 Marta Simone
Marta Simone Antonio Trabacca
Antonio Trabacca Elena Panzeri
Elena Panzeri Luciana Losito
Luciana Losito Andrea Citterio
Andrea Citterio Maria Teresa Bassi
Maria Teresa Bassi- 1Unit for Severe Disabilities in Developmental Age and Young Adults, Developmental Neurology and Neurorehabilitation, Scientific Institute IRCCS E. Medea, Brindisi, Italy
- 2Laboratory of Molecular Biology, Scientific Institute IRCCS E. Medea, Lecco, Italy
This paper describes the clinical evolution and the novel genetic findings in a KIF5A mutated family previously reported as affected by spastic paraparesis only. The additional evidence we report here, a homozygous ALS2 mutation detected in the proband, and the clinical evolution observed in the affected members of the family, are in line with the evidence of an overlap between Hereditary Spastic Paraplegias and Amyotrophic Lateral Sclerosis associated with variants in these genes. The proband, a 14-years-old boy, started manifesting a pure form of HSP at age 14 months. The disease rapidly progressed to a juvenile form of ALS. This boy carries a heterozygous missense variant in KIF5A p.(Glu755Lys), inherited from the father, and a homozygous missense variant in the alsin protein encoded by the ALS2 gene p.(Pro192Leu). The father shows a family history of ALS. In the last few years, he has been developing signs and symptoms of both upper and lower motor neuron degeneration, with mild bulbar motor involvement and emotional lability. The patients described in this family, confirm the continuum and partial overlap of the two clinical entities, HSP and ALS, historically viewed as distinct entities. The genetic findings in this family further substantiate the genetic bases underlying the overlap, broadening the clinical spectrum associated with KIF5A mutations.
Introduction
Missense mutations within the kinesin family member 5A gene (KIF5A) are known causes of a dominant form of hereditary spastic paraparesis (spastic paraplegia type 10- SPG10, OMIM: 604187) and of Charcot-Marie-Tooth disease type 2 (CMT2) (1, 2). KIF5A encodes the neuronal kinesin heavy chain (KHC) implicated in the anterograde axonal transport (3). Recently, using a large-scale genome-wide association study and exome sequencing, KIF5A was described as a novel gene associated with Amiotrofic Lateral Sclerosis (ALS) (4, 5).
Mutations in ALS2, the gene coding for alsin protein, have been demonstrated (6) to be associated with a spectrum of rare autosomal recessive disorders including infantile ascending hereditary spastic paralysis (IAHPS), juvenile primary sclerosis (JPLS) with retrograde degeneration of the upper motor neurons, and juvenile ALS with both upper and lower motor neuron involvement. Alsin protein is a member of the guanine nucleotide exchange factors for the small GTPase RAB5 which affects endosome trafficking. Further evidence in one of the four different mouse models generated, indicate that the lack of alsin leads to selective defects both in mitochondria and Golgi apparatus, of the Corticospinal motor neurons (CSMN), thereby revealing the unique importance of alsin function for CSMN health and stability (7).
Here we report the clinical evolution and the novel genetic findings in a two generation family previously reported, with members affected by spastic paraparesis only and carrying a KIF5A mutation (1). The proband, a 14-years-old boy started manifesting an early onset (age 14 months) pure form of HSP rapidly progressing to a juvenile form of ALS. The boy has a family history for ALS, with the father manifesting, at present, only mild symptoms of ALS and a paternal uncle who died for ALS. The boy carries a homozygous variant in ALS2 c.575C>T,p.(Pro192Leu) and a heterozygous variant in KIF5A c.2263G>A, p.(Glu755Lys); the latter is of paternal origin.
