two patients. Eur J Int Med. (2006) 17:500–2. doi: 10.1016/j.ejim.2006.02.018
86. Lima I, Melo A, Brandi IV, Costa O, Santiago M. Lupoid sclerosis: what is the role of antiphospholipid antibodies? J Clin Rheumatol. (2007) 13:85–6. doi: 10.1097/01.rhu.0000260637.95695.40
87. Mayer M, Cerovec M, Rados M, Cikes N. Antiphospholipid syndrome and central nervous system. Clin Neurol Neurosurg. (2010) 112:602–8. doi: 10.1016/j.clineuro.2010.03.023
88. JW IJ, Conti-Kelly AM, Greco P, Abedi M, Amos M, Provenzale JM, et al. Anti-phospholipid antibodies in patients with multiple sclerosis and MS-like illnesses: MS or APS? Lupus (1999) 8:109–15. doi: 10.1191/096120399678847461
89. Arnson Y, Shoenfeld Y, Alon E, Amital H. The antiphospholipid syndrome as a neurological disease. Semin Arthritis Rheum. (2010) 40:97–108. doi: 10.1016/j.semarthrit.2009.05.001
90. Stosic M, Ambrus J, Garg N, Weinstock-Guttman B, Ramanathan M, Kalman B, et al. MRI characteristics of patients with antiphospholipid syndrome and multiple sclerosis. J Neurol. (2010) 257:63–71. doi: 10.1007/s00415-009-5264-6
91. Ferreira S, D'Cruz DP, Hughes GR. Multiple sclerosis, neuropsychiatric lupus and antiphospholipid syndrome: where do we stand? Rheumatology (Oxford, England). (2005) 44:434–42. doi: 10.1093/rheumatology/keh532
92. Rodrigues CE, de Carvalho JF. Clinical, radiologic, and therapeutic analysis of 14 patients with transverse myelitis associated with antiphospholipid syndrome: report of 4 cases and review of the literature. Semin Arthritis Rheum. (2011) 40:349–57. doi: 10.1016/j.semarthrit.2010.05.004
93. Sherer Y, Hassin S, Shoenfeld Y, Levy Y, Livneh A, Ohry A, et al. Transverse myelitis in patients with antiphospholipid antibodies–the importance of early diagnosis and treatment. Clin Rheumatol. (2002) 21:207–10. doi: 10.1007/s10067-002-8287-2
94. Komolafe MA, Komolafe EO, Sunmonu TA, Olateju SO, Asaleye CM, Adesina OA, et al. New onset neuromyelitis optica in a young Nigerian woman with possible antiphospholipid syndrome: a case report. J Med Case Rep. (2008) 2:348. doi: 10.1186/1752-1947-2-348
95. Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity (2014) 47:154–61. doi: 10.3109/08916934.2014.883501
96. Tobin WO, Weinshenker BG, Lucchinetti CF. Longitudinally extensive transverse myelitis. Curr Opin Neurol. (2014) 27:279–89. doi: 10.1097/WCO.0000000000000093
97. Brey RL, Muscal E, Chapman J. Antiphospholipid antibodies and the brain: a consensus report. Lupus (2011) 20:153–7. doi: 10.1177/0961203310396748
98. Jacobson MW, Rapport LJ, Keenan PA, Coleman RD, Tietjen GE. Neuropsychological deficits associated with antiphospholipid antibodies. J Clin Exp Neuropsychol. (1999) 21:251–64. doi: 10.1076/jcen.21.2.251.931
99. Tektonidou MG, Varsou N, Kotoulas G, Antoniou A, Moutsopoulos HM. Cognitive deficits in patients with antiphospholipid syndrome: association with clinical, laboratory, and brain magnetic resonance imaging findings. Arch Inter Med. (2006) 166:2278–84. doi: 10.1001/archinte.166.20.2278
100. Mosek A, Yust I, Treves TA, Vardinon N, Korczyn AD, Chapman J. Dementia and antiphospholipid antibodies. Dement Geriat Cogn Dis. (2000) 11:36–8. doi: 10.1159/000017211
101. Islam MA, Alam F, Kamal MA, Gan SH, Sasongko TH, Wong KK. Presence of anticardiolipin antibodies in patients with dementia: a systematic review and meta-analysis. Front Aging Neurosci. (2017) 9:250. doi: 10.3389/fnagi.2017.00250
102. Appenzeller S, Lapa AT, Guirau CR, de Carvalho JF, Shoenfeld Y. Cognitive impairment in antiphospholipid syndrome: evidence from animal models. Clin Rheumatol. (2012) 31:403–6. doi: 10.1007/s10067-011-1922-z
