 Mary Kay Floeter
Mary Kay Floeter Tania F. Gendron
Tania F. Gendron- 1National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, United States
- 2Department of Neuroscience, Mayo Clinic, Jacksonville, FL, United States
Now that genetic testing can identify persons at risk for developing amyotrophic lateral sclerosis (ALS) many decades before symptoms begin, there is a critical need for biomarkers that signal the onset and progression of degeneration. The search for candidate disease biomarkers in patients with mutations in the gene C9orf72 has included imaging, physiology, and biofluid measurements. In cross-sectional imaging studies, C9+ ALS patients display diffuse reductions of gray and white matter integrity compared to ALS patients without mutations. This structural imaging signature overlaps with frontotemporal dementia (FTD), reflecting the frequent co-occurrence of cognitive impairment, even frank FTD, in C9+ ALS patients. Changes in functional connectivity occur as critical components of the networks associated with cognition and behavior degenerate. In presymptomatic C9+carriers, subtle differences in volumes of subcortical structures and functional connectivity can be detected, often decades before the typical family age of symptom onset. Dipeptide repeat proteins produced by the repeat expansion mutation are also measurable in the cerebrospinal fluid (CSF) of presymptomatic gene carriers, possibly throughout their lives. In contrast, a rise in the level of neurofilament proteins in the CSF appears to presage the onset of degeneration in presymptomatic carriers in one longitudinal study. Cross-sectional studies indicate that neurofilament protein levels may provide prognostic information for survival in C9+ ALS patients. Longitudinal studies will be needed to validate the candidate biomarkers discussed here. Understanding how these candidate biomarkers change over time is critical if they are to be used in future therapeutic decisions.
Introduction
A repeat expansion mutation in the C9orf72 gene is the most common cause of familial amyotrophic lateral sclerosis (ALS) in people of Northern European ancestry and accounts for 5-10% of sporadic ALS cases in Europe and the USA (1, 2). The C9orf72 mutation (C9+) is also a common cause of familial frontotemporal dementia (FTD) (3). The clinical phenotype is often mixed, and many C9+ ALS patients have some degree of cognitive impairment, ranging from mild executive dysfunction to frank FTD (4). Because mutation carriers can be identified by genetic testing many decades before symptoms begin, there is considerable interest in biomarkers to identify when degeneration begins and to monitor disease progression. Currently, development of such biomarkers is at the early stage of identifying measures that differ in group comparisons. This review will discuss the current status of studies of non-invasive biomarkers such as imaging and physiology, and minimally invasive biomarkers derived from biofluids.
Imaging Studies
There is particular interest in neuroimaging as a biomarker because it offers a way to visualize pathological changes in the brains of living patients. In autopsy studies, brains from C9+ patients exhibited the neuronal loss, gliosis, and TDP-43 inclusions characteristic of sporadic ALS and some FTD patients (5), as well as the nuclear RNA foci and cytoplasmic aggregates of dipeptide repeat (DPR) proteins specific to the C9orf72 mutation (5, 6). The distribution of these pathologic findings differs between C9+ ALS and C9+ FTD patient brains (7, 8). The story emerging from neuroimaging studies is that the diversity of clinical phenotypes reflects the extent to which the most affected brain regions contribute to networks that underlie cognitive, behavioral, motor, and language function (9, 10).
Structural MRI—Gray Matter Atrophy
In structural MRI scans, C9+ ALS patients displayed extensive, relatively symmetric volume loss and cortical thinning compared to similarly aged healthy subjects (1, 11–14). Compared to C9– ALS patients (i.e., without the C9orf72 mutation), C9+ ALS patients had greater atrophy of extra-motor cortical regions, particularly parieto-occipital cortical areas, including the cuneus and precuneus (11–13), and relatively less atrophy of the precentral motor cortex (13, 14). Correlations between volumetric changes and cognitive testing measures have led several investigators to conclude that the predominant gray matter imaging pattern in C9+ ALS patients is associated with cognitive changes (11–14). A similar pattern of diffuse, relatively symmetric cortical volume loss is found in C9+ FTD patients (15–19).
Several studies report more atrophy of subcortical structures in C9+ ALS than in C9– ALS patients. The topographic specificity of connections between these subcortical structures and specific cortical regions can lead to discrete functional deficits. Nearly all volumetric studies to date have reported thalamic atrophy in C9+ carriers. Thalamic atrophy has been reported in C9+ ALS patients (11–13), C9+ FTD patients (15, 16, 18–22), and presymptomatic C9+ carriers (23–26). Although C9+ ALS patients may have more thalamic atrophy compared to C9– ALS patients with a similar degree of cognitive impairment (11), the association between thalamic atrophy and cognitive impairment can be seen in FTD patients with other gene mutations (27) and C9– ALS patients with cognitive impairment (28). Because there is topographic specificity of corticothalamic circuits, degeneration of particular thalamic nuclei should produce different functional impairments. However, most MRI studies measured the hemi-thalamus in its entirety. Using a more refined segmentation scheme in a cohort of C9+ FTD patients, Lee and colleagues (20) found atrophy specifically in the medial pulvinar nucleus of the thalamus, a multisensory nucleus with connections to posterior parietal, prefrontal, and cingulate cortical areas (29). Schonecker and colleagues reported greater atrophy of motor sub-regions of the thalamus in symptomatic C9+ carriers (30).
