 Yu-pei Guo
Yu-pei Guo Bei-sha Tang
Bei-sha Tang Ji-feng Guo
Ji-feng Guo- 1Center for Brain Disorders Research, Capital Medical University and Beijing Institute of Brain Disorders, Beijing, China
- 2Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 3National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 4Center for Medical Genetics, Central South University, Changsha, China
- 5Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
Phospholipase A2 group VI (PLA2G6)-associated neurodegeneration (PLAN) includes a series of neurodegenerative diseases that result from the mutations in PLA2G6. PLAN has genetic and clinical heterogeneity, with different mutation sites, mutation types and ethnicities and its clinical phenotype is different. The clinical phenotypes and genotypes of PLAN are closely intertwined and vary widely. PLA2G6 encodes a group of VIA calcium-independent phospholipase A2 proteins (iPLA2β), an enzyme involved in lipid metabolism. According to the age of onset and progressive clinical features, PLAN can be classified into the following subtypes: infantile neuroaxonal dystrophy (INAD), atypical neuroaxonal dystrophy (ANAD) and parkinsonian syndrome which contains adult onset dystonia parkinsonism (DP) and autosomal recessive early-onset parkinsonism (AREP). In this review, we present an overview of PLA2G6-associated neurodegeneration in the context of current research.
Introduction
PLA2G6-associated neurodegeneration (PLAN) is a complex group of neurodegenerative diseases that result from mutations in a gene known as PLA2G6. According to the age of onset and clinical features, PLAN can be mainly classified into four subtypes: infantile neuroaxonal dystrophy (INAD), atypical neuroaxonal dystrophy (ANAD), adult-onset dystonia-parkinsonism (DP) and autosomal recessive early-onset parkinsonism (AREP). The onset of INAD and ANAD occurs in childhood and these diseases manifest as progressive psychomotor deterioration, axial dystonia, spasticity, and ataxia, as well as optic atrophy in some children. Cerebellar cortical atrophy and iron deposition in the globus pallidus and substantia nigra can be detected by Magnetic Resonance Imaging (MRI) in most patients (1, 2). Past research has suggested that PLAN can be classified as neurodegeneration with brain iron accumulation II (NBIA II) (3, 4). However, although there is a phenotypical intersection between NBIA and PLAN, we propose that neither disease can completely include the other. The onset of DP and AREP occurs in adulthood and patients often have normal birth, achieve early age mile-stones and have a normal childhood. Patients with DP or AREP show clinical manifestations of parkinsonian syndrome. These patients are characterized by bradykinesia and tremors with occurrence of dystonia, in addition to cognitive regression as well as gait instability. Some symptoms are similar to those of parkinsonism, but cerebellar cortical atrophy and iron deposition do not occur in these patients. A high clinical variability is exhibited in these phenotypes, but age of onset and clinical manifestations are the main criteria used to make distinctions between the subtypes of PLAN.
In 2006, the PLA2G6 gene was initially cloned in two unrelated Israeli INAD families, both of which included consanguineous marriages (5). In 2010, for the first time, the PLA2G6 gene mutation was associated with parkinsonism (6). At present, the pathogenesis of the PLA2G6 mutation in neurodegenerative diseases remains unclear. Different mutations and even mutations at the same site may cause phenotypic disparities. For example, the presence of pathogenic PLA2G6 gene mutation sites (p. D331Y) reported in AREP patients can be associated with a 70% decrease in enzyme activity (7), and the (His597fx69) frameshift mutation can cause the activity of enzymes to differ from previous cases (8). Here, we speculate whether different mutations can result in the diversity of enzymatic activity, thus causing different clinical phenotypes. In this review, we demonstrate clinical phenotypes with different genotypes in PLAN and discuss the underlying relationships of these symptoms with evidence from genetic studies, with a primary focus on the clinical manifestations and genotypic features supported by neuropsychology research, neuroimaging and molecular genetics. Finally, we explore the link between phenotypes and genotypes for PLAN in the light of current PLA2G6 gene research.
The Clinical Phenotypes Of PLA2G6-Associated Neurodegeneration
The most common phenotype of the PLA2G6 gene mutation is NBIA II. PLAN mainly includes INAD, ANAD, and two other diseases are present in parkinsonian syndrome, DP and AREP (9). In addition, some patients present sporadic parkinsonism similar to AREP, known as sporadic early-onset parkinsonism (EOP) (10). PLA2G6 mutations that cause phenotypical clinical characteristics are shown in Table 1. Moreover, these mutations are associated with hypothyroidism, schizophrenia, diabetes and other diseases (11–13). On magnetic resonance images, most patients carrying the PLA2G6 mutation showed an iron accumulation in the globus pallidus and/or the substantia nigra in T2-weighted images (13, 14). In pathological examinations of individuals with the PLA2G6 mutation, abnormal α-synuclein proteins and hyperphosphorylation of tau proteins were found, and may progress to become Lewy bodies (LBs), neurofibrillary tangles and neuropil threads (15, 16). Neuronal biopsies of patients' central nervous systems and peripheral nervous systems tissue provided evidence of the presence of axonal distension, swellings and spheroid bodies (17). In other cases, brain tissue sections with Perl's staining showed iron deposition in the globus pallidus or substantia nigra (18), and oculogyric crises were also found in PLAN patients (19). Here, we describe the clinical features of several PLA2G6-associated neurodegenerative diseases.