103. Abreu MM, Danowski A, Wahl DG, Amigo MC, Tektonidou M, Pacheco MS, et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th international congress on antiphospholipid antibodies technical task force report on antiphospholipid syndrome clinical features. Autoimmun Rev. (2015) 14:401–14. doi: 10.1016/j.autrev.2015.01.002
104. Gris JC, Cyprien F, Bouvier S, Cochery-Nouvellon E, Lavigne-Lissalde G, Mercier E, et al. Antiphospholipid antibodies are associated with positive screening for common mental disorders in women with previous pregnancy loss. The NOHA-PSY observational study. World J Biol Psychiatry (2017). 2017:1–13. doi: 10.1080/15622975.2017.1333146
105. Santos MS, de Carvalho JF, Brotto M, Bonfa E, Rocha FA. Peripheral neuropathy in patients with primary antiphospholipid (Hughes') syndrome. Lupus (2010) 19:583–90. doi: 10.1177/0961203309354541
106. Sanna G, Bertolaccini ML, Cuadrado MJ, Khamashta MA, Hughes GR. Central nervous system involvement in the antiphospholipid (Hughes) syndrome. Rheumatology (2003) 42:200–13. doi: 10.1093/rheumatology/keg080
107. Schofield JR. Autonomic neuropathy-in its many guises-as the initial manifestation of the antiphospholipid syndrome. Immunol Res. (2017) 65:532–42. doi: 10.1007/s12026-016-8889-4
108. Resseguier AS, Pereira B, Rieu V, Le Guenno G, Grobost V, Ruivard M. Direct oral anticoagulants: an alternative treatment for thrombotic antiphospholipid syndrome? Lupus (2017) 26:1297–303. doi: 10.1177/0961203317701841
109. Saraiva Sda S, Custodio IF, Mazetto Bde M, Collela MP, de Paula EV, Appenzeller S, et al. Recurrent thrombosis in antiphospholipid syndrome may be associated with cardiovascular risk factors and inflammatory response. Thromb Res. (2015) 136:1174–8. doi: 10.1016/j.thromres.2015.10.029
110. Julkunen H, Hedman C, Kauppi M. Thrombolysis for acute ischemic stroke in the primary antiphospholipid syndrome. J Rheumatol. (1997) 24:181–3.
111. Camara-Lemarroy CR, Infante-Valenzuela A, Andrade-Vazquez CJ, Enriquez-Noyola RV, Garcia-Valadez EA, Gongora-Rivera F. Successful intravenous thrombolysis in a patient with antiphospholipid syndrome, acute ischemic stroke and severe thrombocytopenia. Blood Coagul Fibrinol. (2016) 27:354–6. doi: 10.1097/MBC.0000000000000432
112. Stadler K, Mutzenbach JS, Kalss G, Sellner J, Al-Schameri AR, Trinka E, et al. Therapeutic challenges after successful thrombectomy in a patient with an antiphospholipid syndrome associated M1-occlusion: a case report. Int Neuroradiol. (2015) 21:598–602. doi: 10.1177/1591019915590371
113. Keeling D, Mackie I, Moore GW, Greer IA, Greaves M. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol. (2012) 157:47–58. doi: 10.1111/j.1365-2141.2012.09037.x
114. Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, Brey R, Crowther M, Derksen R, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus (2011) 20:206–18. doi: 10.1177/0961203310395803
115. Rosa RF, Ugolini-Lopes MR, Zeinad-Valim AK, D'Amico E, Andrade D. Difficult clinical situations in the antiphospholipid syndrome. Curr Rheumatol Rep. (2015) 17:29. doi: 10.1007/s11926-015-0502-7
116. Ruiz-Irastorza G, Khamashta MA, Hunt BJ, Escudero A, Cuadrado MJ, Hughes GR. Bleeding and recurrent thrombosis in definite antiphospholipid syndrome: analysis of a series of 66 patients treated with oral anticoagulation to a target international normalized ratio of 3.5. Arch Int Med. (2002) 162:1164–9. doi: 10.1001/archinte.162.10.1164
117. Giron-Gonzalez JA, Garcia del Rio E, Rodriguez C, Rodriguez-Martorell J, Serrano A. Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals. J Rheumatol. (2004) 31:1560–7.