Case Report
The proband of the family we report here, was born after an uneventful pregnancy to healthy, unrelated parents. At the 40th day of life, he underwent a cardiac surgery for severe aortic coarctation. A paternal uncle was affected by ALS (the diagnosis was supported by available clinical record reporting clinical and electrophysiological data) and he died at the age of 50 years before the patient came to our attention. The patient sat up unsupported at the age of 10 months and was able to walk at the age of 14 months, but he had abnormal gait with a progressive tendency to skidding and subsequent frequent falls. The child complained of easy fatigue and cramping at the lower limbs especially during the night. At the neurological examination, at age 4 years, he showed a paraparetic gait with lower limbs spastic hypertonia, enhanced deep tendon reflexes and bilateral ankle clonus and Babinski sign. Language skills developmental and cognitive functions were preserved. He underwent brain Magnetic Resonance Imaging, electromyography, somatosensory evoked potentials, blood tests, extensive search for metabolic disorders that resulted all negative, except for altered levels of muscle enzymes. At age 6 years, he underwent the first genetic screening for mutations in the most frequently mutated HSP genes available at that time (SPG4, SPG7, KIF5A, REEP1, SPG11). A missense mutation in the KIF5A gene was identified c.2263G>A, leading to an aminoacid change within the stalk domain, p.(Glu755Lys) (rs387907286, freq. 0.001219% in gnomAD Exomes). This variant was not found in 800 ethnically-matched controls from our internal database (48% females, 52% males) and this is in agreement with the low frequency of this variant in ExAC browser (8.256e-06, found only in European non-Finnish/Latino/South Asian females).The KIF5A variant is of paternal origin (Figure 1). At that time, the neurological examination of the father (aged 50 years) showed only subclinical sign of the disease with increased reflex at both upper and lower limbs. During the next few years, the disease slowly worsened in both the proband and his father.
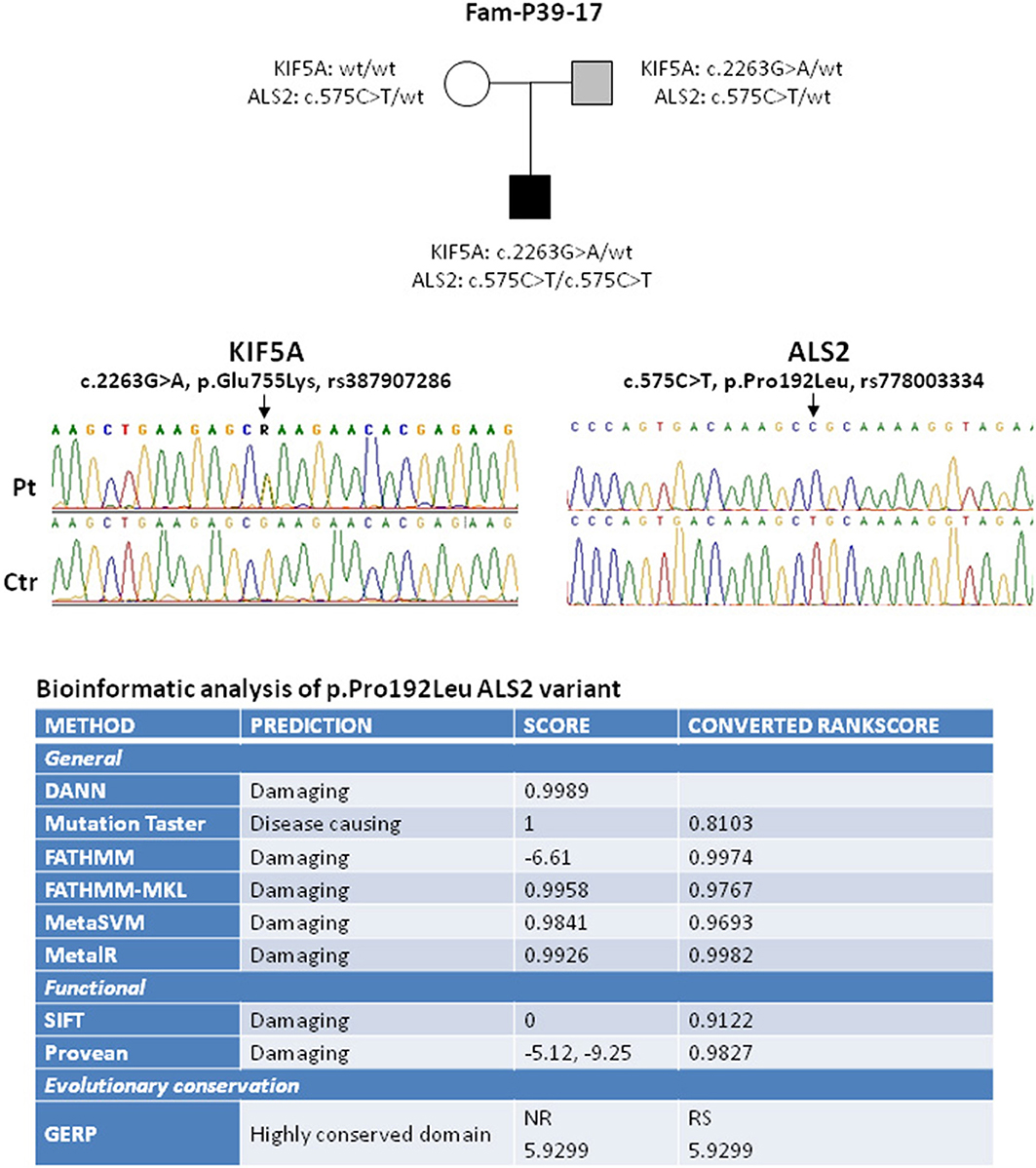
Figure 1. Elechtropherograms of the variants in KIF5A and ALS2 identified in the proband and his family. In the upper panel the black box indicates the proband affected by the juvenile onset of ALS while the light gray box indicates the mild ALS clinical phenotype of the proband father. A summary of the bioinformatic analysis results of the ALS2 variant is reported in the bottom part. As listed in the figure, the software used provide prediction of negative effects at the general and functional level. GERP Software provide information about the evolutionary conservation of the substituted amino acid residue.