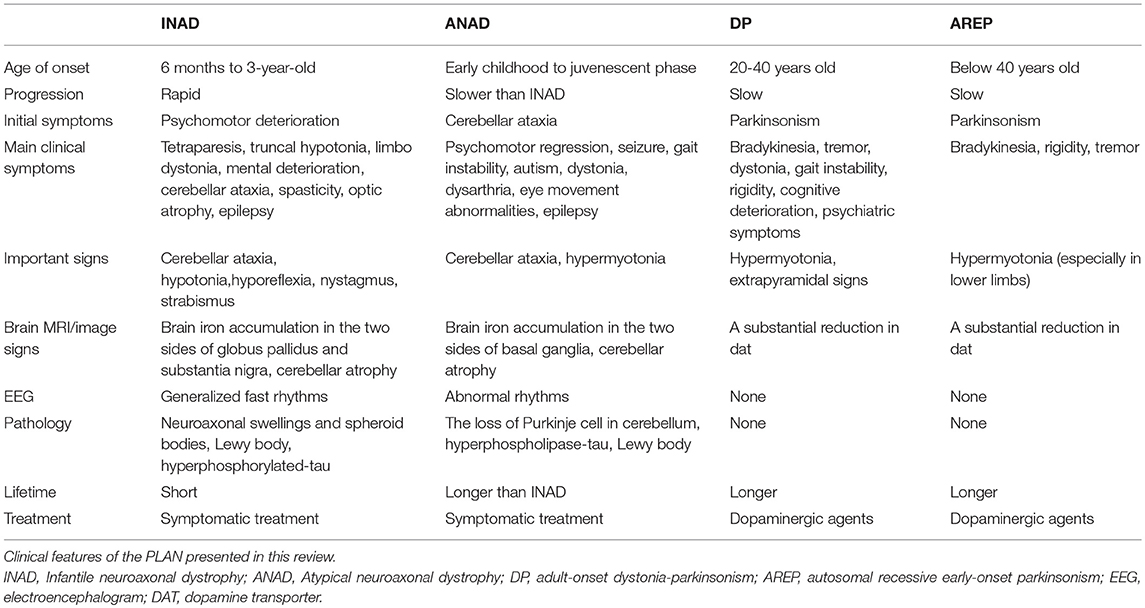
Table 1. Summary of the clinical features in the PLA2G6-associated neurodegeneration.
Infantile Neuroaxonal Dystrophy (INAD)
INAD was first discovered and described by Seitelberger in 1952, and was initially known as Seitelberger's disease (20). INAD is an autosomal recessive neurodegenerative disease (20). The age of onset is around 2 years old, mostly occurring before the age of 18 (21). Before the onset of the disease, compared to normal infants, some patients may present a delay in psychomotor development, while most cases present no indication (22). This rare neurological disease is mainly characterized by progressive psychomotor deterioration, truncal hypotonia, cerebellar ataxia, extrapyramidal signs, and early visual failure caused by optic atrophy. Generalized fast rhythms are frequently observed in electroencephalogram (EEG) and seizures may also occur (5, 13, 23–25). Patients often show slight psychomotor and dystonia disorders during infancy and childhood. The other clinical manifestations are bilateral limb spasticity, bulbar signs (impaired swallowing and dyspnoea), pendular nystagmus, strabismus, distal contractures, optic atrophy, and hearing impairment (2, 18, 26–30). Cognitive impairment might also be observed in the disease evaluations. In most INAD cases, an MRI shows signs of iron accumulation in the globus pallidus and/or the substantia nigra (21). At the early stages of the disease, the MRI might not detect the iron accumulation, but as the disease progresses, iron accumulation can often be detected by the MRI between the ages of 3 and 25 years old (18). Another typical sign of INAD is a fast progression of cerebellar atrophy, which is shown by the MRI (31). In addition, some MRIs also show thin optic chiasma, signal hyperintensity of the dentate nuclei and white matter, and cerebral cortical atrophy (32). An electromyography (EMG) also shows denervation in the peripheral nervous system (29), and an EEG can reveal the widespread high-amplitude fast activity at 16–22 Hz after 2 years of age (33). Visual evoked potentials (VEPs) and electroretinograms (ERGs) appear normal in the early stage of the disease, followed by an increase in abnormal signs over time (27). In a Chinese population, axonal spheroids were discovered in the biopsy specimens of skin and sural nerves among ten patients with INAD (34). In addition, neuroimaging showed cerebellar atrophy occurring in the early stages of INAD, but not in other late-onset diseases. Most patients with INAD have progressively worsening symptoms throughout infancy and early childhood and have a shorter survival period (2, 34). The main features of the pathology are axon spheroids and vacuoles, which are widely present in the central and peripheral nervous systems. Brain tissue pathology can also reveal the presence of iron deposits in the bilateral basal ganglia and globus pallidus (35), as well as phosphorylated α-synuclein-positive LBs. Phosphorylated tau-positive neurofibrillary tangles can also be found in some cases (16). Currently, there are no effective treatments, only palliative methods that can relieve symptoms and prevent secondary complications.
Atypical Neuroaxonal Dystrophy (ANAD)
ANAD is another subtype of PLAN, with atypical clinical characteristics. When the onset of the PLA2G6 mutation occurs later, the phenotype may be atypical. Different from INAD, the age of onset for ANAD ranges from 3 years old to the late teens. Before the onset of clinical symptoms, motor and intelligence development is relatively normal in these patients (16). Patient symptoms include ataxia, rigidity, spasticity, dystonia, and even myoclonic epilepsy. ANAD is also associated with mental impairment and often visual failure. Some patients develop symptoms before 3 years of age, similar to classical INAD, but neurological deterioration during the course of the disease is often delayed (26). In some cases, an MRI can reveal advanced cerebellar atrophy and iron accumulation in the substantia nigra (18, 21), albeit the absence of cerebellar ataxia (36). Iron deposits in the substantia nigra are present in some atypical cases (15, 21), but it is not a universal feature of PLAN. The majority of late-onset cases lack signs of iron accumulation, and MRIs may even appear completely normal. Other cases may show cortical atrophy or white matter changes; for instance, in one study, obvious cerebellar atrophy was detected during head imaging examination and the bilateral basal ganglia showed signs of iron deposition (2). Pathological examination also revealed the loss of the cerebellar Purkinje cells, the deposition of highly phosphorylated tau proteins that formed neurofibrillary tangles and the deposition of phosphorylated α-synuclein that formed LBs (16). Compared to patients with INAD, patients with ANAD present a slower progression and longer survival times (37). The treatment of ANAD is similar to that of INAD.