118. Ruffatti A, Del Ross T, Ciprian M, Nuzzo M, Rampudda M, Bertero MT, et al. Risk factors for a first thrombotic event in antiphospholipid antibody carriers. A multicentre, retrospective follow-up study. Ann Rheumat Dis. (2009) 68:397–9. doi: 10.1136/ard.2008.096669
119. Ruffatti A, Del Ross T, Ciprian M, Bertero MT, Sciascia S, Scarpato S, et al. Risk factors for a first thrombotic event in antiphospholipid antibody carriers: a prospective multicentre follow-up study. Ann Rheumat Dis. (2011) 70:1083–6. doi: 10.1136/ard.2010.142042
120. Pengo V, Testa S, Martinelli I, Ghirarduzzi A, Legnani C, Gresele P, et al. Incidence of a first thromboembolic event in carriers of isolated lupus anticoagulant. Thromb Res. (2015) 135:46–9. doi: 10.1016/j.thromres.2014.10.013
121. Rodriguez-Pinto I, Espinosa G, Erkan D, Shoenfeld Y, Cervera R. The effect of triple therapy on the mortality of catastrophic anti-phospholipid syndrome patients. Rheumatology (Oxford, England). (2018) 57:1264–70. doi: 10.1093/rheumatology/key082
122. Cervera R, Rodriguez-Pinto I, Colafrancesco S, Conti F, Valesini G, Rosario C, et al. 14th international congress on antiphospholipid antibodies task force report on catastrophic antiphospholipid syndrome. Autoimmun Rev. (2014) 13:699–707. doi: 10.1016/j.autrev.2014.03.002
123. Khattri S, Zandman-Goddard G, Peeva E. B-cell directed therapies in antiphospholipid antibody syndrome–new directions based on murine and human data. Autoimmun Rev. (2012) 11:717–22. doi: 10.1016/j.autrev.2011.12.011
124. Erdozain JG, Ruiz-Irastorza G, Egurbide MV, Aguirre C. Sustained response to rituximab of autoimmune hemolytic anemia associated with antiphospholipid syndrome. Haematologica (2004) 89:Ecr34.
125. Trappe R, Loew A, Thuss-Patience P, Dorken B, Riess H. Successful treatment of thrombocytopenia in primary antiphospholipid antibody syndrome with the anti-CD20 antibody rituximab–monitoring of antiphospholipid and anti-GP antibodies: a case report. Ann Hematol. (2006) 85:134–5. doi: 10.1007/s00277-005-0028-6
126. Gamoudi D, Cutajar M, Gamoudi N, Camilleri DJ, Gatt A. Achieving a satisfactory clinical and biochemical response in antiphospholipid syndrome and severe thrombocytopenia with rituximab: two case reports. Clin Case Rep. (2017) 5:845–8. doi: 10.1002/ccr3.946
127. Dioszegi A, Tarr T, Nagy-Vincze M, Nanasy-Vass M, Veisz R, Bidiga L, et al. Microthrombotic renal involvement in an SLE patient with concomitant catastrophic antiphospholipid syndrome: the beneficial effect of rituximab treatment. Lupus (2018) 2018:961203318768890. doi: 10.1177/0961203318768890
128. Wang CR, Liu MF. Rituximab usage in systemic lupus erythematosus-associated antiphospholipid syndrome: a single-center experience. Semin Arthritis Rheum. (2016) 46:102–8. doi: 10.1016/j.semarthrit.2016.02.002
129. Emmi G, Urban ML, Scalera A, Becatti M, Fiorillo C, Silvestri E, et al. Repeated low-dose courses of rituximab in SLE-associated antiphospholipid syndrome: data from a tertiary dedicated centre. Semin Arthritis Rheum. (2017) 46:e21–3. doi: 10.1016/j.semarthrit.2016.08.002
130. Bakshi J, Stevens R. Rituximab therapy for recurrent thromboembolic disease in antiphospholipid syndrome. Lupus (2013) 22:865–7. doi: 10.1177/0961203313491023
131. Asherson RA, Giampaulo D, Singh S, Sulman L. Dramatic response of severe headaches to anticoagulation in a patient with antiphospholipid syndrome. J Clin Rheumatol. (2007) 13:173–4. doi: 10.1097/RHU.0b013e3180690af6
132. Roie EV, Labarque V, Renard M, Van Geet C, Gabriels L. Obsessive-compulsive behavior as presenting symptom of primary antiphospholipid syndrome. Psychosomat Med. (2013) 75:326–30. doi: 10.1097/PSY.0b013e31828acfbc
133. D'Cruz DP, Mellor-Pita S, Joven B, Sanna G, Allanson J, Taylor J, et al. Transverse myelitis as the first manifestation of systemic lupus erythematosus or lupus-like disease: good functional outcome and relevance of antiphospholipid antibodies. J Rheumatol. (2004) 31:280–5.