Atrophy of other subcortical structures has also been reported. The cerebellum has been of particular interest because high levels of DPR proteins (8, 31, 32) and RNA foci were found in cerebellar Purkinje and granule cells in C9+ patients (33), and levels of cerebellar DPR proteins in C9+ ALS were correlated with cognitive impairment (31). While a pathological study reported no appreciable neuronal loss in the cerebellum (15), cerebellar atrophy has been reported in some, but not all, imaging studies. Detection differences largely reflect whether the whole cerebellum or focal cerebellar regions were measured. Changes in focal cerebellar regions, such as in lobule VIIa/crus I, were found in several studies of C9+ ALS and C9+ FTD patients (11, 17, 21, 27, 34). This region of the cerebellum has been mapped in functional MRI studies to cortical association networks, including the dorsolateral prefrontal cortex and parietal association areas that play a role in executive function (35). Volume loss has also been reported in various nuclei of the basal ganglia in C9+ ALS and C9+ FTD patients (20, 28, 36), a finding associated with cognitive and behavioral scores across the spectrum of ALS and FTD, and thought to result from disruption of corticostriatal circuits (37). Two studies also reported hippocampal atrophy in C9+ ALS (11, 38), a finding consistent with the occurrence of hippocampal sclerosis in some C9+ ALS-FTD brains (5) and memory deficits.
The diffuse nature of the brain atrophy, involving cortical and subcortical structures, has led to the suggestion that changes in ventricular volume be used to follow longitudinal disease progression in C9+ carriers (13, 17, 34) (Figure 1).
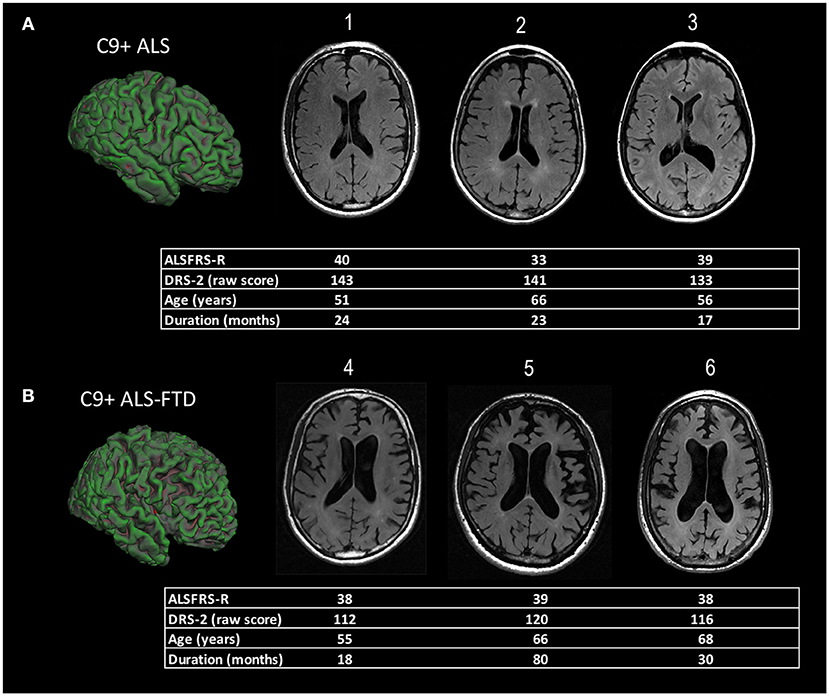
Figure 1. Representative examples of diffuse cortical atrophy in MRI scans of ALS patients with C9orf72 mutations. The demographic information and scores on motor and cognitive scales are listed below each patient's scan. (A) Compared to age-matched controls, mild ventricular enlargement was seen in C9+ patients 1, 2, and 3 who had ALS, but good cognitive function, as evidenced by their scores on the Mattis Dementia Rating Scale−2 (DRS-2). The surface rendering of one patient [left side of panel (A)] shows sulci in frontal lobe sulci are also mildly enlarged compared to the occipital lobe. (B) C9+ patients 4, 5, and 6 had ALS-FTD with a similar degree of motor dysfunction to those in panel (A), as measured by their ALS functional rating scale revised (ALSFRS-R) scores, but marked cognitive impairment with low DRS-2 scores. There is marked enlargement of ventricles evident in axial slices, as well as enlargement of frontal and temporal sulci in the surface rendering at left of panel (B).
Pathological Correlates
The distribution of atrophy in structural MRI scans of C9+ ALS and FTD patients mirrors the distribution of neuronal loss and TDP-43 pathology in brains of C9+ ALS-FTD patients (5) and sporadic ALS and FTD patients (39). However, the relationship between these hallmarks of degeneration–neuronal loss, gliosis, and TDP-43 inclusions—and the RNA foci and DPR protein aggregates specific for the C9+ genotype is still evolving. Unlike TDP-43 pathology, which closely parallels neurodegeneration, the distribution of RNA foci (33) and DPR protein pathology do not (6–8, 33, 40, 41), although reports on the latter have been somewhat conflicting. A moderate association between the amount of poly(GA) dystrophic neurites and degeneration in the frontal cortex was observed (40), and inclusions of poly(GR), which is especially toxic in in vitro models (42), correlated with TDP-43 pathology and neurodegeneration in C9+ FTD-ALS brains (7, 41). Nevertheless, the presence of DPR protein aggregates and RNA foci did not lead to TDP-43 accumulation in a neurologically healthy mosaic carrier (43), and DPR protein pathology with little, if any, TDP-43 pathology was observed in a c9FTD kindred with early intellectual disability (44) and three C9orf72 mutation carriers who developed relatively rapid cognitive decline but died prematurely due to unrelated illness (45).
