 Foteini Christidi1*
Foteini Christidi1* Efstratios Karavasilis2
Efstratios Karavasilis2 Michail Rentzos1Nikolaos Kelekis2
Michail Rentzos1Nikolaos Kelekis2 Ioannis Evdokimidis1
Ioannis Evdokimidis1 Peter Bede3
Peter Bede3- 1First Department of Neurology, Aeginition Hospital, National and Kapodistrian University of Athens, Athens, Greece
- 2Second Department of Radiology, University General Hospital Attikon, National and Kapodistrian University of Athens, Athens, Greece
- 3Computational Neuroimaging Group, Academic Unit of Neurology, Trinity College Dublin, Dublin, Ireland
Amyotrophic lateral sclerosis (ALS) is now universally recognized as a complex multisystem disorder with considerable extra-motor involvement. The neuropsychological manifestations of frontotemporal, parietal, and basal ganglia involvement in ALS have important implications for compliance with assistive devices, survival, participation in clinical trials, caregiver burden, and the management of individual care needs. Recent advances in neuroimaging have been instrumental in characterizing the biological substrate of heterogeneous cognitive and behavioral deficits in ALS. In this review we discuss the clinical and radiological aspects of cognitive and behavioral impairment in ALS focusing on the recognition, assessment, and monitoring of these symptoms.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease (MND), a progressive neurodegenerative condition defined by concomitant lower and upper motor neuron degeneration (1). Motor symptoms include muscle weakness, fasciculations, cramps, as well as spasticity and brisk reflexes that accrue to considerable limb and bulbar disability over time, and eventually respiratory failure (1). The identification of TAR DNA-binding protein 43 (TDP-43) positive ubiquitinated cytoplasmic inclusions in almost all patients with ALS and more than half of patients with frontotemporal dementia (FTD) has placed ALS on the so-called “ALS-FTD continuum,” highlighting the considerable clinical, pathophysiological, and neuroimaging overlap between the two neurodegenerative conditions (2).
Although mentioned in early descriptions of ALS (3, 4), cognitive and behavioral deficits and frank dementia were previously considered atypical of ALS. It is not until the end of the twentieth century that clinical and research interest shifted to the extra-motor features of ALS and it has been gradually recognized as a genuine multisystem disease (5–8).
Neuropsychological deficits in ALS range from mild impairment to full-blown FTD. Up to 65% of ALS patients exhibit some cognitive or behavioral impairment (9–12) and 6–15% of sporadic ALS patients meet diagnostic criteria for FTD (10–13). While hexanucleotide repeat expansions in C9ORF72 are often associated with ALS-FTD (14), extra-motor symptoms are not unique to this mutation and extra-motor neuroimaging findings can also be readily identified in a significant proportion of C9 negative patients (15, 16). The early recognition of extra-motor involvement in ALS is crucial due to its impact on functional decline (17), survival (18), compliance with assistive devices (19), decision-making, and engagement in end-of-life and legal decisions (20).
Cognitive Dysfunction
Much attention has been initially focused on executive dysfunction (21, 22) in ALS which has been gradually complemented by the characterization of language (23, 24), memory (25, 26), praxis (27), and theory of mind deficits (28) (Table 1). Population-based studies have confirmed distinct cognitive phenotypes without executive impairment (10, 11, 29).
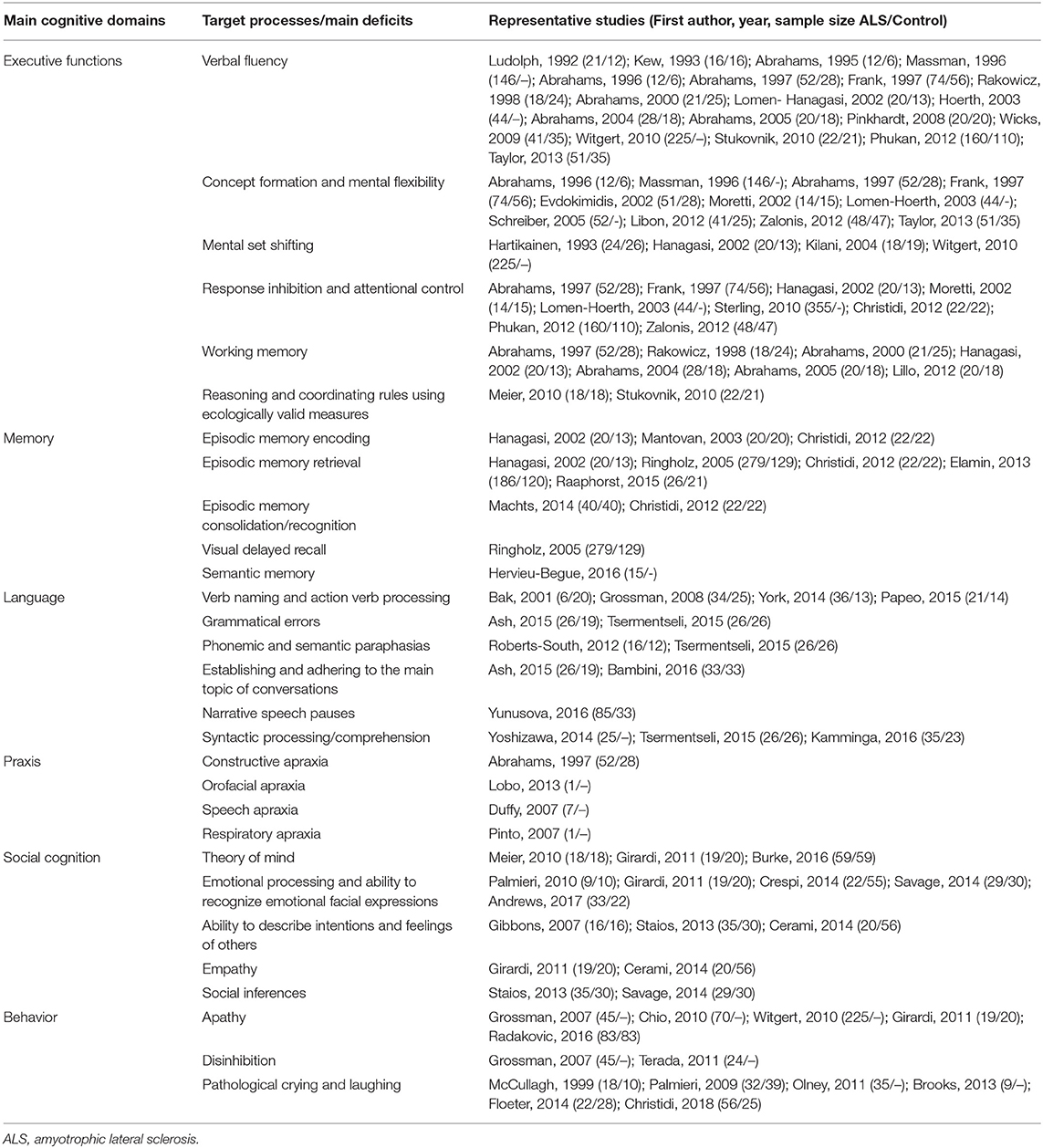
Table 1. Most characteristic neuropsychological deficits in ALS categorised per cognitive domain.
Executive Dysfunction
Executive dysfunction is the most commonly cited facet of cognitive impairment in ALS. Executive function however is an umbrella term encompassing several relatively distinct higher-order processes, such as planning, organization, goal-directed activity, working memory, initiation, behavioral regulation, and inhibitory control, as well as situation-appropriate decision-making on the basis of projected positive and negative outcomes in novel, complex or ambiguous situations (30). In addition, tests of verbal (i.e., phonemic and semantic/category) and figural/design fluency are also often conceptualized as proxies of executive performance (31).
Verbal fluency impairment has been consistently reported in ALS (11, 22, 24, 27, 32–46). Coexisting phonemic and semantic fluency dysfunction or phonemic fluency deficits alone are often linked to executive dysfunction, while isolated semantic fluency deficits are associated with impaired semantic memory processing. Semantic (24, 34, 40, 44, 46, 47) and figural (34, 46) fluency are not typically impaired in ALS. A verbal fluency index has been proposed and is now widely utilized to account for patients' motor disability (32, 48). Other executive processes are also affected in ALS, such as concept formation and mental flexibility (24, 27, 33, 35, 36, 41, 49–53) which is typically examined by the Wisconsin Card Sorting Test or the Dellis-Kaplan Executive Function System Card Sorting Test (31). However, not all neuropsychology studies corroborate these findings (34, 38, 39, 45, 46, 54–56). Several studies have specifically evaluated mental set shifting ability in ALS using the Trail Making Test; most of them identifying considerable dysfunction (37, 42, 47, 55, 57), while others have not captured such deficits (58, 59). Response inhibition and attentional control are typically examined by the Stroop test, and are often impaired in ALS (11, 27, 35–37, 51, 53, 57, 60), but unaffected cohorts have also been reported (39, 40, 44, 49). ALS patients also often exhibit difficulties in maintaining, manipulating and retrieving information relying on working memory (27, 32, 34, 37, 43, 46, 61), but preserved working memory has also been observed (39, 44, 51, 54, 55, 58, 62). Subtle deficits in reasoning and coordinating rules have been found using ecologically valid measures of executive functions (44, 63).
Memory Deficits
Following inconsistent initial reports, memory dysfunction in ALS has received increasing attention recently (7, 64). While autobiographic memory seems to be preserved in ALS (65), semantic memory is often affected (66). Episodic memory is the most commonly evaluated memory domain in ALS, typically tested by list-learning tests, associate-learning tests, prose memory, as well as visual memory tests (7). Several studies have reported mild to moderate episodic memory impairments which are often interpreted as the corollary of underlying executive deficits (27, 35, 37, 39, 41, 67–69). Memory impairment in ALS is rarely identified in isolation (11), but using data-driven taxonomy approaches a subgroup of patients may show non-executive memory dysfunction (29). Several studies have found impaired encoding (37, 60, 68), retrieval (12, 17, 37, 60, 70) consolidation and recognition (26, 60), although recognition deficits in ALS are not universally recognized (11, 37, 41). Visual memory dysfunction has also been noted in ALS (12), although visual recall is typically less affected than delayed verbal recall (7). Neuroimaging studies have contributed to the characterization of ALS-associated memory impairment highlighting mesial temporal lobe involvement irrespective of frontal lobe pathology (64).