134. Ayalew Y, Khattak F. Antiphospholipid antibody syndrome presenting with hemichorea. Case Rep Rheumatol. (2012) 2012:471543. doi: 10.1155/2012/471543
135. Demonty J, Gonce M, Ribai P, Verellen-Dumoulin C, Hustinx R. Chorea associated with anti-phospholipid antibodies: case report. Acta Clin Belg. (2010) 65:350–3. doi: 10.1179/acb.2010.075
136. Espinosa G, Cervera R. Current treatment of antiphospholipid syndrome: lights and shadows. Nat Rev Rheumatol. (2015) 11:586–96. doi: 10.1038/nrrheum.2015.88
137. Yokoyama K, Mori M, Yoshida A. Mycophenolate mofetil therapy for two cases of antiphospholipid antibody-associated chorea. Mod Rheumatol. (2018) 28:709–11. doi: 10.3109/14397595.2015.1134035
138. Brogna C, Mariotti P, Manna R. Conventional and intravenous immunoglobulin therapy in paediatric antiphospholipid antibodies-related chorea. Lupus (2014) 23:1449–51. doi: 10.1177/0961203314551084
139. Lai JY, Wu PC, Chen HC, Lee MB. Early neuropsychiatric involvement in antiphospholipid syndrome. General Hosp Psychiatry (2012) 34:579.e1–3. doi: 10.1016/j.genhosppsych.2012.02.005
140. Erkan D, Vega J, Ramon G, Kozora E, Lockshin MD. A pilot open-label phase II trial of rituximab for non-criteria manifestations of antiphospholipid syndrome. Arthritis Rheum. (2013) 65:464–71. doi: 10.1002/art.37759
141. Alberio L. The new direct oral anticoagulants in special indications: rationale and preliminary data in cancer, mechanical heart valves, anti-phospholipid syndrome, and heparin-induced thrombocytopenia and beyond. Semin Hematol. (2014) 51:152–6. doi: 10.1053/j.seminhematol.2014.03.002
142. Signorelli F, Balbi GGM, Domingues V, Levy RA. New and upcoming treatments in antiphospholipid syndrome: a comprehensive review. Pharmacol Res. (2018) 133:108–20. doi: 10.1016/j.phrs.2018.04.012
143. Cohen H, Hunt BJ, Efthymiou M, Arachchillage DR, Mackie IJ, Clawson S, et al. Rivaroxaban versus warfarin to treat patients with thrombotic antiphospholipid syndrome, with or without systemic lupus erythematosus (RAPS): a randomised, controlled, open-label, phase 2/3, non-inferiority trial. Lancet Haematol. (2016) 3:e426–36. doi: 10.1016/S2352-3026(16)30079-5
144. Dufrost V, Risse J, Zuily S, Wahl D. Direct oral anticoagulants use in antiphospholipid syndrome: are these drugs an effective and safe alternative to warfarin? A systematic review of the literature. Curr Rheumatol Rep. (2016) 18:74. doi: 10.1007/s11926-016-0623-7
145. Pengo V, Denas G. Rivaroxaban vs. warfarin in high-risk patients with antiphospholipid syndrome. Blood (2018) 132:1365–71. doi: 10.1182/blood-2018-04-848333
146. Pierangeli SS, Vega-Ostertag M, Harris EN. Intracellular signaling triggered by antiphospholipid antibodies in platelets and endothelial cells: a pathway to targeted therapies. Thromb Res. (2004) 114:467–76. doi: 10.1016/j.thromres.2004.06.031
147. Gerosa M, Meroni PL, Erkan D. Recognition and management of antiphospholipid syndrome. Curr Opin Rheumatol. (2016) 28:51–9. doi: 10.1097/BOR.0000000000000240
148. Chambers JD Jr Haire HD, Deligonul U. Multiple early percutaneous transluminal coronary angioplasty failures related to lupus anticoagulant. Am Heart J. (1996) 132(1 Pt. 1):189–90. doi: 10.1016/S0002-8703(96)90409-3
149. Marti V, Seixo F, Santalo M, Serra A. Antiphospholipid syndrome and acute myocardial infarction: treatment with thrombectomy and abciximab. Rev Port Cardiol. (2014) 33:465.e1–4. doi: 10.1016/j.repc.2014.01.020