Diffusion Tensor Imaging of White Matter Tracts
In diffusion tensor imaging (DTI) studies, C9+ ALS patients showed more widespread loss of white matter integrity compared to healthy controls and C9– ALS patients, most commonly in the frontal white matter, as measured by decreased fractional anisotropy, increased radial diffusivity, or increased mean diffusivity (11, 12, 14, 38, 46). Several white matter tracts affected in C9+ ALS are not typically affected in cognitively intact C9– ALS patients, including the genu of the corpus callosum, anterior limbs of the internal capsule, thalamic radiations, and long association tracts such as the uncinate fasciculus, superior longitudinal fasciculus, and inferior longitudinal fasciculus (11, 12, 14, 38, 46). These frontal and association tracts were also affected in diffusion studies of C9+ FTD patients (17, 20, 36), and presymptomatic C9+ carriers in some studies (47). Motor tracts, including the corticospinal tract and motor segment of the corpus callosum, were affected in C9+ ALS patients compared to healthy controls (11, 46), but exhibited less disruption than in C9– ALS patients (14). In a group of C9+ carriers with a mixture of phenotypes, changes in diffusion indices of specific tracts correlated with clinical measures: frontal white matter correlated with lexical fluency and behavioral scores, and changes in motor tracts correlated with the ALS functional rating scale (46).
Unresolved Questions About Structural Imaging as a Biomarker
Several questions arise from the findings in structural MRI scans. First, does a genotype-specific C9+ MRI signature exist? To address this question, Westeneng and colleagues (38) identified a candidate “genotype-specific MRI signature” in a model comparing 92 volumetric and DTI variables in scans of 28 C9+ to 28 C9– ALS patients. Although 11 imaging variables identified a C9+ specific signature in the training dataset, nearly 20% of C9– ALS patients in a large validation dataset were classified as having the C9+ MRI signature. Misclassified patients scored more poorly on a measure of executive function, thus underscoring the close association between neuroanatomical atrophy patterns and clinical phenotypes. A second question is whether the volumetric differences in adult C9+ carriers arise during development or are a consequence of degeneration. This question was addressed in imaging studies comparing relatively young presymptomatic C9+ carriers (< age 40) to non-carriers from the same families. Although older presymptomatic C9+ carriers had clear evidence of atrophy compared to similarly-aged C9– family members, so did younger C9+ presymptomatic carriers when compared to C9– family members of the same age (24–26, 47, 48). Cortical and subcortical structures were smaller, particularly the thalamus, in younger C9+ carriers. The common genetic background of family members with and without the C9orf72 mutation facilitated detection of small differences in these studies. Lee and colleagues found that smaller gray matter volumes occurred across a range of ages in presymptomatic C9+ carriers and had a similar age-related decline as in C9– controls, suggesting a developmental origin (47). Longitudinal studies in individual C9+ carriers before and after the onset of symptoms will be needed to truly determine whether congenitally small brain structures begin accelerated volume loss with the onset of degeneration in adulthood or whether the C9orf72 mutation leads to slow, lifelong accumulation of subclinical pathology. Lastly, because the distribution of atrophy mirrors the distribution of TDP-43 in pathological studies (5), longitudinal structural imaging, in combination with clinical phenotyping, can be used to test hypotheses that TDP-43 pathology spreads through axonal connections. Pathological studies in sporadic ALS have led to the proposal that TDP-43 pathology spreads through corticofugal projections (49). In contrast, in behavioral-variant FTD, TDP-43 pathology has been proposed to spread from orbitofrontal cortex to posterior regions through axonal tracts (50).
Functional Connectivity
Changes in functional connectivity using task-based or resting state fMRI have been reported prior to development of clinical symptoms in patients with GRN or MAPT mutations at risk for FTD (51). Three studies examined changes in functional connectivity in resting state networks in C9+ carriers. One study in symptomatic carriers found that C9+ and C9– behavioral variant FTD patients had disruption of salience network connectivity that was associated with neuropsychiatric severity, as well as disruption of sensorimotor connectivity (20). The disruption of the salience network occurred with atrophy of different nodes within the salience network in individual patients (20). Disruption of the salience network and a network generated from a medial pulvinar nucleus seed was also observed in young presymptomatic C9+ carriers (47). Another study reported increased connectivity in the visual network of C9+ carriers with a mixture of motor and cognitive phenotypes compared to sporadic cases with similar phenotypes (11).
Proton Emission Tomography
Hypometabolism in the frontal lobes in FDG-PET studies is considered supportive of a clinical diagnosis of FTD (52). The few reports of PET imaging in C9+ carriers had slightly different findings. In one study, C9+ ALS patients had more widespread hypometabolism occurring in the cingulate, insula, caudate, and thalamus, with clusters of hypermetabolism in occipital, left precentral, left postcentral, and superior temporal cortex when compared to C9– ALS patients with or without FTD (53). In contrast, the other study reported that C9+ ALS and C9– ALS patients exhibited hypometabolism in peri-rolandic cortex; several prefrontal regions had hypometabolism in both groups, but C9+ ALS patients alone had focal hypometabolism in the thalamus and posterior cingulate cortex (54). One case study also reported frontal and temporal hypometabolism in a C9+ ALS patient who subsequently developed FTD (55). Another reported that amyloid imaging, but not FDG-PET, distinguished FTD from Alzheimer disease in a C9+ carrier (56).