Parkinsonian Syndrome In Plan
Adult-Onset Dystonia-Parkinsonism (DP)
From the perspective of onset age, DP differs from INAD or ANAD, with a much later onset, occurring from 20 to 40 years old, and exhibiting some typical symptoms, including marked cognitive impairment and some parkinsonian manifestations, such as bradykinesia, ataxia, limb tremors, dystonia, dysarthria, and epilepsy (38, 39). Moreover, oculogyric crises are induced by levodopa in some cases (40). In addition to motor disturbances, non-motor symptoms, such as depression and other adolescent-like behavior changes are observed in DP (41). An MRI of patients with DP reveals some abnormal signals from the frontal lobe, corresponding to severe cognitive impairment. Compared with the MRI results observed in patients with INAD, the MRI results of patients with DP exhibit a rate and degree of severity of iron accumulation and cerebellum atrophy, indicating that these features play a minor role in the manifestation of DP (39, 42). The disease progresses rapidly in patients and is effectively treated with levodopa or polyamine receptor agonists. Patients will often temporarily have dyskinesia after treatment with dopa preparations (6, 38, 43).
Autosomal Recessive Early-Onset Parkinsonism (AREP)
Based on our previous study, the PLA2G6 gene was confirmed to be associated with AREP (7). Patients with PLA2G6-related AREP exhibited tremors and bradykinesia in the lower limbs, postural instability and hypomimia. Additionally, cerebellar ataxia and autonomic dysfunction were recognized in the late stages of the disease. In the MRI, no evidence of iron deposition was found on T2-weighted images. Levodopa treatment can also be beneficial to AREP patients. In recent years, the PLA2G6 mutation was found to be closely related to sporadic early-onset parkinsonism (EOP) (6). Unlike AREP, the genetic characterization of EOP is sporadic. The age of onset of EOP is around 20 years old. The disease is characterized by extrapyramidal signs, cognitive decline, dystonia, dysarthria/dysphonia, swallowing problems, limb tremors and abnormal gait, which are sensitive to dopaminergic agents. The MRIs of EOP patients may show iron accumulations in the brain but not in all individuals; frontal lobe and general white matter atrophy can also be observed (44). EEG and EMG examinations are normal in some individuals. Epileptic seizures also occur during the progression of the disease. Currently, some researchers believe that the PLA2G6 mutation is not a major risk factor for Parkinson's disease in Asian populations (45–47). Moreover, patients with EOP are sensitive to dopamine treatment.
In addition to DP and AREP, there is also a new view regarding the link between PLAN and hereditary spastic paraplegia (HSP). Reports from different countries show that some clinical symptoms of patients with PLAN do not solely include those related to parkinsonian syndrome (48, 49). The clinical features of these patients are mental retardation, extrapyramidal symptoms, lower limb spasticity, cerebellar ataxia, and peripheral neuropathy. Thin corpus callosa and iron accumulations can also be found on MRI images (48, 50).
The Genotypes Of PLA2G6—Associated Neurodegeneration
PLA2G6 mutations have both clinical and genetic heterogeneity. Patients have different types of PLA2G6 mutations, including missense mutations, truncation mutations, and copy number variations. Individuals carrying the PLA2G6 mutation can also have different clinical phenotypes depending on specific genotypic features. The previous reported mutation sites of PLA2G6 are listed in Figure 1 (1, 2, 6–8, 13, 15–18, 22, 23, 30, 34, 38, 41–43, 45, 47, 51–69). Reportedly, in two PLA2G6 mutation families, all three patients carried PLA2G6 mutations (p.R632W) but presented different clinical manifestations than previously reported (13, 43). Shi et al. (7) also suggested that the incomplete loss of enzymatic activity causes AREP. Moreover, expansive copy number variants (CNVs) have been detected in the development of PLAN (27, 57). The crystal structure of iPLA2β is complex, and the different mutation sites are disparately located on the enzyme (70). According to previous studies, mutation sites in the ankyrin repeat (AR) domains, catalytic(CAT) domains, or any other domains may lead to different enzyme activities. We speculate that the coincident mutation of PLAN may initiate the pathological mechanisms. The pivotal factor that affects the relationships between mutations and clinical phenotypes may be enzyme activity. However, more cases are needed to clarify the relationships between genotypes and phenotypes of PLA2G6.