Language Deficits
Language deficits in ALS have traditionally attracted less attention compared to other cognitive domains and have been mostly appraised in association with ALS-FTD (7, 23, 71, 72). However, language dysfunction is increasingly recognized as a core feature of ALS and has been consistently detected in patients without executive dysfunction (24, 29, 73). Patients with ALS show impaired syntactic processing (74), deficits in verb naming and action verb processing (75, 76). Selective impairment in action knowledge (77) has been directly associated with motor cortex degeneration (78) suggesting a link between action execution and action conceptualization (79). Grammatical errors such as incomplete utterances (73, 74) and omission of determiners (73) have been reported in ALS and seem to be dissociable from the patients' motor and executive deficits (73). Phonemic and semantic paraphasias have also been reported (74, 80). Patients with ALS may find narrative discourse particularly challenging due to difficulties to establish (81) and adhere to the main topic of conversation (73, 81). Frequent pauses are another key characteristic of narrative speech in ALS in both demented and non-demented ALS cohorts (82). Syntactic comprehension deficits have also been detected in up to 72% of patients with ALS (83, 84).
Visuo-Perceptive and Visuo-Constructive Deficits
Visuo-perceptive and visuo-constructive functions are seldom specifically examined in ALS. Existing studies tend to focus on visuospatial memory measures and often fail to reach definite conclusions (37, 41, 46, 47, 85). Based on large meta-analyses, these domains are not significantly affected in ALS (7). The relative absence of visuo-perceptual deficits is further supported by the lack of reports on Balint's syndrome in ALS and is consistent with limited occipital involvement on neuroimaging (86) and pathology (87). While praxis deficits are also rarely reported in ALS (27), orofacial (88), speech (89), and respiratory (90) apraxia have been sporadically reported.
Social Cognition Deficits
Social cognition refers to a diverse set of cognitive skills that allow humans to understand themselves, interact with and understand others and are crucial to adopt situation-appropriate, goal-directed behaviors in everyday social interactions (91). Despite considerable variations, deficits in theory of mind, empathy, social perception, social behavior are now recognized as key elements of the ALS-associated cognitive profile (7, 28, 92). It is however still unclear if these deficits are linked to executive dysfunction (29, 93–98) or may be related to non-executive domains, such as episodic memory function and visuospatial abilities (99). Patients with ALS may also exhibit impaired emotional processing and ability to interpret emotional facial expressions, especially with comorbid FTD (96, 100–102). Impairments in complex facial affect recognition, affective prosody recognition and cross-modal integration have also been found in non-demented ALS cohorts (103). Multiple subcomponents of theory of mind seem to be affected in ALS, including the ability to describe the intentions and feelings of others (95, 98, 104), to recognize and provide explanations for social “faux pas” (63) and evaluate object preferences based on the interpretation of eye gaze direction (96, 105). Loss of empathy (96), impaired emotional empathy attribution (95), and erroneous social inferences (98, 100) have also been reported in non-demented ALS cohorts.
Behavioral Deficits
The clinical link between ALS and FTD is exemplified by overlapping behavioral changes which are similar to those observed in behavioral variant of FTD (106). These deficits are typically identified through a structured clinical interview with the caregivers or through validated questionnaires. Perseveration, apathy and disinhibition are the most commonly reported behavioral alterations, followed by loss of disease insight, indifference, loss of interest, aggression, irritability, and lability (107).
Apathy is the most commonly reported behavioral symptom in non-demented ALS (42, 45, 96, 108, 109), which used to be assessed by generic behavioral instruments, such as the Frontal Systems Behavior Scale (110) and the Frontal Behavioral Inventory (111), until the development of ALS-specific scales, such as the Dimensional Apathy Scale (112) which appraises initiation, executive and emotional apathy. Initiation apathy is thought to be particularly prevalent in ALS (113). ALS patients with apathy may require prompts to initiate or follow through with a task, including self-care, feeding, and taking medications. They may appear poorly motivated, aloof or uninterested. Apathy may impact of rehabilitation, hamper gait initiation, and curb communication efforts especially in the presence of bulbar impairment. It can be mistaken for low mood, depression and withdrawal by inexperienced observers. Disinhibition is more readily identified and reported by caregivers, and can precede (108) or follow (114) motor disability. Disinhibited behavior can manifest in rude, offensive, flirtatious comments, puns, “Witzelsucht” often violating social norms, personal space and may result in careless or impulsive decisions. Purchasing expensive items on a whim, hoarding, compulsive behavior, overeating, and developing a preference for sweets have also been reported (115).
Hallucinations have been reported by several groups (116–119) and are sometimes associated with the C9orf72 genotype. Symptomatic treatment includes the judicious use of small dose atypical antipsychotics, if necessary.
Patients with pseudobulbar affect or pathological crying and laughing exhibit sudden situation-inappropriate emotional responses (120–122) which may have a negative impact on their quality of life (123) and lead to social isolation or social stigma. It is most commonly associated with UMN-type bulbar dysfunction (124), but frontal abnormalities, executive dysfunction, basal ganglia pathology and impaired cerebellar gating mechanisms have also been linked this symptom (27, 122, 125–128).
Insights From Neuroimaging
Neuroimaging techniques provide optimal non-invasive tools to characterize extra-motor pathology in ALS underpinning cognitive and behavioral deficits and also permit exploratory correlations with clinical measures (129, 130).
Structural Imaging
Voxel based morphometry (VBM) and surface-based morphometry (SBM) are reproducible, validated and widely-used pipelines that use high resolution 3D T1-weighted MR images to identify focal GM alterations. Beyond the consensus on motor cortex atrophy (131), many studies also detect multifocal frontotemporal and parietal GM changes (132). GM abnormalities have also been identified in subcortical structures (133), such as the hippocampus (134–136), amygdala (137, 138), thalamus (134, 135, 139, 140), and insula (141, 142). Reduced GM density in occipital (139, 143–145) and cerebellar (139, 146) regions is less commonly reported. GM alterations in extra-motor areas have been linked to structure-specific cognitive and behavioral deficits in ALS (147, 148). Recent studies have highlighted extra-motor cortical changes in ALS patients without overt cognitive impairment (134, 135, 146, 149, 150). The anatomical patterns of extra-motor gray matter involvement in ALS further support the notion of the ALS-FTD continuum (72).
White matter integrity in ALS is most commonly evaluated by diffusion tensor imaging (DTI). Reduced fractional anisotropy and increased axial and radial diffusivity in the corticospinal tracts and corpus callosum are hallmark features of ALS (151, 152). Extra-motor white matter pathology has been consistently detected in frontal (139, 153–160), temporal (53, 154, 161), cingular (162), parahippocampal (25, 157, 160), insular (160), thalamic (141, 159, 163), and cerebellar regions (86, 146, 164). Similarly to gray matter analyses, extra-motor white matter involvement has also been identified in ALS patients without overt cognitive impairment (146).
Metabolic Imaging
MR spectroscopy in ALS has consistently revealed decreased N-acetyl aspartate (NAA)/choline and NAA/creatine ratios in motor regions (165–167), but whole brain spectroscopy also detected extra-motor NAA reductions in frontal, parietal, thalamic and occipital areas (168, 169).
Most positron emission tomography (PET) studies in ALS use 18F-FDG PET, but TSPO, GABAA (11C-flumazenil) and 5-HT1A receptor (11C-WAY100635) radioligands have also been utilized (170). Hypometabolism in motor regions is a characteristic FDG-PET finding in ALS (171–174), but extra-motor changes in dorsolateral prefrontal, orbitofrontal, anterior frontal, anterior temporal, fusiform, and occipital regions have also been reported (171–174). Frontotemporal hypometabolism has been linked to cognitive performance (22, 39, 172), is thought to precede atrophy (175) and has been linked to shorter survival (176). There is also evidence of hypermetabolism in the hippocampus, amygdala midbrain, pons and cerebellum (173, 174, 177). PET imaging has identified microglial activation in frontotemporal, thalamic, midbrain, and pontine regions suggestive of extra-motor inflammation (178–181). Widespread reduction of 11C-Flumazenil binding to GABAA in sporadic ALS has been interpreted as inhibitory dysfunction (182) and is regarded as a one of cornerstones of ALS pathogenesis (183). Reduced serotonin receptor binding has also been reported in ALS using the 11C-WAY100635 radio-ligand (184).
Functional Imaging
Resting state fMRI enables the assessment of functional connectivity between different brain regions by evaluating synchronized neuronal activity at rest. Reduced (185–189) and increased (183, 190) functional connectivity have both been reported in sensorimotor networks of ALS patients which may be explained by the different sub-regions evaluated (191–193) and also by the inclusion of patients in different disease-stages. Similarly, both reduced and increased functional connectivity alterations have been reported in extra-motor areas which mediate cognitive and behavioral functions (187, 188, 193, 194). The functional connectivity of the default mode network (DMN) has been reported to be both decreased (187, 189, 193) and increased (193, 195). Increased functional connectivity has been detected in the DMN using graph theory-based analyses (196). Increased (193) and decreased (186, 189, 193) fronto-parietal network integrity has been both reported. Reduced “executive control network” (middle frontal cortex) and “salience network” (medial prefrontal cortex, insula) connectivity has been described in ALS cohorts without dementia (189). Increased connectivity in ALS has either been interpreted as evidence of attempted compensation for structural degeneration (197, 198) or proof of inhibitory dysfunction (183, 190, 199).
Task-based fMRI studies in ALS have consistently revealed the recruitment of pre- and supplementary motor regions when executing motor tasks. Additional activation has also been observed in areas associated with motor learning areas, such as the basal ganglia and cerebellum (200, 201). Despite difference in study protocols, an activation shift to premotor (202, 203), temporal and parietal regions (203–205) has been often noted. Cognitive paradigms have been particularly helpful in capturing frontotemporal network alterations. Impaired verbal fluency was linked to reduced frontotemporal, parietal, and cingulate activation in non-demented ALS patients (46). Impaired frontal inhibitory control was confirmed by a number of fMRI paradigms, such as Stroop, negative priming, antisaccade tasks, go/no-go tasks etc. Increased activation during the Stroop paradigm and decreased activation in negative priming conditions has been reported mostly in left hemispheric regions (206). Increased activation in supplementary and frontal eye fields and reduced activation in dorsolateral prefrontal cortex have been noted in antisaccade tasks (207). Furthermore, in go/no-go paradigms, ALS patients show increased inhibition-related activation in frontal and basal ganglia regions and increased execution-related activity in contralateral sensorimotor regions (208). Few studies have specifically examined the functional correlates of social cognition to date. Patients with ALS tend to show increased activation compared to healthy participants in the right supramarginal, anterior cingulate and bilateral dorsolateral prefrontal cortex in response to socio-emotional stimuli (56, 209). The combined use of motor and memory tasks on fMRI enables the longitudinal characterization of divergent motor and extra-motor functional changes. Increased motor activation was found in ALS compared to controls at baseline, which has decreased on the follow-up assessment, suggestive of failing compensation. Contrary to the functional motor changes, hippocampal activation increased on follow-up when novel stimuli was presented (210).