The boy started developing weakness and spasticity in the upper limbs with distal fasciculations and painful muscle cramps, mild dysphagia and dysarthria. Electromyography showed signs of active denervation (fibrillation and positive sharp waves) and unstable fasciculation potentials. At present, the proband is 14 years old and reaches a score of 21 at the Spastic Paraplegia Rating Scale (SPRS), thus suggestive of moderate disability (8). In addition to the motor neuron signs, optical coherence tomography examination revealed the presence of a bilateral optic atrophy. Due to the progression of the disease and the symptomatic bulbar motor involvement, he underwent further genetic screening, by using a targeted next generation sequencing approach with a specific panel of 185 genes (including all known genes for HSP, ataxia, neuropathies and motor neuron disease including all known genes for familial ALS—the gene list is provided upon request). The screening detected a homozygous variant in the ALS2 gene, c.575C>T, p.(Pro192Leu) (Figure 1). The variant frequency in gnomAD Exomes is 0.00164% (rs778003334). It has never been described in homozygous state and it was not found in 800 ethnically-matched controls from our database (48% females, 52% males), in agreement with its low frequency in ExAC browser (8.366e-06, found only in Latino/Other males and not present in the European population so far). Nine different prediction programs (DANN, Mutation taster, FATHMM, FATHMM-MKL, MetaSVM, MetaLR, SIFT, Provean, GERP) predict that the substitution of this evolutionary conserved residue in the N-terminus of the protein (RCC1-like domain) negatively affects protein function. Indeed, in vitro studies of another missense mutation in the RCC1-like domain, p.(Cys156Tyr), indicated that the aminoacid change reduced protein stability and led to loss of the protein (9). Further search for variant through a focused exome screening, performed on the proband and on both parents, failed to reveal additional nucleotide sequence changes with pathogenic significance. Both parents are obligate carriers of the ALS2 variant and, as expected, the mother is healthy. The father carries both the ALS2 and the KIF5A heterozygous variants; in the last 4–5 years, he started developing, initially focal, asymmetric signs and symptoms of affection of both the upper and lower motor neuron systems (fasciculations and muscular atrophy in upper limbs and spasticity and hyperreflexia in lower limbs), mild bulbar motor involvement (dysphagia) and emotional lability with no vegetative nor sensory symptoms. Electromyography showed signs of spontaneous electrical activity. He reached a total score of 36 at the ALS Functional Rating Scale-Revised (ALSFRS-R) which confirmed the initial progression of the disease (10).