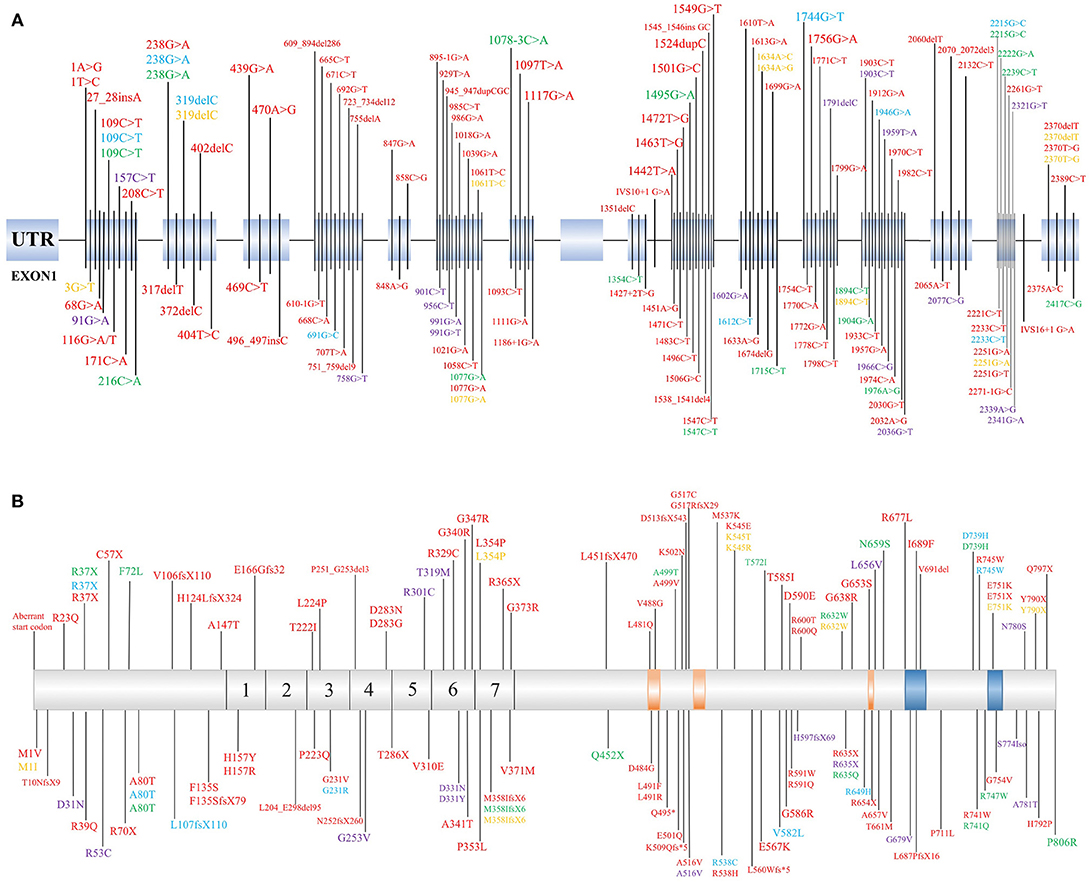
Figure 1. Identified mutation sites of PLA2G6 gene. (A) There are 17 exons of the PLA2G6 gene, and mutation sites were distributed on the exons. (B) Schematic representation in scale of the full-length iPLA2β protein with functional regions and position of the variant protein. The mutations which cause dystonia-parkinsonism, previously identified by others, are shown above. 1–7 = ankyrin repeat region. The position of calmodulin domains and motifs was provided by https://www.uniprot.org/uniprot/O60733. Red, INAD; Blue, ANAD; Green, DP; Yellow, NBIA; Purple, parkinsonian syndrome.
Genetic Function Of PLA2G6
The PLA2G6 gene is located on 22q13.11 (71), with 17 exons. The protein, encoded by PLA2G6, is a member of the A2 phospholipase family (PLA2), known as group VI calcium-independent phospholipase A2 (iPLA2β). It is an enzyme functioning in inflammation, immune responses, cell proliferation, apoptosis and remodeling of membrane phospholipids (36, 72).
iPLA2β is an intracellular and calcium ion-independent protein. This protein was first isolated from the P388D1 cell line and described in 1994 (73). iPLA2β contains 806 amino acids; human iPLA2β is 88 KDa and contains an N-terminal domain, ARs domain and CAT domains (70, 74, 75). The iPLA2β protein encoded by the PLA2G6 gene is an important lipase in the human body which is widely distributed in the tissues of human organs (http://www.proteinatlas.org). In the human brain, iPLA2β is highly expressed in the substantia nigra, cortex and the hippocampus (76–78). iPLA2β can hydrolyse the sn-2 acyl chain of phospholipids and the major decomposition products are docosahexaenoic acid (DHA) and lysophospholipids (75). Under the action of cyclooxygenase and lipoxygenase, DHA produces neuroprotectin D1 (NPD1), and NPD1 plays a crucial role in anti-inflammatory processes and immune responses in the brain (79, 80). NPD1 is specifically involved in the catabolism of fatty acids and arachidonic acid (AA)-related inflammatory reactions (81, 82). iPLA2β exerts an anti-inflammatory function through the action of NPD1, against AA, and has a protective effect on cells in inflammatory reactions. The loss of iPLA2β's function may affect proteins and processes normally involved in regulating the movement of membranes within axons and dendrites, subsequently leading to mitochondrial abnormalities and synaptic transmission impairment (83–86). Moreover, animal models of PLA2G6 showed neurodegeneration (87) and revealed that iPLA2β is closely related to dopaminergic cells, axonal development (88), endoplasmic reticulum stress, mitophagy impairment (89), and changes in Ca(2+) signaling (90). Thus, these animals provide great disease models for PLAN.
iPLA2β is associated with a variety of diseases and medical emergencies, including strokes, spinal cord injuries and neurodegenerative diseases (91–93). However, the pathogenesis of PLA2G6 in neurodegenerative diseases remains unclear and the function of iPLA2β, resulting from different mutation sites and types, may be the vital factor. It was reported that pathogenic PLA2G6 gene mutation sites (p.A341T, p.G517C) in patients with INAD\NBIA can cause significant decreases in enzymatic activity (94). Moreover, the activity of iPLA2β was reduced by 70% compared to normal functioning, owing to the p.D331Y homozygous mutation (7) and the (His597fx69) frameshift mutation, making the activity of enzymes <6% compared to that of WT iPLA2β (8). Differential enzymatic activity caused by multifarious mutations may be a key factor in explaining the high clinical variability in PLAN.
Conclusion
PLA2G6 mutations have both genotypic and phenotypic heterogeneity. Here, we summarized the major subtypes of PLAN and analyzed their potential relationships. Mutated forms of PLA2G6 include missense mutations, truncated mutants, fragment deletions, and CNVs. Individuals carrying different PLA2G6 mutations may also display varied clinical symptoms. The subtypes of PLA2G6 mutation-related disorders are INAD, ANAD, DP, and AREP, with distinct characteristics associated with each disorder. In recent years, HSP has also been found to be associated with the PLA2G6 gene mutations. Some mutation sites of HSP also coincide with the mutation sites of the previous four phenotypes. Whether HSP can be considered a PLAN phenotype requires additional research. In the cases of INAD/ANAD, an MRI exhibited iron accumulation in the basal ganglia and globus pallidus, as well as abnormal α-synuclein and hyperphosphorylation of tau proteins in brain tissues, while other cases had relatively moderate MRI features. Moreover, α-synuclein and neurofibrillary tangles pathologies indicate that PLAN may be consistent with idiopathic Parkinson's disease (iPD) to some extent. Interestingly, the view that later onset cases tend to have less tau involvement but still severe α-synuclein pathology may need further discussion. (3, 15, 16, 95).