Relevance to Clinical Care
The detection (48), expert evaluation (11), categorization (211), and follow-up (17) of extra-motor deficits in ALS is crucially important for individualized patient care. While screening tests (Table 2) are useful for the detection of gross deficits, expert review by neuropsychologists is indicated for accurate patient classification. Adherence to treatment, compliance with assistive devices, participation in clinical trials, making informed financial and end-of-life decisions, choices in participating in non-licensed treatments are just some of the aspects of a patient journey which may be significantly affected by cognitive or behavioral deficits (19, 212). Cognitive impairment in ALS is widely regarded as a negative prognostic indicator and linked to reduced survival (17, 18, 213). Neuropsychological deficits in ALS are thought to be associated with increased caregiver burden (214, 215) and reduced quality of life (216). The recognition of the far-reaching effects of neuropsychological deficits on nearly all aspects of ALS care, caregiver support, resource allocation, and prognosis, led to the inclusion of specialist neuropsychologists as core members of ALS multidisciplinary teams worldwide (217, 218). The careful evaluation of motor deficits which are not directly linked to the corticospinal axis and are not reflected in the ALSFRS-R score, such as extra-pyramidal deficits are also crucial (219). Extra-pyramidal deficits may contribute to falls and gait impairment and are increasingly investigated in neuroimaging studies (220, 221). These symptoms may present early in the course of the disease, and contribute the clinical heterogeneity of the condition (220, 222). Postural instability and rigidity may be associated with other extra-motor deficits, and potentially linked to poor survival (205, 223). There is some controversy about the chronology of motor and extra-motor involvement in ALS. Extra-motor manifestations, such as dementia (224, 225), psychiatric features (226), and extra-pyramidal symptoms (227) have been reported to precede motor symptoms in some cases, and there is also compelling evidence of early extra-motor pathology in cognitively normal ALS patients (134, 135, 146).
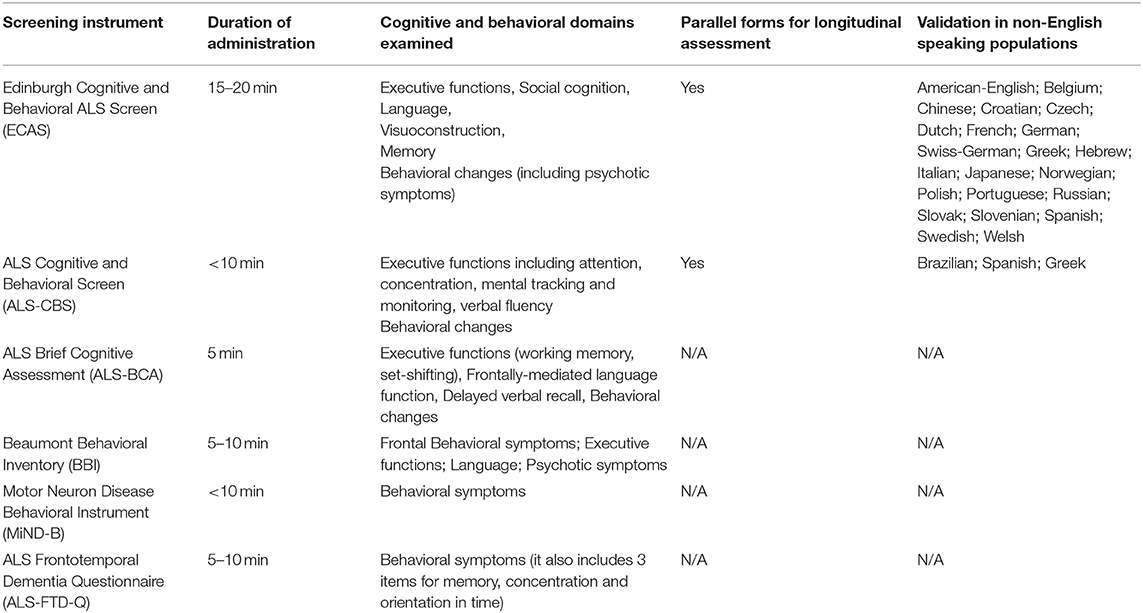
Table 2. ALS-specific instruments to screen for cognitive and behavioral changes at baseline and during the course of the disease.
Research Opportunities and Future Directions
Even though the high incidence of cognitive impairment and its impact on individualized patient care are now universally recognized, the neuropsychological aspects of ALS are seldom considered for patient stratification in clinical trials (228). Several ALS-specific cognitive screening tests have now been validated, but generic tests, such as MOCA and MMSE are still in use in some clinics. While neuropsychological scores are often adjusted for motor-disability and depression, medication-effects, fatigue, and hypoxia are seldom considered when interpreting cognitive performance on various instruments. Despite sporadic reports, the full spectrum of psychiatric manifestations and the precise incidence of psychosis remain to be established in ALS (119, 229, 230). Certain cognitive domains, such as memory and praxis have not been exhaustively characterized in ALS to date. Relatively little is known of the neuropsychological profile of ALS-causing mutation carriers before they develop motor symptoms (231–233). The gaps in our current understanding of extra-motor pathology in ALS shape future study designs. Novel technologies such as online assessments, internet-based data collection, mobile phone apps, and wearable devices are emerging research resources. Irrespective of specific neuropsychological instruments, the early detection, and careful of monitoring of cognitive deficits in ALS is pivotal for optimized patient and caregiver support and tailoring precision management strategies to individual patient needs.
Author Contributions
The paper was drafted by FC, EK, and PB and has been reviewed for intellectual content by MR, NK, and IE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
FC is supported by the State Scholarships Foundation (I.K.Y.; Postdoctoral Support; EP ANADEDBM/ESPA 2014–2020). Peter Bede's group is supported by the Health Research Board (HRB–Ireland; HRB EIA-2017-019), the Andrew Lydon scholarship, the Irish Institute of Clinical Neuroscience IICN–Novartis Ireland Research Grant, the Iris O'Brien Foundation, the Perrigo Clinician-Scientist Research Fellowship, and the Research Motor Neuron (RMN–Ireland) Foundation.
References
1. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet (2011) 377:942–55. doi: 10.1016/S0140-6736(10)61156-7
2. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP−43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (2006) 314:130–3. doi: 10.1126/science.1134108
3. Gallassi R, Montagna P, Ciardulli C, Lorusso S, Mussuto V, Stracciari A. Cognitive impairment in motor neuron disease. Acta Neurol Scand. (1985) 71:480–4. doi: 10.1111/j.1600-0404.1985.tb03231.x
4. David AS, Gillham RA. Neuropsychological study of motor neuron disease. Psychosomatics (1986) 27:441–5. doi: 10.1016/S0033-3182(86)72673-X
5. Silani V, Ludolph A, Fornai F. The emerging picture of ALS: a multisystem, not only a "motor neuron disease. Arch Ital Biol. (2017) 155:99–109.
6. Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain (1981) 104:217–47. doi: 10.1093/brain/104.2.217
7. Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ. The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry (2016) 87:611–9. doi: 10.1136/jnnp-2015-310734
8. Beeldman E, Raaphorst J, Klein Twennaar M, Govaarts R, Pijnenburg YAL, de Haan RJ, et al. The cognitive profile of behavioural variant FTD and its similarities with ALS: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry (2018) 89:995–1002. doi: 10.1136/jnnp-2017-317459
9. Goldstein LH, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol. (2013) 12:368–80. doi: 10.1016/S1474-4422(13)70026-7
10. Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, et al. Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry (2015) 86:168–73. doi: 10.1136/jnnp-2013-307223
11. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry (2012) 83:102–8. doi: 10.1136/jnnp-2011-300188
12. Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology (2005) 65:586–90. doi: 10.1212/01.wnl.0000172911.39167.b6
13. Trojsi F, Siciliano M, Femiano C, Santangelo G, Lunetta C, Calvo A, et al. Comorbidity of dementia with amyotrophic lateral sclerosis (ALS): insights from a large multicenter Italian cohort. J Neurol. (2017) 264:2224–31. doi: 10.1007/s00415-017-8619-4
14. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
15. Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology (2018) 91:e1370–80. doi: 10.1212/WNL.0000000000006317
16. Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. (2012) 11:232–40. doi: 10.1016/S1474-4422(12)70014-5
17. Elamin M, Bede P, Byrne S, Jordan N, Gallagher L, Wynne B, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology (2013) 80:1590–7. doi: 10.1212/WNL.0b013e31828f18ac
18. Elamin M, Phukan J, Bede P, Jordan N, Byrne S, Pender N, et al. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology (2011) 76:1263–9. doi: 10.1212/WNL.0b013e318214359f
19. Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology (2005) 65:1774–7. doi: 10.1212/01.wnl.0000188759.87240.8b
20. Merrilees J, Klapper J, Murphy J, Lomen-Hoerth C, Miller BL. Cognitive and behavioral challenges in caring for patients with frontotemporal dementia and amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2010) 11:298–302. doi: 10.3109/17482961003605788
21. Abrahams S, Goldstein LH, Lloyd CM, Brooks DJ, Leigh PN. Cognitive deficits in non-demented amyotrophic lateral sclerosis patients: a neuropsychological investigation. J Neurol Sci. (1995) 129(Suppl.):54–5. doi: 10.1016/0022-510X(95)00063-8
22. Abrahams S, Leigh PN, Kew JJ, Goldstein LH, Lloyd CM, Brooks DJ. A positron emission tomography study of frontal lobe function (verbal fluency) in amyotrophic lateral sclerosis. J Neurol Sci. (1995) 129(Suppl.):44–6. doi: 10.1016/0022-510X(95)00060-F
23. Pinto-Grau M, Hardiman O, Pender N. The study of language in the amyotrophic lateral sclerosis - frontotemporal spectrum disorder: a systematic review of findings and new perspectives. Neuropsychol Rev. (2018) 28:251–68. doi: 10.1007/s11065-018-9375-7
24. Taylor LJ, Brown RG, Tsermentseli S, Al-Chalabi A, Shaw CE, Ellis CM, et al. Is language impairment more common than executive dysfunction in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry (2013) 84:494–8. doi: 10.1136/jnnp-2012-303526
25. Christidi F, Karavasilis E, Zalonis I, Ferentinos P, Giavri Z, Wilde EA, et al. Memory-related white matter tract integrity in amyotrophic lateral sclerosis: an advanced neuroimaging and neuropsychological study. Neurobiol Aging (2017) 49:69–78. doi: 10.1016/j.neurobiolaging.2016.09.014
26. Machts J, Bittner V, Kasper E, Schuster C, Prudlo J, Abdulla S, et al. Memory deficits in amyotrophic lateral sclerosis are not exclusively caused by executive dysfunction: a comparative neuropsychological study of amnestic mild cognitive impairment. BMC Neurosci. (2014) 15:83. doi: 10.1186/1471-2202-15-83
27. Abrahams S, Goldstein LH, Al-Chalabi A, Pickering A, Morris RG, Passingham RE, et al. Relation between cognitive dysfunction and pseudobulbar palsy in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (1997) 62:464–72. doi: 10.1136/jnnp.62.5.464
28. Bora E. Meta-analysis of social cognition in amyotrophic lateral sclerosis. Cortex (2017) 88:1–7. doi: 10.1016/j.cortex.2016.11.012
29. Consonni M, Catricala E, Dalla Bella E, Gessa VC, Lauria G, Cappa SF. Beyond the consensus criteria: multiple cognitive profiles in amyotrophic lateral sclerosis? Cortex (2016) 81:162–7. doi: 10.1016/j.cortex.2016.04.014