Physiology
Physiological methods have been used to assess cortical function in C9+ carriers. Transcranial magnetic stimulation (TMS) is a non-invasive technique for assessing cortical excitability. Numerous TMS studies in sporadic ALS patients have provided evidence for hyperexcitability of the motor cortex early in disease (57), with loss of excitability at late stages (58). C9+ ALS patients were similarly found to have increased cortical excitability according to several different TMS indices, but presymptomatic C9+ carriers did not (59–61). Evoked potential measures have been used to explore particular cognitive functions in C9+ patients (62), but have not been routinely used to identify disease onset or severity. Electroimpedance myography (63) and Motor Unit Number Index (MUNIX) (64) are non-invasive methods that have been used to follow lower motor neuron dysfunction in ALS patients in clinical trials but, to date, have not been reported in C9+ ALS patients.
Energy Metabolism
Patients with ALS develop defects in energy metabolism that include low body mass index (BMI), hypermetabolism, and hyperlipidemia (65, 66). While the contribution of dysregulated energy homeostasis to ALS pathogenesis remains to be resolved, such defects correlate with prognostic factors. For instance, weight loss and hypermetabolism are associated with faster disease progression and shorter survival in ALS (66–68). The cause of these metabolic changes is unknown, but may result from hypothalamic atrophy. Gorges et al. (69) have shown that the hypothalamus is atrophied in ALS patients and in presymptomatic ALS mutation carriers (the latter were comprised predominantly of C9+ individuals). Furthermore, they found a modest but significant correlation between hypothalamic volume and BMI, especially in patients with familial ALS, and observed that anterior hypothalamic volumes correlate with age of disease onset (69). While these findings are not specific to C9+ carriers, they do suggest that hypothalamic atrophy, BMI, and disturbances in energy homeostasis could provide prognostic insight.
CSF and Biofluid Studies
Fluid-based biomarker discovery efforts for ALS have most often been conducted using cerebrospinal fluid (CSF) due to its proximity to affected neuroanatomical regions. However, progress has been made using plasma and serum, and studies using urine and saliva are emerging (70). Among the more widely studied biomarker candidates are inflammatory mediators, metabolic markers and neurofilament proteins; the latter, however, have arguably garnered the most attention (70, 71). Neurofilament proteins, which include neurofilament heavy chain (NfH), neurofilament medium chain and neurofilament light chain (NfL), are abundantly and exclusively expressed in neurons where they form the neuronal cytoskeleton. Because neurofilament proteins are released from neurons upon axonal damage or degeneration, they are considered indicators of neuronal injury for various neurological disorders.
Neurofilament Proteins
In C9+ carriers, levels of CSF phosphorylated NfH (pNfH) were significantly higher in patients with ALS or FTD compared to asymptomatic individuals, and strongly associated with survival in patients with C9+ ALS (72). Notably, C9+ ALS patients had significantly higher pNFH levels than C9– ALS patients, which presumably reflected increased neurodegeneration, consistent with reports that patients with C9+ ALS develop greater brain atrophy, particularly in extra-motor regions, compared to C9– ALS patients (11–13). More diffuse degeneration may account for the shorter survival of C9+ ALS patients compared to C9– ALS patients (1, 72–75). Similar to pNfH, CSF NfL levels were elevated in symptomatic compared to presymptomatic C9+ carriers (76, 77), and higher NfL levels in symptomatic individuals correlated with greater disease severity and shorter survival (77). Furthermore, elevated CSF NfL in C9+ carriers was associated with lower gray matter volumes in the ventral and dorsomedial prefrontal cortex, ventral, and dorsal insula, anterior cingulate, caudate, medial thalamus, and other frontotemporoparietal regions (77).
These findings supporting CSF pNfH and NfL as prognostic markers for C9+ patients could significantly impact drug development. For instance, the heterogeneity of disease course in C9+ ALS could result in different proportions of fast and slow progressors in clinical treatment arms. Using pNfH and NfL levels as surrogates for progression rate could facilitate stratification of patients into balanced groups to reduce variability in treatment outcomes. Early evidence also suggests that NfL in CSF and serum can inform the potential phenoconversion of individuals from an asymptomatic to a symptomatic state (78). Through the study of individuals that carry a mutation in C9orf72 or other ALS-associated genes, Benatar and colleagues found that NfL in asymptomatic mutation carriers was elevated above the range seen in healthy individuals as early as 12 months prior to the earliest clinical symptoms (78). Should these findings be validated in additional cohorts, NfL could provide insight on when neurodegeneration begins. This would facilitate the timely diagnosis of C9+ ALS, and increase the likelihood of enrolling patients in clinical trials at an early stage of disease when they are most likely to benefit from therapeutic intervention.
Dipeptide Repeat Proteins
In addition to prognostic biomarkers, markers of target engagement would improve the interpretation of clinical trials for C9+ ALS and FTD. As mentioned above, a characteristic neuropathological feature of C9+ ALS and FTD is the presence of neuronal inclusions formed of DPR proteins synthesized from expanded C9orf72 repeats. One of these proteins, poly(GP), is abundantly expressed in the brain of C9+ carriers and is detected in CSF (72, 77, 79, 80). While several studies observed that CSF poly(GP) did not associate with age at disease onset, survival, or markers of neurodegeneration (e.g., CSF pNfH or NfL, or measures of brain atrophy) (72, 77, 79), poly(GP) shows promise as a pharmacodynamic biomarker (81).