Materials and Methods
After obtaining an informed consent from the proband and the parents, genomic DNA was prepared from blood with standard method. The proband's DNA was screened by using a targeted next generation sequencing approach with a gene panel including 185 genes: all known causative genes for HSP, the known genes for recessive ataxia and for spinocerebellar ataxias, the most frequently mutated genes in neuropathies and the known genes for familial ALS (the gene list is available upon request). The targeted regions were designed to include coding exons with intronic 50 bp flanking sites and 3′ and 5′ untranslated regions (UTRs) by using the SureDesign system (Agilent Technologies, Santa Clara, CA, USA). The sequencing libraries were prepared from genomic DNA by using a Sure Select enrichment system (Agilent Technologies). Targeted libraries were run on MiSeq platform according to the manufacturer's instructions (Illumina, San Diego, CA, USA). ANNOVAR was used for annotation against the RefSeq database and the Single Nucleotide Polymorphism databases. The filtering strategy we applied led us to select only variants located in the coding regions, including the splice site, (synonymous variants were excluded), variants that exhibited a minor allele frequency of <1% or were not present in variant databases including those of the 1,000 Genomes Project, the Exome Aggregation Consortium (ExAC), the NHLBI exome sequencing project ESP6500. On average, 98.66 and 99.3% of bases were covered by at least 10 and 20 sequence reads, respectively. The mean read depth of the targeted regions was 824X. We used DANN, Mutation taster, FATHMM, FATHMM-MKL, MetaSVM, MetaLR to assess the general effects of the variants, SIFT and Provean for the functional effects and GERP for the evolutionary conservation of the variants. DANN is a pathogenicity scoring methodology based on deep neural networks (11). MUTATION TASTER (http://www.mutationtaster.org/) is an in silico prediction tool for the pathogenicity of a variant. FATHMM (Functional Analysis through Hidden Markov Models, http://fathmm.biocompute.org.uk/) is an in silico tool that predicts the effects of protein missense mutations based on a combination of sequence conservation and “pathogenicity weights”. FATHMM-MKL (http://fathmm.biocompute.org.uk/fathmmMKL.htm) predicts non-coding effects by integrating functional annotation information from the ENCODE. MetaSVM (12) is an ensemble score using Support Vector Machine (SVM) to integrate nine prediction scores and allele frequencies in 1KG database. MetaLR (13) is an ensemble score using Logistic Regression (LR) to integrate nine prediction scores and allele frequencies in 1KG database. SIFT (Sorts Intolerant From Tolerant, http://sift.bii.a-star.edu.sg/) is an in silico prediction tool for non-synonymous variants based on sequence homology derived from closely related sequences collected through PSI-BLAST. Provean (Protein Variation Effect Analyzer, http://provean.jcvi.org) is an in silico tool that predicts how non-synonymous or in-frame indel variant will affect a protein's biological function. GERP (Genomic Evolutionary Rate Profiling, http://mendel.stanford.edu/SidowLab/downloads/gerp/) is a conservation score calculated by quantifying substitution deficits across multiple alignments of orthologs using the genomes of 35 mammals. After filtering, we performed Sanger sequencing to confirm the variants detected through targeted sequencing analysis. The family trio (proband and both parents) was also rescreened for exome analysis by using the SureSelect Focused Exome (Agilent Technologies) with 6,100 known genes. The libraries of the trio were run on a NextSEQ500 (Illumina). Variant filtering and analysis were done as described above. A recessive model was first used and de novo variants were also searched.