31. Lezak MD. Neuropsychological Assessment. 5th ed. ed Oxford New York, NY: Oxford University Press (2012) p. 1161.
32. Abrahams S, Leigh PN, Harvey A, Vythelingum GN, Grise D, Goldstein LH. Verbal fluency and executive dysfunction in amyotrophic lateral sclerosis (ALS). Neuropsychologia (2000) 38:734–47. doi: 10.1016/S0028-3932(99)00146-3
33. Abrahams S, Goldstein LH, Kew JJ, Brooks DJ, Lloyd CM, Frith CD, et al. Frontal lobe dysfunction in amyotrophic lateral sclerosis. A PET study. Brain (1996) 119( Pt. 6):2105–20.
34. Abrahams S, Leigh PN, Goldstein LH. Cognitive change in ALS: a prospective study. Neurology (2005) 64:1222–6. doi: 10.1212/01.WNL.0000156519.41681.27
35. Frank B, Haas J, Heinze HJ, Stark E, Munte TF. Relation of neuropsychological and magnetic resonance findings in amyotrophic lateral sclerosis: evidence for subgroups. Clin Neurol Neurosurg. (1997) 99:79–86. doi: 10.1016/S0303-8467(97)80001-7
36. Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology (2003) 60:1094–7. doi: 10.1212/01.WNL.0000055861.95202.8D
37. Hanagasi HA, Gurvit IH, Ermutlu N, Kaptanoglu G, Karamursel S, Idrisoglu HA, et al. Cognitive impairment in amyotrophic lateral sclerosis: evidence from neuropsychological investigation and event-related potentials. Brain Res Cogn Brain Res. (2002) 14:234–44. doi: 10.1016/S0926-6410(02)00110-6
38. Kew JJ, Goldstein LH, Leigh PN, Abrahams S, Cosgrave N, Passingham RE, et al. The relationship between abnormalities of cognitive function and cerebral activation in amyotrophic lateral sclerosis. A neuropsychological and positron emission tomography study. Brain (1993) 116(Pt 6):1399–423.
39. Ludolph AC, Langen KJ, Regard M, Herzog H, Kemper B, Kuwert T, et al. Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. (1992) 85:81–9. doi: 10.1111/j.1600-0404.1992.tb04003.x
40. Pinkhardt EH, Jurgens R, Becker W, Molle M, Born J, Ludolph AC, et al. Signs of impaired selective attention in patients with amyotrophic lateral sclerosis. J Neurol. (2008) 255:532–8. doi: 10.1007/s00415-008-0734-9
41. Massman PJ, Sims J, Cooke N, Haverkamp LJ, Appel V, Appel SH. Prevalence and correlates of neuropsychological deficits in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (1996) 61:450–5. doi: 10.1136/jnnp.61.5.450
42. Witgert M, Salamone AR, Strutt AM, Jawaid A, Massman PJ, Bradshaw M, et al. Frontal-lobe mediated behavioral dysfunction in amyotrophic lateral sclerosis. Eur J Neurol. (2010) 17:103–10. doi: 10.1111/j.1468-1331.2009.02801.x
43. Rakowicz WP, Hodges JR. Dementia and aphasia in motor neuron disease: an underrecognised association? J Neurol Neurosurg Psychiatry (1998) 65:881–9. doi: 10.1136/jnnp.65.6.881
44. Stukovnik V, Zidar J, Podnar S, Repovs G. Amyotrophic lateral sclerosis patients show executive impairments on standard neuropsychological measures and an ecologically valid motor-free test of executive functions. J Clin Exp Neuropsychol. (2010) 32:1095–109. doi: 10.1080/13803391003749236
45. Wicks P, Abrahams S, Papps B, Al-Chalabi A, Shaw CE, Leigh PN, et al. SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis. J Neurol. (2009) 256:234–41. doi: 10.1007/s00415-009-0078-0
46. Abrahams S, Goldstein LH, Simmons A, Brammer M, Williams SC, Giampietro V, et al. Word retrieval in amyotrophic lateral sclerosis: a functional magnetic resonance imaging study. Brain (2004) 127(Pt 7):1507–17. doi: 10.1093/brain/awh170
47. Hartikainen P, Helkala EL, Soininen H, Riekkinen P Sr. Cognitive and memory deficits in untreated Parkinson's disease and amyotrophic lateral sclerosis patients: a comparative study. J Neural Transm Park Dis Dement Sect. (1993) 6:127–37. doi: 10.1007/BF02261006
48. Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:9–14. doi: 10.3109/21678421.2013.805784
49. Evdokimidis I, Constantinidis TS, Gourtzelidis P, Smyrnis N, Zalonis I, Zis PV, et al. Frontal lobe dysfunction in amyotrophic lateral sclerosis. J Neurol Sci. (2002) 195:25–33. doi: 10.1016/S0022-510X(01)00683-9
50. Libon DJ, McMillan C, Avants B, Boller A, Morgan B, Burkholder L, et al. Deficits in concept formation in amyotrophic lateral sclerosis. Neuropsychology (2012) 26:422–9. doi: 10.1037/a0028668
51. Moretti R, Torre P, Antonello RM, Carraro N, Cazzato G, Bava A. Complex cognitive disruption in motor neuron disease. Dement Geriatr Cogn Disord. (2002) 14:141–50. doi: 10.1159/000063600
52. Schreiber H, Gaigalat T, Wiedemuth-Catrinescu U, Graf M, Uttner I, Muche R, et al. Cognitive function in bulbar- and spinal-onset amyotrophic lateral sclerosis. A longitudinal study in 52 patients. J Neurol. (2005) 252:772–81. doi: 10.1007/s00415-005-0739-6
53. Zalonis I, Christidi F, Paraskevas G, Zabelis T, Evdokimidis I, Kararizou E. Can executive cognitive measures differentiate between patients with spinal- and bulbar-onset amyotrophic lateral sclerosis? Arch Clin Neuropsychol. (2012) 27:348–54. doi: 10.1093/arclin/acs031
54. Hammer A, Vielhaber S, Rodriguez-Fornells A, Mohammadi B, Munte TF. A neurophysiological analysis of working memory in amyotrophic lateral sclerosis. Brain Res. (2011) 1421:90–9. doi: 10.1016/j.brainres.2011.09.010
55. Kilani M, Micallef J, Soubrouillard C, Rey-Lardiller D, Demattei C, Dib M, et al. A longitudinal study of the evolution of cognitive function and affective state in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2004) 5:46–54. doi: 10.1080/14660820310017560
56. Lule D, Diekmann V, Anders S, Kassubek J, Kubler A, Ludolph AC, et al. Brain responses to emotional stimuli in patients with amyotrophic lateral sclerosis (ALS). J Neurol. (2007) 254:519–27. doi: 10.1007/s00415-006-0409-3
57. Sterling LE, Jawaid A, Salamone AR, Murthy SB, Mosnik DM, McDowell E, et al. Association between dysarthria and cognitive impairment in ALS: a prospective study. Amyotroph Lateral Scler. (2010) 11:46–51. doi: 10.3109/17482960903207997
58. Jelsone-Swain L, Persad C, Votruba KL, Weisenbach SL, Johnson T, Gruis KL, et al. The relationship between depressive symptoms, disease state, and cognition in amyotrophic lateral sclerosis. Front Psychol. (2012) 3:542. doi: 10.3389/fpsyg.2012.00542
59. Mioshi E, Lillo P, Yew B, Hsieh S, Savage S, Hodges JR, et al. Cortical atrophy in ALS is critically associated with neuropsychiatric and cognitive changes. Neurology (2013) 80:1117–23. doi: 10.1212/WNL.0b013e31828869da
60. Christidi F, Zalonis I, Smyrnis N, Evdokimidis I. Selective attention and the three-process memory model for the interpretation of verbal free recall in amyotrophic lateral sclerosis. J Int Neuropsychol Soc. (2012) 18:809–18. doi: 10.1017/S1355617712000562
61. Lillo P, Mioshi E, Burrell JR, Kiernan MC, Hodges JR, Hornberger M. Grey and white matter changes across the amyotrophic lateral sclerosis-frontotemporal dementia continuum. PLoS ONE (2012) 7:e43993. doi: 10.1371/journal.pone.0043993
62. Newsom-Davis IC, Lyall RA, Leigh PN, Moxham J, Goldstein LH. The effect of non-invasive positive pressure ventilation (NIPPV) on cognitive function in amyotrophic lateral sclerosis (ALS): a prospective study. J Neurol Neurosurg Psychiatry (2001) 71:482–7. doi: 10.1136/jnnp.71.4.482
63. Meier SL, Charleston AJ, Tippett LJ. Cognitive and behavioural deficits associated with the orbitomedial prefrontal cortex in amyotrophic lateral sclerosis. Brain (2010) 133:3444–57. doi: 10.1093/brain/awq254
64. Christidi F, Karavasilis E, Velonakis G, Ferentinos P, Rentzos M, Kelekis N, et al. The clinical and radiological spectrum of hippocampal pathology in amyotrophic lateral sclerosis. Front Neurol. (2018) 9:523. doi: 10.3389/fneur.2018.00523
65. Hsieh S, Irish M, Foxe D, Caga J, Devenney E, Ahmed R, et al. My memories are important to me: changes in autobiographical memory in amyotrophic lateral sclerosis. Neuropsychology (2016) 30:920–30. doi: 10.1037/neu0000291
66. Hervieu-Begue M, Rouaud O, Graule Petot A, Catteau A, Giroud M. Semantic memory assessment in 15 patients with amyotrophic lateral sclerosis. Revue Neurol. (2016) 172:307–12. doi: 10.1016/j.neurol.2015.10.009
67. Iwasaki Y, Kinoshita M, Ikeda K, Takamiya K, Shiojima T. Cognitive impairment in amyotrophic lateral sclerosis and its relation to motor disabilities. Acta Neurol Scand. (1990) 81:141–3. doi: 10.1111/j.1600-0404.1990.tb00950.x
68. Mantovan MC, Baggio L, Dalla Barba G, Smith P, Pegoraro E, Soraru G, et al. Memory deficits and retrieval processes in ALS. Eur J Neurol. (2003) 10:221–7. doi: 10.1046/j.1468-1331.2003.00607.x
69. Abe K, Fujimura H, Toyooka K, Sakoda S, Yorifuji S, Yanagihara T. Cognitive function in amyotrophic lateral sclerosis. J Neurol Sci. (1997) 148:95–100. doi: 10.1016/S0022-510X(96)05338-5
70. Raaphorst J, van Tol MJ, de Visser M, van der Kooi AJ, Majoie CB, van den Berg LH, et al. Prose memory impairment in amyotrophic lateral sclerosis patients is related to hippocampus volume. Eur J Neurol. (2015) 22:547–54. doi: 10.1111/ene.12615
71. Bede P, Omer T, Finegan E, Chipika RH, Iyer PM, Doherty MA, et al. Connectivity-based characterisation of subcortical grey matter pathology in frontotemporal dementia and ALS: a multimodal neuroimaging study. Brain Imag Behav. (2018). doi: 10.1007/s11682-018-9837-9. [Epub ahead of print].