Since RNA transcripts of expanded C9orf72 repeats are believed to play a key role in C9+ ALS and FTD (82), therapeutic strategies that target C9orf72 repeat RNA are being developed. Given that levels of poly(GP) correlated with levels of repeat-containing RNA in the cerebellum of C9+ carriers (31, 83), poly(GP) was investigated as a marker of target engagement for repeat RNA-based therapies. Antisense oligonucleotides (ASOs), small molecules and genetic modifiers that target C9orf72 repeat RNA attenuated poly(GP) levels in various preclinical models including yeast, worms, mice, and C9+ ALS patient cell lines (81, 84, 85). For example, poly(GP) was detected in CSF of mice expressing an expanded C9orf72 repeat in the brain, and CSF poly(GP) was decreased following treatment with a repeat RNA-targeting ASO. Of note, CSF poly(GP) levels correlated with DPR protein pathology, repeat RNA levels and RNA foci burden in the brains of mice (81). These data suggest that monitoring CSF poly(GP) before and during treatment of patients participating in clinical trials presents a feasible approach to gauge target engagement.
Summary
The search for biomarkers of disease onset and progression in C9orf72 repeat expansion carriers has yielded promising candidate biomarkers (Table 1). Clinically, cognitive, behavioral, and motor impairment occur on a continuum in patients with the C9orf72 mutation. Non-invasive imaging studies in C9+ carriers have identified structural and functional changes in critical components of the networks associated with cognition and behavior. Early thalamic involvement has been detected in structural, functional, and metabolic imaging studies in C9+ carriers across different clinical phenotypes, in both prospective and retrospective studies. Diffusion changes in frontal white matter may also occur early in disease. These non-invasive imaging measures warrant further study in asymptomatic carriers as early markers of degeneration. Among the minimally invasive biomarker measures, CSF pNfH or NfL may allow identification of disease onset in asymptomatic carriers and forecast survival in symptomatic carriers (72, 77, 78). Now that C9orf72 mutation carriers can be identified by genetic testing many decades before symptoms begin, and efforts to develop gene-directed therapy are underway, it is possible to imagine that biomarkers will play important roles in future therapeutic decisions. For example, in the future, persons known to carry the C9orf72 mutation could undergo periodic screening with non-invasive tests such as MRI or physiology, followed by minimally invasive testing to measure CSF or blood biomarkers when findings suspicious for neurodegeneration arise.
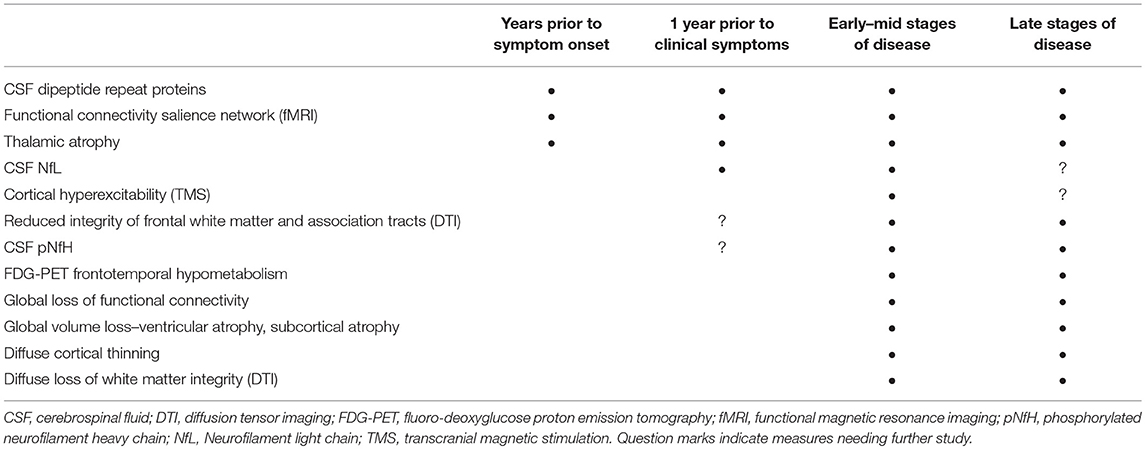
Table 1. Timeframes for detecting changes in selected candidate biomarkers in C9orf72 carriers.
Author Contributions
MF and TG drafted sections of the manuscript and proofread the entire manuscript.
Funding
MF is supported by the intramural program of the National Institutes of Health, NINDS Z01 NS003146 for research on neurological disorders. This funding pays for the open access fee. TG's research is supported by the Muscular Dystrophy Association, the Target ALS Foundation, and the National Institutes of Health, NINDS P01 NS099114.