150. Andrade D, Tektonidou M. Emerging therapies in antiphospholipid syndrome. Curr Rheumatol Rep. (2016) 18:22. doi: 10.1007/s11926-016-0566-z
151. Ciccone A, Motto C, Abraha I, Cozzolino F, Santilli I. Glycoprotein IIb-IIIa inhibitors for acute ischaemic stroke. Cochrane Database Syst Rev. (2014) 2014:Cd005208. doi: 10.1002/14651858.CD005208.pub3
152. Raschi E, Borghi MO, Grossi C, Broggini V, Pierangeli S, Meroni PL. Toll-like receptors: another player in the pathogenesis of the anti-phospholipid syndrome. Lupus (2008) 17:937–42. doi: 10.1177/0961203308095140
153. Sperber K, Quraishi H, Kalb TH, Panja A, Stecher V, Mayer L. Selective regulation of cytokine secretion by hydroxychloroquine: inhibition of interleukin 1 alpha (IL−1-alpha) and IL−6 in human monocytes and T cells. J Rheumatol. (1993) 20:803–8.
154. Achuthan S, Ahluwalia J, Shafiq N, Bhalla A, Pareek A, Chandurkar N, et al. Hydroxychloroquine's Efficacy as an antiplatelet agent study in healthy volunteers: a proof of concept study. J Cardiovasc Pharmacol Therapeut. (2015) 20:174–80. doi: 10.1177/1074248414546324
155. Rand JH, Wu XX, Quinn AS, Ashton AW, Chen PP, Hathcock JJ, et al. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood (2010) 115:2292–9. doi: 10.1182/blood-2009-04-213520
156. Schmidt-Tanguy A, Voswinkel J, Henrion D, Subra JF, Loufrani L, Rohmer V, et al. Antithrombotic effects of hydroxychloroquine in primary antiphospholipid syndrome patients. J Thromb Haemost. (2013) 11:1927–9. doi: 10.1111/jth.12363
157. Meroni PL, Raschi E, Testoni C, Tincani A, Balestrieri G, Molteni R, et al. Statins prevent endothelial cell activation induced by antiphospholipid (anti-beta2-glycoprotein I) antibodies: effect on the proadhesive and proinflammatory phenotype. Arthritis Rheumat. (2001) 44:2870–8. doi: 10.1002/1529-0131(200112)44:12<2870::AID-ART475>3.0.CO;2-Y
158. Erkan D, Willis R, Murthy VL, Basra G, Vega J, Ruiz-Limon P, et al. A prospective open-label pilot study of fluvastatin on proinflammatory and prothrombotic biomarkers in antiphospholipid antibody positive patients. Ann Rheumat Dis. (2014) 73:1176–80. doi: 10.1136/annrheumdis-2013-203622
159. Yazici A, Yazirli B, Erkan D. Belimumab in primary antiphospholipid syndrome. Lupus (2017) 26:1123–4. doi: 10.1177/0961203316682102
160. Sciascia S, Rubini E, Radin M, Cecchi I, Rossi D, Roccatello D. Anticardiolipin and anti-beta 2 glycoprotein-I antibodies disappearance in patients with systemic lupus erythematosus and antiphospholipid syndrome while on belimumab. Ann Rheumat Dis. (2018) 77:1694–5. doi: 10.1136/annrheumdis-2018-213496
161. Lonze BE, Singer AL, Montgomery RA. Eculizumab and renal transplantation in a patient with CAPS. N Engl J Med. (2010) 362:1744–5. doi: 10.1056/NEJMc0910965
162. Shapira I, Andrade D, Allen SL, Salmon JE. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. (2012) 64:2719–23. doi: 10.1002/art.34440
163. Canaud G, Kamar N, Anglicheau D, Esposito L, Rabant M, Noel LH, et al. Eculizumab improves posttransplant thrombotic microangiopathy due to antiphospholipid syndrome recurrence but fails to prevent chronic vascular changes. Am J Transpl. (2013) 13:2179–85. doi: 10.1111/ajt.12319
164. Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transpl. (2014) 14:459–65. doi: 10.1111/ajt.12540
165. Bakhtar O, Thajudeen B, Braunhut BL, Yost SE, Bracamonte ER, Sussman AN, et al. A case of thrombotic microangiopathy associated with antiphospholipid antibody syndrome successfully treated with eculizumab. Transplantation (2014) 98:e17–8. doi: 10.1097/TP.0000000000000267