72. Omer T, Finegan E, Hutchinson S, Doherty M, Vajda A, McLaughlin RL, et al. Neuroimaging patterns along the ALS-FTD spectrum: a multiparametric imaging study. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:611–23. doi: 10.1080/21678421.2017.1332077
73. Ash S, Olm C, McMillan CT, Boller A, Irwin DJ, McCluskey L, et al. Deficits in sentence expression in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:31–9. doi: 10.3109/21678421.2014.974617
74. Tsermentseli S, Leigh PN, Taylor LJ, Radunovic A, Catani M, Goldstein LH. Syntactic processing as a marker for cognitive impairment in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 17:69–76. doi: 10.3109/21678421.2015.1071397
75. Bak TH, O'Donovan DG, Xuereb JH, Boniface S, Hodges JR. Selective impairment of verb processing associated with pathological changes in Brodmann areas 44 and 45 in the motor neurone disease-dementia-aphasia syndrome. Brain (2001) 124(Pt 1):103–20. doi: 10.1093/brain/124.1.103
76. Papeo L, Cecchetto C, Mazzon G, Granello G, Cattaruzza T, Verriello L, et al. The processing of actions and action-words in amyotrophic lateral sclerosis patients. Cortex (2015) 64:136–47. doi: 10.1016/j.cortex.2014.10.007
77. Grossman M, Anderson C, Khan A, Avants B, Elman L, McCluskey L. Impaired action knowledge in amyotrophic lateral sclerosis. Neurology (2008) 71:1396–401. doi: 10.1212/01.wnl.0000319701.50168.8c
78. York C, Olm C, Boller A, McCluskey L, Elman L, Haley J, et al. Action verb comprehension in amyotrophic lateral sclerosis and Parkinson's disease. J Neurol. (2014) 261:1073–9. doi: 10.1007/s00415-014-7314-y
79. Bak TH, Chandran S. What wires together dies together: verbs, actions and neurodegeneration in motor neuron disease. Cortex (2012) 48:936–44. doi: 10.1016/j.cortex.2011.07.008
80. Roberts-South A, Findlater K, Strong MJ, Orange JB. Longitudinal changes in discourse production in amyotrophic lateral sclerosis. Semin Speech Language (2012) 33:79–94. doi: 10.1055/s-0031-1301165
81. Bambini V, Arcara G, Martinelli I, Bernini S, Alvisi E, Moro A, et al. Communication and pragmatic breakdowns in amyotrophic lateral sclerosis patients. Brain Language (2016) 153–154:1–12. doi: 10.1016/j.bandl.2015.12.002
82. Yunusova Y, Graham NL, Shellikeri S, Phuong K, Kulkarni M, Rochon E, et al. Profiling speech and pausing in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). PLoS ONE (2016) 11:e0147573. doi: 10.1371/journal.pone.0147573
83. Kamminga J, Leslie FVC, Hsieh S, Caga J, Mioshi E, Hornberger M, et al. Syntactic comprehension deficits across the FTD-ALS continuum. Neurobiol Aging (2016) 41:11–8. doi: 10.1016/j.neurobiolaging.2016.02.002
84. Yoshizawa K, Yasuda N, Fukuda M, Yukimoto Y, Ogino M, Hata W, et al. Syntactic comprehension in patients with amyotrophic lateral sclerosis. Behav Neurol. (2014) 2014:230578. doi: 10.1155/2014/230578
85. Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology (1999) 53:1665–70. doi: 10.1212/WNL.53.8.1665
86. Bede P, Iyer PM, Schuster C, Elamin M, McLaughlin RL, Kenna K, et al. The selective anatomical vulnerability of ALS: 'disease-defining' and 'disease-defying' brain regions. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:561–70. doi: 10.3109/21678421.2016.1173702
87. Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L, et al. Evidence of multisystem disorder in whole-brain map of pathological TDP−43 in amyotrophic lateral sclerosis. Arch Neurol. (2008) 65:636–41. doi: 10.1001/archneur.65.5.636
88. Lobo PP, Pinto S, Rocha L, Reimao S, de Carvalho M. Orofacial apraxia in motor neuron disease. Case Rep Neurol. (2013) 5:47–51. doi: 10.1159/000349895
89. Duffy JR, Peach RK, Strand EA. Progressive apraxia of speech as a sign of motor neuron disease. Am J Speech Language Pathol. (2007) 16:198–208. doi: 10.1044/1058-0360(2007/025)
90. Pinto S, Pinto A, Atalaia A, Peralta R, de Carvalho M. Respiratory apraxia in amyotrophic lateral sclerosis. Amyotr Lateral Scler. (2007) 8:180–4. doi: 10.1080/17482960701249340
91. Adolphs R. The social brain: neural basis of social knowledge. Ann Rev Psychol. (2009) 60:693–716. doi: 10.1146/annurev.psych.60.110707.163514
92. Christidi F, Migliaccio R, Santamaria-Garcia H, Santangelo G, Trojsi F. Social cognition dysfunctions in neurodegenerative diseases: neuroanatomical correlates and clinical implications. Behav Neurol. (2018) 2018:1849794. doi: 10.1155/2018/1849794
93. Carluer L, Mondou A, Buhour MS, Laisney M, Pelerin A, Eustache F, et al. Neural substrate of cognitive theory of mind impairment in amyotrophic lateral sclerosis. Cortex (2015) 65:19–30. doi: 10.1016/j.cortex.2014.12.010
94. Cavallo M, Adenzato M, Macpherson SE, Karwig G, Enrici I, Abrahams S. Evidence of social understanding impairment in patients with amyotrophic lateral sclerosis. PLoS ONE (2011) 6:e25948. doi: 10.1371/journal.pone.0025948
95. Cerami C, Dodich A, Canessa N, Crespi C, Iannaccone S, Corbo M, et al. Emotional empathy in amyotrophic lateral sclerosis: a behavioural and voxel-based morphometry study. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:21–9. doi: 10.3109/21678421.2013.785568
96. Girardi A, MacPherson SE, Abrahams S. Deficits in emotional and social cognition in amyotrophic lateral sclerosis. Neuropsychology (2011) 25:53–65. doi: 10.1037/a0020357
97. Watermeyer TJ, Brown RG, Sidle KC, Oliver DJ, Allen C, Karlsson J, et al. Executive dysfunction predicts social cognition impairment in amyotrophic lateral sclerosis. J Neurol. (2015) 262:1681–90. doi: 10.1007/s00415-015-7761-0
98. Staios M, Fisher F, Lindell AK, Ong B, Howe J, Reardon K. Exploring sarcasm detection in amyotrophic lateral sclerosis using ecologically valid measures. Front Hum Neurosci. (2013) 7:178. doi: 10.3389/fnhum.2013.00178
99. Trojsi F, Siciliano M, Russo A, Passaniti C, Femiano C, Ferrantino T, et al. Theory of mind and its neuropsychological and quality of life correlates in the early stages of amyotrophic lateral sclerosis. Front Psychol. (2016) 7:1934. doi: 10.3389/fpsyg.2016.01934
100. Savage SA, Lillo P, Kumfor F, Kiernan MC, Piguet O, Hodges JR. Emotion processing deficits distinguish pure amyotrophic lateral sclerosis from frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:39–46. doi: 10.3109/21678421.2013.809763
101. Palmieri A, Naccarato M, Abrahams S, Bonato M, D'Ascenzo C, Balestreri S, et al. Right hemisphere dysfunction and emotional processing in ALS: an fMRI study. J Neurol. (2010) 257:1970–8. doi: 10.1007/s00415-010-5640-2
102. Crespi C, Cerami C, Dodich A, Canessa N, Arpone M, Iannaccone S, et al. Microstructural white matter correlates of emotion recognition impairment in Amyotrophic Lateral Sclerosis. Cortex (2014) 53:1–8. doi: 10.1016/j.cortex.2014.01.002
103. Andrews SC, Staios M, Howe J, Reardon K, Fisher F. Multimodal emotion processing deficits are present in amyotrophic lateral sclerosis. Neuropsychology (2017) 31:304–10. doi: 10.1037/neu0000323
104. Gibbons ZC, Snowden JS, Thompson JC, Happe F, Richardson A, Neary D. Inferring thought and action in motor neurone disease. Neuropsychologia (2007) 45:1196–207. doi: 10.1016/j.neuropsychologia.2006.10.008
105. Burke T, Elamin M, Bede P, Pinto-Grau M, Lonergan K, Hardiman O, et al. Discordant performance on the 'Reading the Mind in the Eyes' Test, based on disease onset in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:467–72. doi: 10.1080/21678421.2016.1177088
106. Elamin M, Pinto-Grau M, Burke T, Bede P, Rooney J, O'Sullivan M, et al. Identifying behavioural changes in ALS: validation of the Beaumont Behavioural Inventory (BBI). Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:68–73. doi: 10.1080/21678421.2016.1248976.