Conflict of Interest Statement
TG has a U.S. patent on methods and materials for detecting C9+ ALS and FTD using poly(GP).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. (2012) 11:232–40. doi: 10.1016/S1474-4422(12)70014-5
2. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
3. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
4. Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. (2015) 14:291–301. doi: 10.1016/S1474-4422(14)70233-9
5. Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. (2011) 122:673–90. doi: 10.1007/s00401-011-0907-y
6. Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. (2013) 126:859–79. doi: 10.1007/s00401-013-1181-y
7. Sakae N, Bieniek KF, Zhang YJ, Ross K, Gendron TF, Murray ME, et al. Poly-GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol Commun. (2018) 6:63. doi: 10.1186/s40478-018-0564-7
8. Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Küpper C, et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. (2015) 130:537–55. doi: 10.1007/s00401-015-1450-z
9. Bede P, Omer T, Finegan E, Chipika RH, Iyer PM, Doherty MA, et al. Connectivity-based characterisation of subcortical grey matter pathology in frontotemporal dementia and ALS: a multimodal neuroimaging study. Brain Imaging Behav. (in press). doi: 10.1007/s11682-018-9837-9
10. Sellami L, Bocchetta M, Masellis M, Cash DM, Dick KM, van Swieten J, et al. Distinct neuroanatomical correlates of neuropsychiatric symptoms in the three main forms of genetic frontotemporal dementia in the GENFI cohort. J Alzheimers Dis. (2018) 65:147–63. doi: 10.3233/JAD-180053
11. Agosta F, Ferraro PM, Riva N, Spinelli EG, Domi T, Carrera P, et al. Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiol Aging (2017) 57:206–19. doi: 10.1016/j.neurobiolaging.2017.05.024
12. Bede P, Bokde AL, Byrne S, Elamin M, McLaughlin RL, Kenna K, et al. Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology (2013) 81:361–9. doi: 10.1212/WNL.0b013e31829c5eee
13. Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin. (2016) 12:1035–43. doi: 10.1016/j.nicl.2016.10.014
14. Omer T, Finegan E, Hutchinson S, Doherty M, Vajda A, McLaughlin RL, et al. Neuroimaging patterns along the ALS-FTD spectrum: a multiparametric imaging study. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:611–23. doi: 10.1080/21678421.2017.1332077
15. Irwin DJ, McMillan CT, Brettschneider J, Libon DJ, Powers J, Rascovsky K, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2013) 84:163–9. doi: 10.1136/jnnp-2012-303507
16. Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain (2012) 135(Pt 3):736–50. doi: 10.1093/brain/awr361
17. Mahoney CJ, Downey LE, Ridgway GR, Beck J, Clegg S, Blair M, et al. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther. (2012) 4:41. doi: 10.1186/alzrt144
18. McMillan CT, Russ J, Wood EM, Irwin DJ, Grossman M, McCluskey L, et al. C9orf72 promoter hypermethylation is neuroprotective: neuroimaging and neuropathologic evidence. Neurology (2015) 84:1622–30. doi: 10.1212/WNL.0000000000001495
19. Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain (2012) 135(Pt 3):794–806. doi: 10.1093/brain/aws001
20. Lee SE, Khazenzon AM, Trujillo AJ, Guo CC, Yokoyama JS, Sha SJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain (2014) 137(Pt 11):3047–60. doi: 10.1093/brain/awu248
21. Cash DM, Bocchetta M, Thomas DL, Dick KM, van Swieten JC, Borroni B, et al. Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging (2018) 62:191–6. doi: 10.1016/j.neurobiolaging.2017.10.008
22. Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC, et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology (2012) 79:1002–11. doi: 10.1212/WNL.0b013e318268452e
23. Papma JM, Jiskoot LC, Panman JL, Dopper EG, den Heijer T, Donker Kaat L, et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology (2017) 89:1256–64. doi: 10.1212/WNL.0000000000004393
24. Popuri K, Dowds E, Beg MF, Balachandar R, Bhalla M, Jacova C, et al. Gray matter changes in asymptomatic C9orf72 and GRN mutation carriers. Neuroimage Clin. (2018) 18:591–8. doi: 10.1016/j.nicl.2018.02.017
25. Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. (2015) 14:253–62. doi: 10.1016/S1474-4422(14)70324-2
26. Walhout R, Schmidt R, Westeneng HJ, Verstraete E, Seelen M, van Rheenen W, et al. Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology (2015) 85:1780–8. doi: 10.1212/WNL.0000000000002135
27. Bocchetta M, Gordon E, Cardoso MJ, Modat M, Ourselin S, Warren JD, et al. Thalamic atrophy in frontotemporal dementia - Not just a C9orf72 problem. Neuroimage Clin. (2018) 18:675–81. doi: 10.1016/j.nicl.2018.02.019
28. Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, et al. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology (2013) 81:2107–15. doi: 10.1212/01.wnl.0000437313.80913.2c
29. Cappe C, Rouiller EM, Barone P. Multisensory anatomical pathways. Hear Res. (2009) 258:28–36. doi: 10.1016/j.heares.2009.04.017
30. Schonecker S, Neuhofer C, Otto M, Ludolph A, Kassubek J, Landwehrmeyer B, et al. Atrophy in the thalamus but not cerebellum is specific for C9orf72 FTD and ALS Patients - an atlas-based volumetric MRI study. Front Aging Neurosci. (2018) 10:45. doi: 10.3389/fnagi.2018.00045
31. Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, et al. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. (2015) 130:559–73. doi: 10.1007/s00401-015-1474-4
32. van Blitterswijk MM. DeJesus-Hernandez, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. (2013) 12:978–88. doi: 10.1016/S1474-4422(13)70210-2
33. DeJesus-Hernandez M, Finch NA, Wang X, Gendron TF, Bieniek KF, Heckman MG, et al. In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. (2017) 134:255–69. doi: 10.1007/s00401-017-1725-7
34. Whitwell JL, Boeve BF, Weigand SD, Senjem ML, Gunter JL, Baker MC, et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol. (2015) 22:745–52. doi: 10.1111/ene.12675
35. Buckner RL, Krienen FM, Castellanos A, Diaz JC, Yeo BT. The organization of the human cerebellum estimated by intrinsic functional connectivity. J Neurophysiol. (2011) 106:2322–45. doi: 10.1152/jn.00339.2011
36. Mahoney CJ, Simpson IJ, Nicholas JM, Fletcher PD, Downey LE, Golden HL, et al. Longitudinal diffusion tensor imaging in frontotemporal dementia. Ann Neurol. (2015) 77:33–46. doi: 10.1002/ana.24296
37. Machts J, Loewe K, Kaufmann J, Jakubiczka S, Abdulla S, Petri S, et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology (2015) 85:1301–9. doi: 10.1212/WNL.0000000000002017
38. Westeneng HJ, Walhout R, Straathof M, Schmidt R, Hendrikse J, Veldink JH, et al. Widespread structural brain involvement in ALS is not limited to the C9orf72 repeat expansion. J Neurol Neurosurg Psychiatry (2016) 87:1354–60. doi: 10.1136/jnnp-2016-313959
39. Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. (2009) 66:180–9. doi: 10.1001/archneurol.2008.558
40. Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. (2015) 130:845–61. doi: 10.1007/s00401-015-1476-2
41. Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D, et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. (2018) 135:459–74. doi: 10.1007/s00401-017-1793-8
42. Flores BN, Dulchavsky ME, Krans A, Sawaya MR, Paulson HL, Todd PK, et al. Distinct C9orf72-associated dipeptide repeat structures correlate with neuronal toxicity. PLoS ONE (2016) 11:e0165084. doi: 10.1371/journal.pone.0165084
43. McGoldrick P, Zhang M, van Blitterswijk M, Sato C, Moreno D, Xiao S, et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology (2018) 90:e323–31. doi: 10.1212/WNL.0000000000004865
44. Proudfoot M, Gutowski NJ, Edbauer D, Hilton DA, Stephens M, Rankin J, et al. Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol. (2014) 127:451–8. doi: 10.1007/s00401-014-1245-7
45. Baborie A, Griffiths TD, Jaros E, Perry R, McKeith IG, Burn DJ, et al. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol Appl Neurobiol. (2015) 41:601–12. doi: 10.1111/nan.12178
46. Floeter MK, Danielian LE, Braun LE, Wu T. Longitudinal diffusion imaging across the C9orf72 clinical spectrum. J Neurol Neurosurg Psychiatry (2018) 89:53–60. doi: 10.1136/jnnp-2017-316799
47. Lee SE, Sias AC, Mandelli ML, Brown JA, Brown AB, Khazenzon AM, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. (2017) 14:286–97. doi: 10.1016/j.nicl.2016.12.006
48. Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. (2018) 75:236–45. doi: 10.1001/jamaneurol.2017.4266
49. Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K. Amyotrophic lateral sclerosis-a model of corticofugal axonal spread. Nat Rev Neurol. (2013) 9:708–14. doi: 10.1038/nrneurol.2013.221
50. Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. (2014) 127:423–39. doi: 10.1007/s00401-013-1238-y
51. Dopper EG, Rombouts SA, Jiskoot LC, den Heijer T, de Graaf JR, de Koning I, et al. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology (2014) 83:e19–26. doi: 10.1212/WNL.0000000000000583
52. Shivamurthy VK, Tahari AK, Marcus C, Subramaniam RM. Brain FDG PET and the diagnosis of dementia. AJR Am J Roentgenol. (2015) 204:W76–85. doi: 10.2214/AJR.13.12363
53. Cistaro A, Pagani M, Montuschi A, Calvo A, Moglia C, Canosa A, et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging (2014) 41:844–52. doi: 10.1007/s00259-013-2667-5
54. Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, et al. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. (2014) 71:553–61. doi: 10.1001/jamaneurol.2014.62
55. Adeli A, Savica R, Lowe VJ, Vemuri P, Knopman DS, Dejesus-Hernandez M, et al. The GGGGCC repeat expansion in C9ORF72 in a case with discordant clinical and FDG-PET findings: PET trumps syndrome. Neurocase (2014) 20:110–20. doi: 10.1080/13554794.2012.732090
56. Martikainen MH, Gardberg M, Jansson L, Röyttä M, Rinne JO, Kaasinen V. Brain F-FDG and C-PiB PET findings in two siblings with FTD/ALS associated with the C9ORF72 repeat expansion. Neurocase (2014) 20:150–7. doi: 10.1080/13554794.2012.741252
57. Vucic S, Kiernan MC. Transcranial magnetic stimulation for the assessment of neurodegenerative disease. Neurotherapeutics (2017) 14:91–106. doi: 10.1007/s13311-016-0487-6
58. Triggs WJ, Menkes D, Onorato J, Yan RS, Young MS, Newell K, et al. Transcranial magnetic stimulation identifies upper motor neuron involvement in motor neuron disease. Neurology (1999) 53:605–11. doi: 10.1212/WNL.53.3.605
59. Geevasinga N, Menon P, Nicholson GA, Ng K, Howells J, Kril JJ, et al. Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol. (2015) 72:1268–74. doi: 10.1001/jamaneurol.2015.1872