The link between the phenotypes and genotypes of PLAN suggests that different mutation sites lead to various protein activities. Mutation sites in different domains, for example, ARs or the CAT domains, may have different effects to physiological processes. Based on a previously described case, a PLA2G6 gene mutation site can result in patients presenting several different phenotypes. Potentially, enzyme activity as well as DNA methylation, synergistic genetic processes, or environmental factors may participate in the pathogenesis of PLAN. Based on the information presented in Figure 1, we speculate that there is no obvious rule for mutation site distribution of PLAN, but further studies are required to clarify why there is no mutation localized in exon 9 and whether the mutations can be categorized according to the 3D structure of iPLA2β (70).
In recent decades, the study on PLA2G6-related disorders has been performed in terms of pathogenesis and iPLA2β function, which provides hope of a detailed understanding of this disease. iPLA2β is a vital protein involved in immune responses, inflammatory processes, fatty acid metabolism, oxidative stress, and apoptosis, which may be the pathogenic mechanisms underlying the progression of neurodegenerative diseases. iPLA2β is involved in the metabolism of DHA and NPD1, which are closely related to human neurocognitive development and anti-inflammatory properties, respectively (96). Whether mental retardation manifesting in young children is caused as a result of iPLA2β affecting the metabolism of DHA is unclear; a better understanding would perhaps provide new information regarding the treatment of PLAN. In addition, iron metabolism may offer clues regarding disease therapy. In summary, the features of PLAN may provide information regarding the etiology of other neurodegenerative diseases.
Author Contributions
YG conceived the study and wrote the manuscript. BT and JG discussed and revised the manuscript. YG prepared the tables. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grant 2016YFC1306000 from the National Key Plan for Scientific Research and Development of China (to BT), grant 2017YFC0909100 from the National Key Plan for Scientific Research and Development of China (to JG), grants 81371405 and 81873785 from the National Natural Science Foundation of China (to JG), and grant 2017JJ1037 from Hunan Science Funds for Distinguished Young Scholar (to JG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology (2008) 71:1402–9. doi: 10.1212/01.wnl.0000327094.67726.28
2. Kurian MA, Morgan NV, Macpherson L, Foster K, Peake D, Gupta R, et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology (2008) 70:1623–9. doi: 10.1212/01.wnl.0000310986.48286.8e
3. Schneider SA, Hardy J, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord. (2012) 27:42–53. doi: 10.1002/mds.23971
4. Hayflick SJ, Kurian MA, Hogarth P. Neurodegeneration with brain iron accumulation. Handb Clin Neurol. (2018) 147:293–305. doi: 10.1016/B978-0-444-63233-3.00019-1
5. Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. (2006) 79:942–8. doi: 10.1086/508572
6. Paisan-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. (2010) 25:1791–800. doi: 10.1002/mds.23221
7. Shi CH, Tang BS, Wang L, Lv ZY, Wang J, Luo LZ, et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology (2011) 77:75–81. doi: 10.1212/WNL.0b013e318221acd3
8. Gui YX, Xu ZP, Wen L, Liu HM, Zhao JJ, Hu XY. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkins Relat Disord. (2013) 19:21–6. doi: 10.1016/j.parkreldis.2012.07.016
9. Gregory A, Kurian MA, Maher ER, Hogarth P, Hayflick SJ. PLA2G6-Associated Neurodegeneration. In: Adam MP, Ardinger HH, Pagon RA, Wallace, SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews®. Seattle WA: University of Washington (1993).
10. Xie F, Cen Z, Ouyang Z, Wu S, Xiao J, Luo W. Homozygous p.D331Y mutation in PLA2G6 in two patients with pure autosomal-recessive early-onset parkinsonism: further evidence of a fourth phenotype of PLA2G6-associated neurodegeneration. Parkinsonism Relat Disord. (2015) 21:420–2. doi: 10.1016/j.parkreldis.2015.01.012
11. Smalheiser NR, Swanson DR. Calcium-independent phospholipase A2 and schizophrenia. Arch Gen Psychiatry (1998) 55:752–3.
12. Ramanadham S, Song H, Hsu FF, Zhang S, Crankshaw M, Grant GA, et al. Pancreatic islets and insulinoma cells express a novel isoform of group VIA phospholipase A2 (iPLA2 beta) that participates in glucose-stimulated insulin secretion and is not produced by alternate splicing of the iPLA2 beta transcript. Biochemistry (2003) 42:13929–40. doi: 10.1021/bi034843p
13. Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. (2006) 38:752–4. doi: 10.1038/ng1826
14. Khan AO, Aldrees A, Elmalik SA, Hassan HH, Michel M, Stevanin G, et al. Ophthalmic features of PLA2G6-related paediatric neurodegeneration with brain iron accumulation. Br J Ophthalmol. (2014) 98:889–93. doi: 10.1136/bjophthalmol-2013-304527
15. Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging (2012) 33:814–23. doi: 10.1016/j.neurobiolaging.2010.05.009
16. Riku Y, Ikeuchi T, Yoshino H, Mimuro M, Mano K, Goto Y, et al. Extensive aggregation of alpha-synuclein and tau in juvenile-onset neuroaxonal dystrophy: an autopsied individual with a novel mutation in the PLA2G6 gene-splicing site. Acta Neuropathol Commun. (2013) 1:12. doi: 10.1186/2051-5960-1-12
17. Kapoor S, Shah MH, Singh N, Rather MI, Bhat V, Gopinath S, et al. Genetic Analysis of PLA2G6 in 22 Indian Families with Infantile Neuroaxonal Dystrophy, Atypical Late-Onset Neuroaxonal Dystrophy and Dystonia Parkinsonism Complex. PLoS ONE (2016) 11:e0155605. doi: 10.1371/journal.pone.0155605
18. Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R, et al. PLA2G6-associated neurodegeneration (PLAN): Further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab. (2014) 112:183–9. doi: 10.1016/j.ymgme.2014.03.008
19. Rohani M, Shahidi G, Vali F, Lang AE, Slow E, Gahl WA, et al. Oculogyric crises in PLA2G6 associated neurodegeneration. Parkins Relat Disord. (2018) 52:111–2. doi: 10.1016/j.parkreldis.2018.03.010
20. Seitelberger F. Neuropathological conditions related to neuroaxonal dystrophy. Acta Neuropathol. (1971) (Suppl. 5) 17–29.
21. Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. (2009) 46:73–80. doi: 10.1136/jmg.2008.061929
22. Tonelli A, Romaniello R, Grasso R, Cavallini A, Righini A, Bresolin N, et al. Novel splice-site mutations and a large intragenic deletion in PLA2G6 associated with a severe and rapidly progressive form of infantile neuroaxonal dystrophy. Clin Genet. (2010) 78:432–40. doi: 10.1111/j.1399-0004.2010.01417.x
23. Salih MA, Mundwiller E, Khan AO, Aldrees A, Elmalik SA, Hassan HH, et al. New findings in a global approach to dissect the whole phenotype of PLA2G6 gene mutations. PLoS ONE (2013) 8:e76831. doi: 10.1371/annotation/cb01a74a-3330-4412-8040-2a94842420ed
24. Darling A, Aguilera-Albesa S, Tello CA, Serrano M, Tomas M, Camino-Leon R, et al. PLA2G6-associated neurodegeneration: new insights into brain abnormalities and disease progression. Parkinsonism Relat Disord (2018). doi: 10.1016/j.parkreldis.2018.10.013. [Epub ahead of print].
25. Gitiaux C, Kaminska A, Boddaert N, Barcia G, Gueden S, The Tich SN, et al. PLA2G6-associated neurodegeneration: Lessons from neurophysiological findings. Eur J Paediatr Neurol. (2018) 22:854–61. doi: 10.1016/j.ejpn.2018.05.005
26. Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, et al. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology (1999) 52:1472–8. doi: 10.1212/WNL.52.7.1472
27. Crompton D, Rehal PK, Macpherson L, Foster K, Lunt P, Hughes I, et al. Multiplex ligation-dependent probe amplification (MLPA) analysis is an effective tool for the detection of novel intragenic PLA2G6 mutations: implications for molecular diagnosis. Mol Genet Metab. (2010) 100:207–12. doi: 10.1016/j.ymgme.2010.02.009
28. Davids M, Kane MS, He M, Wolfe LA, Li X, Raihan MA, et al. Disruption of Golgi morphology and altered protein glycosylation in PLA2G6-associated neurodegeneration. J Med Genet. (2016) 53:180–9. doi: 10.1136/jmedgenet-2015-103338
29. Li H, Zou Y, Bao X, Wang H, Wang J, Jin H, et al. Monozygotic twins with infantile neuroaxonal dystrophy: A case report and literature review. Exp Ther Med. (2016) 12:3387–9. doi: 10.3892/etm.2016.3761
30. Iannello G, Graziano C, Cenacchi G, Cordelli DM, Zuntini R, Papa V, et al. A new PLA2G6 mutation in a family with infantile neuroaxonal dystrophy. J Neurol Sci. (2017) 381:209–12. doi: 10.1016/j.jns.2017.08.3260
31. Mascalchi M, Mari F, Berti B, Bartolini E, Lenge M, Bianchi A, et al. Fast progression of cerebellar atrophy in pla2g6-associated infantile neuronal axonal dystrophy. Cerebellum (2017) 16:742–5. doi: 10.1007/s12311-017-0843-z
32. Farina L, Nardocci N, Bruzzone MG, D'incerti L, Zorzi G, Verga L, et al. Infantile neuroaxonal dystrophy: neuroradiological studies in 11 patients. Neuroradiology (1999) 41:376–80. doi: 10.1007/s002340050768
33. Iodice A, Spagnoli C, Salerno GG, Frattini D, Bertani G, Bergonzini P, et al. Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. (2017) 39:93–100. doi: 10.1016/j.braindev.2016.08.012
34. Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y, Xiong H, et al. Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy. Eur J Neurol. (2009) 16:240–5. doi: 10.1111/j.1468-1331.2008.02397.x
35. Bernardi B, Pini A, Santucci M, Cenacchi C, Garavaglia B, Ucchino V, et al. MRI findings in patients with clinical onset consistent with Infantile Neuroaxonal Dystrophy (INAD), literature review, clinical and MRI Follow-up. Neuroradiol J. (2011) 24:202–14. doi: 10.1177/197140091102400207
36. Aoun M, Tiranti V. Mitochondria: A crossroads for lipid metabolism defect in neurodegeneration with brain iron accumulation diseases. Int J Biochem Cell Biol. (2015) 63:25–31. doi: 10.1016/j.biocel.2015.01.018
37. Sadeh M. Neurodegeneration associated with genetic defects in phospholipase A2. Neurology (2009) 73:819. doi: 10.1212/WNL.0b013e3181b2851b
38. Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. (2009) 65:19–23. doi: 10.1002/ana.21415
39. Karkheiran S, Shahidi GA, Walker RH, Paisan-Ruiz C. PLA2G6-associated dystonia-parkinsonism: case report and literature review. Tremor Other Hyperkinet Mov. (2015) 5:317. doi: 10.7916/D84Q7T4W
40. Virmani T, Thenganatt MA, Goldman JS, Kubisch C, Greene PE, Alcalay RN. Oculogyric crises induced by levodopa in PLA2G6 parkinsonism-dystonia. Parkinsonism Relat Disord. (2014) 20:245–7. doi: 10.1016/j.parkreldis.2013.10.016
41. Kamel WA, Al-Hashel JY, Abdulsalam AJ, Damier P, Al-Mejalhem AY. PLA2G6-related parkinsonism presenting as adolescent behavior. Acta Neurol Belg. (2018). doi: 10.1007/s13760-018-1003-z. [Epub ahead of print].