107. Raaphorst J, Beeldman E, De Visser M, De Haan RJ, Schmand B. A systematic review of behavioural changes in motor neuron disease. Amyotroph Lateral Scler. (2012) 13:493–501. doi: 10.3109/17482968.2012.656652
108. Grossman AB, Woolley-Levine S, Bradley WG, Miller RG. Detecting neurobehavioral changes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2007) 8:56–61. doi: 10.1080/17482960601044106
109. Chio A, Vignola A, Mastro E, Giudici AD, Iazzolino B, Calvo A, et al. Neurobehavioral symptoms in ALS are negatively related to caregivers' burden and quality of life. Eur J Neurol. (2010) 17:1298–303. doi: 10.1111/j.1468-1331.2010.03016.x
110. Grace J, Mallow P.F. Frontal Systems Behavior Scale. Lutz, FL: Psychological Assessment Resources (2001).
111. Kertesz A, Davidson W, Fox H. Frontal behavioral inventory: diagnostic criteria for frontal lobe dementia. Can J Neurol Sci. (1997) 24:29–36.
112. Radakovic R, Abrahams S. Developing a new apathy measurement scale: dimensional apathy scale. Psychiatry Res. (2014) 219:658–63. doi: 10.1016/j.psychres.2014.06.010
113. Radakovic R, Stephenson L, Colville S, Swingler R, Chandran S, Abrahams S. Multidimensional apathy in ALS: validation of the Dimensional Apathy Scale. J Neurol Neurosurg Psychiatry (2016) 87:663–9. doi: 10.1136/jnnp-2015-310772
114. Terada T, Obi T, Yoshizumi M, Murai T, Miyajima H, Mizoguchi K. Frontal lobe-mediated behavioral changes in amyotrophic lateral sclerosis: are they independent of physical disabilities? J Neurol Sci. (2011) 309:136–40. doi: 10.1016/j.jns.2011.06.049
115. Turner MR, Talbot K. Sweet food preference in amyotrophic lateral sclerosis. Pract Neurol. (2017) 17:128–9. doi: 10.1136/practneurol-2016-001554
116. Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain (2012) 135(Pt 3):693–708. doi: 10.1093/brain/awr355
117. Kertesz A, Ang LC, Jesso S, MacKinley J, Baker M, Brown P, et al. Psychosis and hallucinations in frontotemporal dementia with the C9ORF72 mutation: a detailed clinical cohort. Cogn Behav Neurol. (2013) 26:146–54. doi: 10.1097/WNN.0000000000000008
118. Nitrini R, Rosemberg S. Psychotic symptoms in dementia associated with motor neuron disease: a pathophysiological hypothesis. J Neuropsychiatry Clin Neurosci. (1998) 10:456–8. doi: 10.1176/jnp.10.4.456
119. Lillo P, Garcin B, Hornberger M, Bak TH, Hodges JR. Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol. (2010) 67:826–30. doi: 10.1001/archneurol.2010.146
120. Noh SM, Chung SJ, Kim KK, Kang DW, Lim YM, Kwon SU, et al. Emotional disturbance in CADASIL: its impact on quality of life and caregiver burden. Cerebrovasc Dis. (2014) 37:188–94. doi: 10.1159/000357798
121. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. (2013) 37:1893–916. doi: 10.1016/j.neubiorev.2013.03.002
122. Bede P, Finegan E. Revisiting the pathoanatomy of pseudobulbar affect: mechanisms beyond corticobulbar dysfunction. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:4–6. doi: 10.1080/21678421.2017.1392578
123. Brooks BR, Crumpacker D, Fellus J, Kantor D, Kaye RE. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS ONE (2013) 8:e72232. doi: 10.1371/journal.pone.0072232
124. Palmieri A, Abrahams S, Soraru G, Mattiuzzi L, D'Ascenzo C, Pegoraro E, et al. Emotional Lability in MND: Relationship to cognition and psychopathology and impact on caregivers. J Neurol Sci. (2009) 278:16–20. doi: 10.1016/j.jns.2008.10.025
125. McCullagh S, Moore M, Gawel M, Feinstein A. Pathological laughing and crying in amyotrophic lateral sclerosis: an association with prefrontal cognitive dysfunction. J Neurol Sci. (1999) 169:43–8. doi: 10.1016/S0022-510X(99)00214-2
126. Olney NT, Goodkind MS, Lomen-Hoerth C, Whalen PK, Williamson CA, Holley DE, et al. Behaviour, physiology and experience of pathological laughing and crying in amyotrophic lateral sclerosis. Brain (2011) 134(Pt 12):3458–69. doi: 10.1093/brain/awr297
127. Floeter MK, Katipally R, Kim MP, Schanz O, Stephen M, Danielian L, et al. Impaired corticopontocerebellar tracts underlie pseudobulbar affect in motor neuron disorders. Neurology (2014) 83:620–7. doi: 10.1212/WNL.0000000000000693
128. Christidi F, Karavasilis E, Ferentinos P, Xirou S, Velonakis G, Rentzos M, et al. Investigating the neuroanatomical substrate of pathological laughing and crying in amyotrophic lateral sclerosis with multimodal neuroimaging techniques. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19(1–2):12–20. doi: 10.1080/21678421.2017.1386689
129. Verstraete E, Turner MR, Grosskreutz J, Filippi M, Benatar M. Mind the gap: The mismatch between clinical and imaging metrics in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:524–9. doi: 10.3109/21678421.2015.1051989
130. Bede P, Querin G, Pradat PF. The changing landscape of motor neuron disease imaging: the transition from descriptive studies to precision clinical tools. Curr Opin Neurol. (2018) 31:431–8. doi: 10.1097/WCO.0000000000000569
131. Shen D, Cui L, Fang J, Cui B, Li D, Tai H. Voxel-wise meta-analysis of gray matter changes in amyotrophic lateral sclerosis. Front Aging Neurosci. (2016) 8:64. doi: 10.3389/fnagi.2016.00064
132. Bede P, Hardiman O. Lessons of ALS imaging: Pitfalls and future directions - A critical review. NeuroImage Clin. (2014) 4:436–43. doi: 10.1016/j.nicl.2014.02.011
133. Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, et al. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology (2013) 81:2107–15. doi: 10.1212/01.wnl.0000437313.80913.2c
134. Bede P, Bokde A, Elamin M, Byrne S, McLaughlin RL, Jordan N, et al. Grey matter correlates of clinical variables in amyotrophic lateral sclerosis (ALS): a neuroimaging study of ALS motor phenotype heterogeneity and cortical focality. J Neurol Neurosurg Psychiatry (2013) 84:766–73. doi: 10.1136/jnnp-2012-302674
135. Bede P, Bokde AL, Byrne S, Elamin M, McLaughlin RL, Kenna K, et al. Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology (2013) 81:361–9. doi: 10.1212/WNL.0b013e31829c5eee
136. Westeneng HJ, Verstraete E, Walhout R, Schmidt R, Hendrikse J, Veldink JH, et al. Subcortical structures in amyotrophic lateral sclerosis. Neurobiol Aging (2015) 36:1075–82. doi: 10.1016/j.neurobiolaging.2014.09.002
137. Pinkhardt EH, van Elst LT, Ludolph AC, Kassubek J. Amygdala size in amyotrophic lateral sclerosis without dementia: an in vivo study using MRI volumetry. BMC Neurol. (2006) 6:48. doi: 10.1186/1471-2377-6-48
138. Machts J, Loewe K, Kaufmann J, Jakubiczka S, Abdulla S, Petri S, et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology (2015) 85:1301–9. doi: 10.1212/WNL.0000000000002017
139. Thivard L, Pradat PF, Lehericy S, Lacomblez L, Dormont D, Chiras J, et al. Diffusion tensor imaging and voxel based morphometry study in amyotrophic lateral sclerosis: relationships with motor disability. J Neurol Neurosurg Psychiatry (2007) 78:889–92. doi: 10.1136/jnnp.2006.101758
140. Chang JL, Lomen-Hoerth C, Murphy J, Henry RG, Kramer JH, Miller BL, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology (2005) 65:75–80. doi: 10.1212/01.wnl.0000167602.38643.29
141. Schuster C, Kasper E, Machts J, Bittner D, Kaufmann J, Benecke R, et al. Focal thinning of the motor cortex mirrors clinical features of amyotrophic lateral sclerosis and their phenotypes: a neuroimaging study. J Neurol. (2013) 260:2856–64. doi: 10.1007/s00415-013-7083-z
142. Bede P, Hardiman O. Longitudinal structural changes in ALS: a three time-point imaging study of white and gray matter degeneration. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:232–41. doi: 10.1080/21678421.2017.1407795
143. Agosta F, Valsasina P, Riva N, Copetti M, Messina MJ, Prelle A, et al. The cortical signature of amyotrophic lateral sclerosis. PLoS ONE (2012) 7:e42816. doi: 10.1371/journal.pone.0042816
144. Mezzapesa DM, Ceccarelli A, Dicuonzo F, Carella A, De Caro MF, Lopez M, et al. Whole-brain and regional brain atrophy in amyotrophic lateral sclerosis. AJNR Am J Neuroradiol. (2007) 28:255–9.