60. Schanz O, Bageac D, Braun L, Traynor BJ, Lehky TJ, Floeter MK. Cortical hyperexcitability in patients with C9ORF72 mutations: relationship to phenotype. Muscle Nerve (2016) 54:264–9. doi: 10.1002/mus.25047
61. Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, et al. Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry (2013) 84:931–5. doi: 10.1136/jnnp-2012-304529
62. Iyer PM, Mohr K, Broderick M, Gavin B, Burke T, Bede P, et al. Mismatch negativity as an indicator of cognitive sub-domain dysfunction in amyotrophic lateral sclerosis. Front Neurol. (2017) 8:395. doi: 10.3389/fneur.2017.00395
63. Rutkove SB, Caress JB, Cartwright MS, Burns TM, Warder J, David WS, et al. Electrical impedance myography correlates with standard measures of ALS severity. Muscle Nerve (2014) 49:441–3. doi: 10.1002/mus.24128
64. Neuwirth C, Barkhaus PE, Burkhardt C, Castro J, Czell D, de Carvalho M, et al. Tracking motor neuron loss in a set of six muscles in amyotrophic lateral sclerosis using the Motor Unit Number Index (MUNIX): a 15-month longitudinal multicentre trial. J Neurol Neurosurg Psychiatry (2015) 86:1172–9. doi: 10.1136/jnnp-2015-310509
65. Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. (2011) 10:75–82. doi: 10.1016/S1474-4422(10)70224-6
66. Steyn FJ, Ioannides ZA, van Eijk RPA, Heggie S, Thorpe KA, Ceslis A, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry (2018) 89:1016–23. doi: 10.1136/jnnp-2017-317887
67. Jawaid A, Murthy SB, Wilson AM, Qureshi SU, Amro MJ, Wheaton M, et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lateral Scler. (2010) 11:542–8. doi: 10.3109/17482968.2010.482592
68. Peter RS, Rosenbohm A, Dupuis L, Brehme T, Kassubek J, Rothenbacher D, et al. Life course body mass index and risk and prognosis of amyotrophic lateral sclerosis: results from the ALS registry Swabia. Eur J Epidemiol. (2017) 32:901–8. doi: 10.1007/s10654-017-0318-z
69. Gorges M, Vercruysse, Muller HP, Huppertz HJ, Rosenbohm A, Nager G, et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2017) 88:1033–41. doi: 10.1136/jnnp-2017-315795
70. Vu LT, Bowser R. Fluid-based biomarkers for amyotrophic lateral sclerosis. Neurotherapeutics (2017) 14:119–134. doi: 10.1007/s13311-016-0503-x
71. Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. (2018) 14:577–89. doi: 10.1038/s41582-018-0058-z
72. Gendron TF, Daughrity LM, Heckman MG, Diehl NN, Wuu J, et al. Phosphorylated neurofilament heavy chain: a biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann Neurol. (2017) 82:139–46. doi: 10.1002/ana.24980
73. Floeter MK, Traynor BJ, Farren J, Braun LE, Tierney M, Wiggs EA, et al. Disease progression in C9orf72 mutation carriers. Neurology (2017) 89:234–41. doi: 10.1212/WNL.0000000000004115
74. García-Redondo A, Dols-Icardo O, Rojas-García R, Esteban-Pérez J, Cordero-Vázquez P, Muñoz-Blanco JL, et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat. (2013) 34:79–82. doi: 10.1002/humu.22211
75. Umoh ME, Fournier C, Li Y, Polak M, Shaw L, Landers JE, et al. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology (2016) 87:1024–30. doi: 10.1212/WNL.0000000000003067
76. Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. (2016) 3:623–36. doi: 10.1002/acn3.325
77. Meeter LHH, Gendron TF, Sias AC, Jiskoot LC, Russo SP, Donker Kaat L, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol. (2018) 5:583–97. doi: 10.1002/acn3.559
78. Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. (2018) 84:130–9. doi: 10.1002/ana.25276
79. Lehmer C, Oeckl P, Weishaupt JH, Volk AE, Diehl-Schmid J, Schroeter ML, et al. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med. (2017) 9:859–68. doi: 10.15252/emmm.201607486
80. Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron (2014) 83:1043–50. doi: 10.1016/j.neuron.2014.07.041
81. Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang YJ, Prudencio M, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. (2017) 9:eaai7866. doi: 10.1126/scitranslmed.aai7866
82. Gendron TF, Petrucelli L. Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb Perspect Med. (2018) 8:a024224. doi: 10.1101/cshperspect.a024224
83. van Blitterswijk M, Gendron TF, Baker MC M. DeJesus-Hernandez, Finch NA, Brown PH, et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. (2015) 130:863–76. doi: 10.1007/s00401-015-1480-6
84. Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron (2016) 90:535–50. doi: 10.1016/j.neuron.2016.04.006
Keywords: C9orf72, cortical thinning, diffusion tensor imaging, dipeptide repeat proteins, functional connectivity, motor neuron disease, neurofilament proteins, biomarker
Citation: Floeter MK and Gendron TF (2018) Biomarkers for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Associated With Hexanucleotide Expansion Mutations in C9orf72. Front. Neurol. 9:1063. doi: 10.3389/fneur.2018.01063
Received: 16 October 2018; Accepted: 21 November 2018;
Published: 05 December 2018.
Edited by:
Peter Bede, Trinity College Dublin, IrelandReviewed by:
Aude-Marie Grapperon, Assistance Publique Hôpitaux de Marseille, FranceMartin Gorges, Universität Ulm, Germany
Copyright © 2018 Floeter and Gendron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Kay Floeter, ZmxvZXRlcm1AbmluZHMubmloLmdvdg==