Discussion
In the family we describe here, the disease history of the boy started as almost pure HSP, affecting both lower and upper limbs, followed by a progressive involvement of the lower motor neuron. These features fit with the clinical picture shown by several patients with homozygous ALS2 mutations. The onset as pure HSP, with involvement of the upper limbs, also correlates with the KIF5A variant we first detected, except for the early age of onset; this is indeed quite atypical for SPG10 patients. The bilateral optic atrophy progressively developed by the proband, is also included in the phenotypic spectrum of KIF5A complicated forms (14). Based on the recently demonstrated role of KIF5A in degeneration of both upper and lower motor neurons, it is hard to dissect the role of ALS2 and KIF5A in the pathogenesis of the disease in the proband. Surely, the proband's father carrying both heterozygous variants in KIF5A and ALS2, and his family history for ALS might help in that. Indeed, the ALS2 heterozygous variant being present also in the healthy mother, by itself, cannot be considered causative of the father's phenotype. Therefore, the KIF5A variant is likely the major contributor to the mild and slowly progressive form of ALS diagnosed in the father. Interestingly, it was demonstrated that KIF5A variants predominantly located in the N-terminal motor domain of KIF5A are causative for SPG10 and CMT2, whereas ALS-associated mutations are primarily located at the C-terminal cargo-binding tail domain (4). However, it is also possible that C-terminal and N-terminal variants act through a common mechanism that may lead to milder (i.e., SPG10) or more severe (i.e., ALS) phenotypes as previously suggested (4). The KIF5A variant p.(Glu755Lys) located in the stalk domain, was previously hypothesized having a destabilizing effect on protein dimerization since the Glu755 residue represents the g residue in the heptad repeats (α-helical coiled-coil repeats of seven amino acids each–abcdefg- n), of the stalk domain and mediates intra- and inter-helices ionic interactions with a stabilizing function (1). The destabilizing effect of the variant p.(Glu755Lys), at the functional level, can easily originate a sort of intermediate effect, in terms of diseases severity, as likely occurring in the father. In the proband, the intermediate effect of the KIF5A variant is instead likely masked by the potential stronger pathogenic effect of the ALS2 variant, that can be therefore considered a likely mutation.
Conclusion
The clinical and genetic findings described in this family, confirm the continuum and partial overlap of the two clinical entities, HSP and ALS, historically viewed as distinct ones. KIF5A represents part of the genetic bases underlying the overlap, based on large patient cohorts studies (4, 5) and single case report (an Asian family with HSP-KIF5A positive and pseudobulbar palsy, muscular fasciculations) (15). However, the genetic bases supporting the overlap of these motor neuron disorders are wider and include also another gene, SPG11-SPATACSIN. Mutations in SPATACSIN are indeed associated with the HSP-subtype, SPG11 and with the AR-juvenile ALS, termed ALS5 (16). More recently, association with an AR axonal Charcot–Marie–Tooth disease which shares some clinical characteristics with HSP and ALS was as also reported (17) suggesting that mutations in the SPATACSIN gene could cause a much wider spectrum of clinical features than previously recognized. In conclusion, this case shows an atypical genotype-phenotype correlation in HSP-KIF5A, broadening the clinical spectrum associated with HSP-KIF5A mutations. Considering the incomplete penetrance and the involvement of both upper and lower neurons, HSP-KIF5A should be considered in differential diagnosis and in the genetic work out of juvenile ALS. In addition, the co-occurence of variants in both genes KIF5A and ALS2, need to be checked as well, in these patients.
Ethics Statement
Written informed consent to publish the report was obtained from each member of the family for the participation in the study and the publication of this report. The report describes the clinical evolution and the novel genetic findings of a family previously reported and approved by the Institutional Review Board and so it did not require a second approval.
Author Contributions
MS and LL acquired the clinical data, reviewed the literature, and drafted the manuscript. AT and MTB designed the study, oversaw data acquisition, supervised the initial drafting, and critically revised the manuscript. EP and AC conducted the genetic analysis with MTB and contributed to manuscript writing analyzed the clinical data and critically revised the manuscript. All authors contributed to the interpretation of results and reviewed the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the patient and his family for participating to the study. This work was supported by the Italian Ministry of health grant n. RC 2017-2018, by the 5XMille Funds to MTB.