145. Mezzapesa DM, D'Errico E, Tortelli R, Distaso E, Cortese R, Tursi M, et al. Cortical thinning and clinical heterogeneity in amyotrophic lateral sclerosis. PLoS ONE (2013) 8:e80748. doi: 10.1371/journal.pone.0080748
146. Christidi F, Karavasilis E, Riederer F, Zalonis I, Ferentinos P, Velonakis G, et al. Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: a combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imaging Behav. (2018) 12:547–63. doi: 10.1007/s11682-017-9722-y
147. Alruwaili AR, Pannek K, Coulthard A, Henderson R, Kurniawan ND, McCombe P. A combined tract-based spatial statistics and voxel-based morphometry study of the first MRI scan after diagnosis of amyotrophic lateral sclerosis with subgroup analysis. J Neuroradiol. (2018) 45:41–8. doi: 10.1016/j.neurad.2017.03.007
148. Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. (2007) 64:530–4. doi: 10.1001/archneur.64.4.530
149. Agosta F, Ferraro PM, Riva N, Spinelli EG, Chio A, Canu E, et al. Structural brain correlates of cognitive and behavioral impairment in MND. Hum Brain Map. (2016) 37:1614–26. doi: 10.1002/hbm.23124
150. Masuda M, Senda J, Watanabe H, Epifanio B, Tanaka Y, Imai K, et al. Involvement of the caudate nucleus head and its networks in sporadic amyotrophic lateral sclerosis-frontotemporal dementia continuum. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:571–9. doi: 10.1080/21678421.2016.1211151
151. Zhang F, Chen G, He M, Dai J, Shang H, Gong Q, et al. Altered white matter microarchitecture in amyotrophic lateral sclerosis: A voxel-based meta-analysis of diffusion tensor imaging. NeuroImage Clin. (2018) 19:122–9. doi: 10.1016/j.nicl.2018.04.005
152. Foerster BR, Dwamena BA, Petrou M, Carlos RC, Callaghan BC, Churchill CL, et al. Diagnostic accuracy of diffusion tensor imaging in amyotrophic lateral sclerosis: a systematic review and individual patient data meta-analysis. Acad Radiol. (2013) 20:1099–106. doi: 10.1016/j.acra.2013.03.017
153. Abe O, Yamada H, Masutani Y, Aoki S, Kunimatsu A, Yamasue H, et al. Amyotrophic lateral sclerosis: diffusion tensor tractography and voxel-based analysis. NMR Biomed. (2004) 17:411–6. doi: 10.1002/nbm.907
154. Agosta F, Pagani E, Rocca MA, Caputo D, Perini M, Salvi F, et al. Voxel-based morphometry study of brain volumetry and diffusivity in amyotrophic lateral sclerosis patients with mild disability. Hum Brain Map. (2007) 28:1430–8. doi: 10.1002/hbm.20364
155. Ciccarelli O, Behrens TE, Johansen-Berg H, Talbot K, Orrell RW, Howard RS, et al. Investigation of white matter pathology in ALS and PLS using tract-based spatial statistics. Hum Brain Map. (2009) 30:615–24. doi: 10.1002/hbm.20527
156. Filippini N, Douaud G, Mackay CE, Knight S, Talbot K, Turner MR. Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis. Neurology (2010) 75:1645–52. doi: 10.1212/WNL.0b013e3181fb84d1
157. Keil C, Prell T, Peschel T, Hartung V, Dengler R, Grosskreutz J. Longitudinal diffusion tensor imaging in amyotrophic lateral sclerosis. BMC Neurosci. (2012) 13:141. doi: 10.1186/1471-2202-13-141
158. Rosskopf J, Muller HP, Dreyhaupt J, Gorges M, Ludolph AC, Kassubek J. Ex post facto assessment of diffusion tensor imaging metrics from different MRI protocols: preparing for multicentre studies in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:92–101. doi: 10.3109/21678421.2014.977297
159. Sach M, Winkler G, Glauche V, Liepert J, Heimbach B, Koch MA, et al. Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain (2004) 127(Pt 2):340–50. doi: 10.1093/brain/awh041
160. Sage CA, Van Hecke W, Peeters R, Sijbers J, Robberecht W, Parizel P, et al. Quantitative diffusion tensor imaging in amyotrophic lateral sclerosis: revisited. Hum Brain Map. (2009) 30:3657–75. doi: 10.1002/hbm.20794
161. Sato K, Aoki S, Iwata NK, Masutani Y, Watadani T, Nakata Y, et al. Diffusion tensor tract-specific analysis of the uncinate fasciculus in patients with amyotrophic lateral sclerosis. Neuroradiology (2010) 52:729–33. doi: 10.1007/s00234-010-0653-1
162. Prell T, Peschel T, Hartung V, Kaufmann J, Klauschies R, Bodammer N, et al. Diffusion tensor imaging patterns differ in bulbar and limb onset amyotrophic lateral sclerosis. Clin Neurol Neurosurg. (2013) 115:1281–7. doi: 10.1016/j.clineuro.2012.11.031
163. Sarica A, Cerasa A, Vasta R, Perrotta P, Valentino P, Mangone G, et al. Tractography in amyotrophic lateral sclerosis using a novel probabilistic tool: a study with tract-based reconstruction compared to voxel-based approach. J Neurosci Methods (2014) 224:79–87. doi: 10.1016/j.jneumeth.2013.12.014
164. Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, et al. Patterns of cerebral and cerebellar white matter degeneration in ALS. J Neurol Neurosur Psychiatry (2015) 86:468–70. doi: 10.1136/jnnp-2014-308172
165. Foerster BR, Carlos RC, Dwamena BA, Callaghan BC, Petrou M, Edden RA, et al. Multimodal MRI as a diagnostic biomarker for amyotrophic lateral sclerosis. Ann Clin Transl Neurol. (2014) 1:107–14. doi: 10.1002/acn3.30
166. Sivak S, Bittsansky M, Kurca E, Turcanova-Koprusakova M, Grofik M, Nosal V, et al. Proton magnetic resonance spectroscopy in patients with early stages of amyotrophic lateral sclerosis. Neuroradiology (2010) 52:1079–85. doi: 10.1007/s00234-010-0685-6
167. Pyra T, Hui B, Hanstock C, Concha L, Wong JC, Beaulieu C, et al. Combined structural and neurochemical evaluation of the corticospinal tract in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2010) 11:157–65. doi: 10.3109/17482960902756473
168. Verma G, Woo JH, Chawla S, Wang S, Sheriff S, Elman LB, et al. Whole-brain analysis of amyotrophic lateral sclerosis by using echo-planar spectroscopic imaging. Radiology (2013) 267:851–7. doi: 10.1148/radiol.13121148
169. Usman U, Choi C, Camicioli R, Seres P, Lynch M, Sekhon R, et al. Mesial prefrontal cortex degeneration in amyotrophic lateral sclerosis: a high-field proton MR spectroscopy study. AJNR Am J Neuroradiol. (2011) 32:1677–80. doi: 10.3174/ajnr.A2590
170. Cistaro A, Cuccurullo V, Quartuccio N, Pagani M, Valentini MC, Mansi L. Role of PET and SPECT in the study of amyotrophic lateral sclerosis. BioMed Res Int. (2014) 2014:237437. doi: 10.1155/2014/237437
171. Pagani M, Chio A, Valentini MC, Oberg J, Nobili F, Calvo A, et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology (2014) 83:1067–74. doi: 10.1212/WNL.0000000000000792
172. Renard D, Collombier L, Castelnovo G, Fourcade G, Kotzki PO, LaBauge P. Brain FDG-PET changes in ALS and ALS-FTD. Acta Neurologica Belgica. (2011) 111:306–9.
173. Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, et al. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. (2014) 71:553–61. doi: 10.1001/jamaneurol.2014.62
174. Cistaro A, Valentini MC, Chio A, Nobili F, Calvo A, Moglia C, et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nuclear Med Mol Imag. (2012) 39:251–9. doi: 10.1007/s00259-011-1979-6
175. Rajagopalan V, Pioro EP. Comparing brain structural MRI and metabolic FDG-PET changes in patients with ALS-FTD: 'the chicken or the egg?' question. J Neurol Neurosurg Psychiatry (2015) 86:952–8. doi: 10.1136/jnnp-2014-308239
176. Van Weehaeghe D, Ceccarini J, Delva A, Robberecht W, Van Damme P, Van Laere K. Prospective validation of 18F-FDG brain PET discriminant analysis methods in the diagnosis of amyotrophic lateral sclerosis. J Nuclear Med. (2016) 57:1238–43. doi: 10.2967/jnumed.115.166272
177. Cistaro A, Pagani M, Montuschi A, Calvo A, Moglia C, Canosa A, et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nuclear Med Mol Imag. (2014) 41:844–52. doi: 10.1007/s00259-013-2667-5
178. Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. (2004) 15:601–9. doi: 10.1016/j.nbd.2003.12.012
179. Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE (2012) 7:e52941. doi: 10.1371/journal.pone.0052941
180. Zurcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with [(11)C]-PBR28. NeuroImage Clin. (2015) 7:409–14. doi: 10.1016/j.nicl.2015.01.009
181. Johansson A, Engler H, Blomquist G, Scott B, Wall A, Aquilonius SM, et al. Evidence for astrocytosis in ALS demonstrated by [11C](L)-deprenyl-D2 PET. J Neurol Sci. (2007) 255:17–22. doi: 10.1016/j.jns.2007.01.057
182. Lloyd CM, Richardson MP, Brooks DJ, Al-Chalabi A, Leigh PN. Extramotor involvement in ALS: PET studies with the GABA(A) ligand [(11)C]flumazenil. Brain (2000) 123( Pt 11):2289–96. doi: 10.1093/brain/123.11.2289
183. Douaud G, Filippini N, Knight S, Talbot K, Turner MR. Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain (2011) 134(Pt 12):3470–9. doi: 10.1093/brain/awr279
184. Turner MR, Rabiner EA, Hammers A, Al-Chalabi A, Grasby PM, Shaw CE, et al. [11C]-WAY100635 PET demonstrates marked 5-HT1A receptor changes in sporadic ALS. Brain (2005) 128(Pt 4):896–905. doi: 10.1093/brain/awh428
185. Jelsone-Swain LM, Fling BW, Seidler RD, Hovatter R, Gruis K, Welsh RC. Reduced interhemispheric functional connectivity in the motor cortex during rest in limb-onset amyotrophic lateral sclerosis. Front Syst Neurosci. (2010) 4:158. doi: 10.3389/fnsys.2010.00158
186. Tedeschi G, Trojsi F, Tessitore A, Corbo D, Sagnelli A, Paccone A, et al. Interaction between aging and neurodegeneration in amyotrophic lateral sclerosis. Neurobiol Aging (2012) 33:886–98. doi: 10.1016/j.neurobiolaging.2010.07.011
187. Mohammadi B, Kollewe K, Samii A, Krampfl K, Dengler R, Munte TF. Changes of resting state brain networks in amyotrophic lateral sclerosis. Exp Neurol. (2009) 217:147–53. doi: 10.1016/j.expneurol.2009.01.025
188. Fekete T, Zach N, Mujica-Parodi LR, Turner MR. Multiple kernel learning captures a systems-level functional connectivity biomarker signature in amyotrophic lateral sclerosis. PLoS ONE (2013) 8:e85190. doi: 10.1371/journal.pone.0085190
189. Trojsi F, Esposito F, de Stefano M, Buonanno D, Conforti FL, Corbo D, et al. Functional overlap and divergence between ALS and bvFTD. Neurobiol Aging (2015) 36:413–23. doi: 10.1016/j.neurobiolaging.2014.06.025