42. Malaguti MC, Melzi V, Di Giacopo R, Monfrini E, Di Biase E, Franco G, et al. A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat Disord (2015) 21:337–9. doi: 10.1016/j.parkreldis.2015.01.001
43. Sina F, Shojaee S, Elahi E, Paisan-Ruiz C. R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol. (2009) 16:101–4. doi: 10.1111/j.1468-1331.2008.02356.x
44. Giri A, Guven G, Hanagasi H, Hauser AK, Erginul-Unaltuna N, Bilgic B, et al. PLA2G6 Mutations Related to Distinct Phenotypes: A New Case with Early-onset Parkinsonism. Tremor Other Hyperkinet Mov. (2016) 6:363. doi: 10.7916/D81G0M12
45. Tomiyama H, Yoshino H, Ogaki K, Li L, Yamashita C, Li Y, et al. PLA2G6 variant in Parkinson's disease. J Hum Genet. (2011) 56:401–3. doi: 10.1038/jhg.2011.22
46. Lv Z, Guo J, Sun Q, Li K, Yu R, Tian J, et al. Association between PLA2G6 gene polymorphisms and Parkinson's disease in the Chinese Han population. Parkinsonism Relat Disord. (2012) 18:641–4. doi: 10.1016/j.parkreldis.2012.02.015
47. Tian JY, Tang BS, Shi CH, Lv ZY, Li K, Yu RL, et al. Analysis of PLA2G6 gene mutation in sporadic early-onset parkinsonism patients from Chinese population. Neurosci Lett. (2012) 514:156–8. doi: 10.1016/j.neulet.2012.02.078
48. Ozes B, Karagoz N, Schule R, Rebelo A, Sobrido MJ, Harmuth F, et al. PLA2G6 mutations associated with a continuous clinical spectrum from neuroaxonal dystrophy to hereditary spastic paraplegia. Clin Genet. (2017) 92:534–9. doi: 10.1111/cge.13008
49. Koh K, Ichinose Y, Ishiura H, Nan H, Mitsui J, Takahashi J, et al. PLA2G6-associated neurodegeneration presenting as a complicated form of hereditary spastic paraplegia. J Hum Genet. (2018) doi: 10.1038/s10038-018-0519-7. [Epub ahead of print].
50. Chen YJ, Chen YC, Dong HL, Li LX, Ni W, Li HF, et al. Novel PLA2G6 mutations and clinical heterogeneity in Chinese cases with phospholipase A2-associated neurodegeneration. Parkinsonism Relat Disord. (2018) 49:88–94. doi: 10.1016/j.parkreldis.2018.02.010
51. Tan EK, Ho P, Tan L, Prakash KM, Zhao Y. PLA2G6 mutations and Parkinson's disease. Ann Neurol. (2010) 67:148. doi: 10.1002/ana.21663
52. Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology (2010) 75:1356–61. doi: 10.1212/WNL.0b013e3181f73649
53. Kauther KM, Hoft C, Rissling I, Oertel WH, Moller JC. The PLA2G6 gene in early-onset Parkinson's disease. Mov Disord. (2011) 26:2415–7. doi: 10.1002/mds.23851
54. Agarwal P, Hogarth P, Hayflick S, Macleod P, Kuriakose R, Mckenzie J, et al. Imaging striatal dopaminergic function in phospholipase A2 group VI-related parkinsonism. Mov Disord. (2012) 27:1698–9. doi: 10.1002/mds.25160
55. Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, et al. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. (2012) 159B:183–91. doi: 10.1002/ajmg.b.32012
56. Koroglu C, Seven M, Tolun A. Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J Med Genet. (2013) 50:515–20. doi: 10.1136/jmedgenet-2013-101634
57. Zhang P, Gao Z, Jiang Y, Wang J, Zhang F, Wang S, et al. Follow-up study of 25 Chinese children with PLA2G6-associated neurodegeneration. Eur J Neurol. (2013) 20:322–30. doi: 10.1111/j.1468-1331.2012.03856.x
58. Goyal M, Bijarnia-Mahay S, Kingsmore S, Farrow E, Saunders C, Saxena R, et al. Molecular diagnosis of infantile neuro axonal dystrophy by next generation sequencing. Indian J Pediatr. (2015) 82:474–7. doi: 10.1007/s12098-014-1608-z
59. Al-Maawali A, Yoon G, Feigenbaum AS, Halliday WC, Clarke JT, Branson HM, et al. Validation of the finding of hypertrophy of the clava in infantile neuroaxonal dystrophy/PLA2G6 by biometric analysis. Neuroradiology (2016) 58:1035–42. doi: 10.1007/s00234-016-1726-6
60. Blake RB, Gilbert DL, Schapiro MB. Child neurology: two sisters with dystonia and regression: PLA2G6-associated neurodegeneration. Neurology (2016) 87:e1–3. doi: 10.1212/WNL.0000000000002804
61. Dastsooz H, Nemati H, Fard MAF, Fardaei M, Faghihi MA. Novel mutations in PANK2 and PLA2G6 genes in patients with neurodegenerative disorders: two case reports. BMC Med Genet. (2017) 18:87. doi: 10.1186/s12881-017-0439-y
62. Guo S, Yang L, Liu H, Chen W, Li J, Yu P, et al. Identification of novel compound mutations in pla2g6-associated neurodegeneration patient with characteristic MRI Imaging. Mol Neurobiol. (2017) 54:4636–43. doi: 10.1007/s12035-016-9991-2
63. Michelis JP, Hattingen E, Gaertner FC, Minnerop M, Traber F, Biskup S, et al. Expanded phenotype and hippocampal involvement in a novel compound heterozygosity of adult PLA2G6 associated neurodegeneration (PARK14). Parkinsonism Relat Disord. (2017) 37:111–3. doi: 10.1016/j.parkreldis.2017.01.005
64. Wirth T, Weibel S, Montaut S, Bigaut K, Rudolf G, Chelly J, et al. Severe early-onset impulsive compulsive behavior and psychosis in PLA2G6-related juvenile Parkinson's disease. Parkinsonism Relat Disord. (2017) 41:127–9. doi: 10.1016/j.parkreldis.2017.05.014
65. Yamashita C, Funayama M, Li Y, Yoshino H, Yamada H, Seino Y, et al. Mutation screening of PLA2G6 in Japanese patients with early onset dystonia-parkinsonism. J Neural Transm. (2017) 124:431–5. doi: 10.1007/s00702-016-1658-7