References
1. Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot–Marie–Tooth type 2. Clin Genet. (2012) 82:157–64. doi: 10.1111/j.1399-0004.2011.01717.x
2. Liu YT, Laurá M, Hersheson J, Horga A, Jaunmuktane Z, Brandner S, et al. Extended phenotypic spectrum of KIF5A mutations from spastic paraplegia to axonal neuropathy. Neurology (2014) 83:612–9. doi: 10.1212/WNL.0000000000000691
3. Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. (2009) 10:682. doi: 10.1038/nrm2774
4. Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron (2018) 97:1268–83.e6. doi: 10.1016/j.neuron.2018.02.027
5. Brenner D, Yilmaz R, Müller K, Grehl T, Petri S, Meyer T, et al. Hot-spot KIF5A mutations cause familial ALS. Brain (2018) 141:688–97. doi: 10.1093/brain/awx370
6. Eymard-Pierre E, Lesca G, Dollet S, Santorelli FM, di Capua M, Bertini E, et al. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am J Hum Genet. (2002) 71:518–27. doi: 10.1086/342359
7. Gautam M, Jara JH, Sekerkova G, Yasvoina MV, Martina M, Özdinler PH. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum Mol Genet. (2016) 25:1074–87. doi: 10.1093/hmg/ddv631
8. Schüle R, Holland-Letz T, Klimpe S, Kassubek J, Klopstock T, Mall V, et al. The Spastic Paraplegia Rating Scale (SPRS) a reliable and valid measure of disease severity. Neurology (2006) 67:430–4. doi: 10.1212/01.wnl.0000228242.53336.90
9. Eymard-Pierre E, Yamanaka K, Haeussler M, Kress W, Gauthier-Barichard F, Combes P, et al. Novel missense mutation in ALS2 gene results in infantile ascending hereditary spastic paralysis. Ann Neurol. (2006) 59:976–80. doi: 10.1002/ana.20879
10. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. (1999) 169:13–21. doi: 10.1016/S0022-510X(99)00210-5
11. Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics (2014) 31:761–3. doi: 10.1093/bioinformatics/btu703
12. Kim S, Jhong JH, Lee J, Koo JY. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. (2017) 10:2. doi: 10.1186/s13040-017-0126-8
13. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. (2014) 24:2125–37. doi: 10.1093/hmg/ddu733
14. Wiethoff S, Zhour A, Schöls L, Fischer MD. Retinal nerve fibre layer loss in hereditary spastic paraplegias is restricted to complex phenotypes. BMC Neurol. (2012) 12:143. doi: 10.1186/1471-2377-12-143
15. Kaji S, Kawarai T, Miyamoto R, Nodera H, Pedace L, Orlacchio A, et al. Late-onset spastic paraplegia type 10 (SPG10) family presenting with bulbar symptoms and fasciculations mimicking amyotrophic lateral sclerosis. J Neurol Sci. (2016) 364:45–9. doi: 10.1016/j.jns.2016.03.001
16. Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain (2010) 133:591–8. doi: 10.1093/brain/awp325
Keywords: hereditary spastic paraplegias, amyotrophic lateral sclerosis, KIF5A, ALS2, Charcot-Marie-Tooth disease, infancy, adulthood
Citation: Simone M, Trabacca A, Panzeri E, Losito L, Citterio A and Bassi MT (2018) KIF5A and ALS2 Variants in a Family With Hereditary Spastic Paraplegia and Amyotrophic Lateral Sclerosis. Front. Neurol. 9:1078. doi: 10.3389/fneur.2018.01078
Received: 21 June 2018; Accepted: 26 November 2018;
Published: 07 December 2018.
Edited by:
Antonio Orlacchio, Fondazione Santa Lucia (IRCCS), ItalyReviewed by:
Toshitaka Kawarai, Tokushima University, JapanFilippo M. Santorelli, Fondazione Stella Maris (IRCCS), Italy
Sadaf Naz, University of the Punjab, Pakistan
Copyright © 2018 Simone, Trabacca, Panzeri, Losito, Citterio and Bassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Trabacca, YW50b25pby50cmFiYWNjYUBvcy5sbmYuaXQ=; YXRyYWJhY2NhQHRpbi5pdA==