190. Agosta F, Canu E, Inuggi A, Chio A, Riva N, Silani V, et al. Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiol Aging (2014) 35:916–25. doi: 10.1016/j.neurobiolaging.2013.09.041
191. Agosta F, Valsasina P, Absinta M, Riva N, Sala S, Prelle A, et al. Sensorimotor functional connectivity changes in amyotrophic lateral sclerosis. Cerebr Cortex (2011) 21:2291–8. doi: 10.1093/cercor/bhr002
192. Zhou F, Xu R, Dowd E, Zang Y, Gong H, Wang Z. Alterations in regional functional coherence within the sensory-motor network in amyotrophic lateral sclerosis. Neurosci Lett. (2014) 558:192–6. doi: 10.1016/j.neulet.2013.11.022
193. Agosta F, Canu E, Valsasina P, Riva N, Prelle A, Comi G, et al. Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol Aging (2013) 34:419–27. doi: 10.1016/j.neurobiolaging.2012.04.015
194. Luo C, Chen Q, Huang R, Chen X, Chen K, Huang X, et al. Patterns of spontaneous brain activity in amyotrophic lateral sclerosis: a resting-state FMRI study. PLoS ONE (2012) 7:e45470. doi: 10.1371/journal.pone.0045470
195. Tietz F HV, Prell T, Penzlin S, Ilse B, Bokemeyer M, et al. The resting state default mode network (DMN) is pathologically hyperactive in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2012) 13:171. doi: 10.3389/fnins.2016.00204
196. Heimrath J, Gorges M, Kassubek J, Muller HP, Birbaumer N, Ludolph AC, et al. Additional resources and the default mode network: Evidence of increased connectivity and decreased white matter integrity in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:537–45. doi: 10.3109/21678421.2014.911914
197. Chiò A, Pagani M, Agosta F, Calvo A, Cistaro A, Filippi M. Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet Neurol. (2014) 13:1228–40. doi: 10.1016/S1474-4422(14)70167-X
198. Bede P. Deciphering neurodegeneration: A paradigm shift from focality to connectivity. Neurology (2017) 89:1758–9. doi: 10.1212/WNL.0000000000004582
199. Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. (2012) 13:245–50. doi: 10.3109/17482968.2011.636050
200. Schoenfeld MA, Tempelmann C, Gaul C, Kuhnel GR, Duzel E, Hopf JM, et al. Functional motor compensation in amyotrophic lateral sclerosis. J Neurol. (2005) 252:944–52. doi: 10.1007/s00415-005-0787-y
201. Konrad C, Jansen A, Henningsen H, Sommer J, Turski PA, Brooks BR, et al. Subcortical reorganization in amyotrophic lateral sclerosis. Exp Brain Res. (2006) 172:361–9. doi: 10.1007/s00221-006-0352-7
202. Konrad C, Henningsen H, Bremer J, Mock B, Deppe M, Buchinger C, et al. Pattern of cortical reorganization in amyotrophic lateral sclerosis: a functional magnetic resonance imaging study. Exp Brain Res. (2002) 143:51–6. doi: 10.1007/s00221-001-0981-9
203. Cosottini M, Pesaresi I, Piazza S, Diciotti S, Cecchi P, Fabbri S, et al. Structural and functional evaluation of cortical motor areas in Amyotrophic Lateral Sclerosis. Exp Neurol. (2012) 234:169–80. doi: 10.1016/j.expneurol.2011.12.024
204. Stanton BR, Williams VC, Leigh PN, Williams SC, Blain CR, Jarosz JM, et al. Altered cortical activation during a motor task in ALS. Evidence for involvement of central pathways. J Neurol. (2007) 254:1260–7. doi: 10.1007/s00415-006-0513-4
205. Poujois A, Schneider FC, Faillenot I, Camdessanche JP, Vandenberghe N, Thomas-Anterion C, et al. Brain plasticity in the motor network is correlated with disease progression in amyotrophic lateral sclerosis. Hum Brain Map. (2013) 34:2391–401. doi: 10.1002/hbm.22070
206. Goldstein LH, Newsom-Davis IC, Bryant V, Brammer M, Leigh PN, Simmons A. Altered patterns of cortical activation in ALS patients during attention and cognitive response inhibition tasks. J Neurol. (2011) 258:2186–98. doi: 10.1007/s00415-011-6088-8
207. Witiuk K, Fernandez-Ruiz J, McKee R, Alahyane N, Coe BC, Melanson M, et al. Cognitive deterioration and functional compensation in ALS measured with fMRI using an inhibitory task. J Neurosci. (2014) 34:14260–71. doi: 10.1523/JNEUROSCI.1111-14.2014
208. Mohammadi B, Kollewe K, Cole DM, Fellbrich A, Heldmann M, Samii A, et al. Amyotrophic lateral sclerosis affects cortical and subcortical activity underlying motor inhibition and action monitoring. Hum Brain Mapp. (2015) 36:2878–89. doi: 10.1002/hbm.22814
209. Passamonti L, Fera F, Tessitore A, Russo A, Cerasa A, Gioia CM, et al. Dysfunctions within limbic-motor networks in amyotrophic lateral sclerosis. Neurobiol Aging (2013) 34:2499–509. doi: 10.1016/j.neurobiolaging.2013.05.016
210. Stoppel CM, Vielhaber S, Eckart C, Machts J, Kaufmann J, Heinze HJ, et al. Structural and functional hallmarks of amyotrophic lateral sclerosis progression in motor- and memory-related brain regions. NeuroImage Clin. (2014) 5:277–90. doi: 10.1016/j.nicl.2014.07.007
211. Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2009) 10:131–46. doi: 10.1080/17482960802654364
212. Bede P, Oliver D, Stodart J, van den Berg L, Simmons Z, D OB, et al. Palliative care in amyotrophic lateral sclerosis: a review of current international guidelines and initiatives. J Neurol Neurosurg Psychiatry (2011) 82:413–8. doi: 10.1136/jnnp.2010.232637
213. Elamin M, Bede P, Montuschi A, Pender N, Chio A, Hardiman O. Predicting prognosis in amyotrophic lateral sclerosis: a simple algorithm. J Neurol. (2015) 262:1447–54. doi: 10.1007/s00415-015-7731-6
214. Burke T, Elamin M, Galvin M, Hardiman O, Pender N. Caregiver burden in amyotrophic lateral sclerosis: a cross-sectional investigation of predictors. J Neurol. (2015) 262:1526–32. doi: 10.1007/s00415-015-7746-z
215. Andrews SC, Pavlis A, Staios M, Fisher F. Which behaviours? Identifying the most common and burdensome behaviour changes in amyotrophic lateral sclerosis. Psychol Health Med. (2017) 22:483–92. doi: 10.1080/13548506.2016.1164871
216. Bock M, Duong YN, Kim A, Allen I, Murphy J, Lomen-Hoerth C. Progression and effect of cognitive-behavioral changes in patients with amyotrophic lateral sclerosis. Neurol Clin Pract. (2017) 7:488–98. doi: 10.1212/CPJ.0000000000000397
217. Van den Berg JP, Kalmijn S, Lindeman E, Veldink JH, de Visser M, Van der Graaff MM, et al. Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology (2005) 65:1264–7. doi: 10.1212/01.wnl.0000180717.29273.12
218. Hardiman O. Multidisciplinary care in ALS: expanding the team. Amyotroph Lateral Scler. (2012) 13:165. doi: 10.3109/17482968.2012.657006
219. Desai J, Swash M. Extrapyramidal involvement in amyotrophic lateral sclerosis: backward falls and retropulsion. J Neurol Neurosurg Psychiatry (1999) 67:214–6. doi: 10.1136/jnnp.67.2.214
220. Feron M, Couillandre A, Mseddi E, Termoz N, Abidi M, Bardinet E, et al. Extrapyramidal deficits in ALS: a combined biomechanical and neuroimaging study. J Neurol. (2018). doi: 10.1007/s00415-018-8964-y. [Epub ahead of print].
221. Chio A, Calvo A, Cammarosano S, Dematteis F, Artusi CA, Pagani M, et al. Extrapyramidal involvement in ALS: a prospective population-based study (P4.314). Neurology (2017) 79:1983–9. doi: 10.1212/WNL.0b013e3182735d36.
222. Pupillo E, Bianchi E, Messina P, Chiveri L, Lunetta C, Corbo M, et al. Extrapyramidal and cognitive signs in amyotrophic lateral sclerosis: a population based cross-sectional study. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:324–30. doi: 10.3109/21678421.2015.1040028
223. McCluskey L, Vandriel S, Elman L, Van Deerlin VM, Powers J, Boller A, et al. ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. Journal of the neurological Sciences (2014) 345:118–24. doi: 10.1016/j.jns.2014.07.022
224. Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A. Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurol Sci. (2011) 32:9–16. doi: 10.1007/s10072-010-0439-6
225. Khan AM. Rare presentation of amyotrophic lateral sclerosis as frontotemporal dementia: a case report. Open Access J Toxicol. (2017) 1:555566. doi: 10.19080/OAJT.2017.01.555566
226. Turner MR, Goldacre R, Talbot K, Goldacre MJ. Psychiatric disorders prior to amyotrophic lateral sclerosis. Ann Neurol. (2016) 80:935–8. doi: 10.1002/ana.24801
227. Qureshi AI, Wilmot G, Dihenia B, Schneider JA, Krendel DA. Motor neuron disease with parkinsonism. Arch Neurol. (1996) 53:987–91. doi: 10.1001/archneur.1996.00550100061015
228. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. (2014) 13:1127–38. doi: 10.1016/S1474-4422(14)70129-2
229. Cui B, Cui L, Liu M, Ma J, Fang J. Amyotrophic lateral sclerosis with frontotemporal dementia presented with prominent psychosis. Chin Med J. (2014) 127:3996–8.
230. Snowden JS, Adams J, Harris J, Thompson JC, Rollinson S, Richardson A, et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:497–505. doi: 10.3109/21678421.2015.1074700
231. Benatar M, Wuu J. Presymptomatic studies in ALS: rationale, challenges, and approach. Neurology (2012) 79:1732–9. doi: 10.1212/WNL.0b013e31826e9b1d
232. Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. (2018) 75:236–45. doi: 10.1001/jamaneurol.2017.4266
Keywords: amyotrophic lateral sclerosis, extra-motor involvement, cognition, behavior, neuropsychological deficits, neuroimaging
Citation: Christidi F, Karavasilis E, Rentzos M, Kelekis N, Evdokimidis I and Bede P (2018) Clinical and Radiological Markers of Extra-Motor Deficits in Amyotrophic Lateral Sclerosis. Front. Neurol. 9:1005. doi: 10.3389/fneur.2018.01005
Received: 03 October 2018; Accepted: 06 November 2018;
Published: 22 November 2018.
Edited by:
Pierre-francois Pradat, Hôpitaux Universitaires Pitié Salpêtrière, FranceReviewed by:
Giorgia Querin, INSERM U1146 Laboratoire d'Imagerie Biomédicale, FranceChristos Bakirtzis, Aristotle University of Thessaloniki, Greece
Copyright © 2018 Christidi, Karavasilis, Rentzos, Kelekis, Evdokimidis and Bede. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Foteini Christidi, Y2hyaXN0aWRpLmYuYUBnbWFpbC5jb20=