166. Strakhan M, Hurtado-Sbordoni M. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. (2014) 2014:704371. doi: 10.1155/2014/704371
167. Kronbichler A, Frank R, Kirschfink M, Szilagyi A, Csuka D, Prohaszka Z, et al. Efficacy of eculizumab in a patient with immunoadsorption-dependent catastrophic antiphospholipid syndrome: a case report. Medicine (2014) 93:e143. doi: 10.1097/MD.0000000000000143
168. Zikos TA, Sokolove J, Ahuja N, Berube C. Eculizumab induces sustained remission in a patient with refractory primary catastrophic antiphospholipid syndrome. J Clin Rheumatol. (2015) 21:311–3. doi: 10.1097/RHU.0000000000000290
169. Gustavsen A, Skattum L, Bergseth G, Lorentzen B, Floisand Y, Bosnes V, et al. Effect on mother and child of eculizumab given before caesarean section in a patient with severe antiphospholipid syndrome: a case report. Medicine (2017) 96:e6338. doi: 10.1097/MD.0000000000006338
170. Geethakumari PR, Mille P, Gulati R, Nagalla S. Complement inhibition with eculizumab for thrombotic microangiopathy rescues a living-donor kidney transplant in a patient with antiphospholipid antibody syndrome. Transfus Apher Sci. (2017) 56:400–3. doi: 10.1016/j.transci.2017.02.007
171. Gallo R, Padurean A, Jayaraman T, Marx S, Roque M, Adelman S, et al. Inhibition of intimal thickening after balloon angioplasty in porcine coronary arteries by targeting regulators of the cell cycle. Circulation (1999) 99:2164–70. doi: 10.1161/01.CIR.99.16.2164
172. Sousa JE, Costa MA, Abizaid AC, Rensing BJ, Abizaid AS, Tanajura LF, et al. Sustained suppression of neointimal proliferation by sirolimus-eluting stents: one-year angiographic and intravascular ultrasound follow-up. Circulation (2001) 104:2007–11. doi: 10.1161/hc4201.098056
173. Canaud G, Bienaime F, Tabarin F, Bataillon G, Seilhean D, Noel LH, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med. (2014) 371:303–12. doi: 10.1056/NEJMoa1312890
174. Comi C, Fleetwood T, Dianzani U. The role of T cell apoptosis in nervous system autoimmunity. Autoimmun Rev. (2012) 12:150–6. doi: 10.1016/j.autrev.2011.08.011
175. Ferrari M, Comi C, Marino F, Magistrelli L, De Marchi F, Cantello R, et al. Polymorphisms of dopamine receptor genes and risk of visual hallucinations in Parkinson's patients. Eur J Clin Pharmacol. (2016) 72:1335–41. doi: 10.1007/s00228-016-2111-4
176. Veres K, Szodoray P, Szekanecz Z, Lakos G, Kiss E, Laczik R, et al. Clinical and immunoserological characteristics of the transition from primary to overlap antiphospholipid syndrome. Lupus (2010) 19:1520–6. doi: 10.1177/0961203310374336
Keywords: APS, antiphospholipid syndrome, aPL, antiphospholipid antibodies, neurological manifestations, pathogenic mechanisms, therapy
Citation: Fleetwood T, Cantello R and Comi C (2018) Antiphospholipid Syndrome and the Neurologist: From Pathogenesis to Therapy. Front. Neurol. 9:1001. doi: 10.3389/fneur.2018.01001
Received: 31 July 2018; Accepted: 06 November 2018;
Published: 26 November 2018.
Edited by:
Tatiana Koudriavtseva, Istituto Nazionale del Cancro Regina Elena, ItalyReviewed by:
Noriko Isobe, Kyushu University, JapanDanieli Castro Oliveira De Andrade, Universidade de São Paulo, Brazil
Luca Prosperini, Azienda Ospedaliera San Camillo-Forlanini, Italy
Copyright © 2018 Fleetwood, Cantello and Comi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristoforo Comi, Y3Jpc3RvZm9yby5jb21pQG1lZC51bml1cG8uaXQ=