66. Elsayed LEO, Mohammed IN, Hamed A, Elseed MA, Salih MM, Yahia A, et al. Case report of a novel homozygous splice site mutation in PLA2G6 gene causing infantile neuroaxonal dystrophy in a Sudanese family. BMC Med Genet. (2018) 19:72. doi: 10.1186/s12881-018-0592-y
67. Ferese R, Scala S, Biagioni F, Giardina E, Zampatti S, Modugno N, et al. Heterozygous PLA2G6 mutation leads to iron accumulation within basal ganglia and Parkinson's disease. Front Neurol. (2018) 9:536. doi: 10.3389/fneur.2018.00536
68. Shen T, Pu J, Lai HY, Xu L, Si X, Yan Y, et al. Genetic analysis of ATP13A2, PLA2G6 and FBXO7 in a cohort of Chinese patients with early-onset Parkinson's disease. Sci Rep. (2018) 8:14028. doi: 10.1038/s41598-018-32217-4
69. Wang B, Wu Tang J. Infantile neuroaxonal dystrophy caused by PLA2G6 gene mutation in a Chinese patient: a case report. Exp Ther Med. (2018) 16:1290–4. doi: 10.3892/etm.2018.6347
70. Malley KR, Koroleva O, Miller I, Sanishvili R, Jenkins CM, Gross RW, et al. The structure of iPLA2beta reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat Commun. (2018) 9:765. doi: 10.1038/s41467-018-03193-0
71. Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13.1. J Biol Chem. (1999) 274:9607–16. doi: 10.1074/jbc.274.14.9607
72. Hooks SB, Cummings BS. Role of Ca2+-independent phospholipase A2 in cell growth and signaling. Biochem Pharmacol. (2008) 76:1059–67. doi: 10.1016/j.bcp.2008.07.044
73. Ackermann EJ, Kempner ES, Dennis EA. Ca(2+)-independent cytosolic phospholipase A2 from macrophage-like P388D1 cells. Isolation and characterization. J Biol Chem. (1994) 269:9227–33.
74. Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. (1997) 272:8567–75. doi: 10.1074/jbc.272.13.8567
75. Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell Signal. (2005) 17:1052–62. doi: 10.1016/j.cellsig.2005.03.002
76. Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot Essent Fatty Acids (2004) 71:161–9. doi: 10.1016/j.plefa.2004.03.004
77. Shirai Y, Ito M. Specific differential expression of phospholipase A2 subtypes in rat cerebellum. J Neurocytol. (2004) 33:297–307. doi: 10.1023/B:NEUR.0000044191.83858.f7
78. Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. (2006) 58:591–620. doi: 10.1124/pr.58.3.7
79. Calandria JM, Bazan NG. Neuroprotectin D1 modulates the induction of pro-inflammatory signaling and promotes retinal pigment epithelial cell survival during oxidative stress. Adv Exp Med Biol. (2010) 664:663–70. doi: 10.1007/978-1-4419-1399-9_76
80. Sun GY, Simonyi A, Fritsche KL, Chuang DY, Hannink M, Gu Z, et al. Docosahexaenoic acid (DHA): An essential nutrient and a nutraceutical for brain health and diseases. Prostaglandins Leukot Essent Fatty Acids (2018) 136:3–13. doi: 10.1016/j.plefa.2017.03.006
81. Cámara-Lemarroy CR, Guzmán-De La Garza FJ, Fernández-Garza NE. Molecular inflammatory mediators in peripheral nerve degeneration and regeneration. Neuroimmunomodulation (2010) 17:314–24. doi: 10.1159/000292020
82. Broersen K, Ruiperez V, Davletov B. Structural and aggregation properties of alpha-synuclein linked to phospholipase A2 Action. Protein Pept Lett. (2018) 25:368–78. doi: 10.2174/0929866525666180326120052
83. Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, et al. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol. (2008) 172:406–16. doi: 10.2353/ajpath.2008.070823
84. Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. (2010) 12:125–69. doi: 10.1089/ars.2009.2668
85. Kinghorn KJ, Castillo-Quan JI, Bartolome F, Angelova PR, Li L, Pope S, et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain (2015) 138:1801–16. doi: 10.1093/brain/awv132
86. Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, et al. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to alpha-Synuclein Gain. Cell Metab. (2018) 28:605–618.e606. doi: 10.1016/j.cmet.2018.05.019
87. Iliadi KG, Gluscencova OB, Iliadi N, Boulianne GL. Mutations in the Drosophila homolog of human PLA2G6 give rise to age-dependent loss of psychomotor activity and neurodegeneration. Sci Rep. (2018) 8:2939. doi: 10.1038/s41598-018-21343-8
88. Sanchez E, Azcona LJ, Paisan-Ruiz C. Pla2g6 deficiency in zebrafish leads to dopaminergic cell death, axonal degeneration, increased beta-synuclein expression, and defects in brain functions and pathways. Mol Neurobiol. (2018) 55:6734–54. doi: 10.1007/s12035-017-0846-2
89. Chiu C-C, Lu C-S, Weng Y-H, Chen Y-L, Huang Y-Z, Chen R-S, et al. PARK14 (D331Y) PLA2G6 causes early-onset degeneration of substantia nigra dopaminergic neurons by inducing mitochondrial dysfunction, ER stress, mitophagy impairment and transcriptional dysregulation in a knockin mouse model. Mol Neurobiol. (2018) doi: 10.1007/s12035-018-1118-5. [Epub ahead of print].
90. Strokin M, Reiser G. Neurons and astrocytes in an infantile neuroaxonal dystrophy (INAD) mouse model show characteristic alterations in glutamate-induced Ca(2+) signaling. Neurochem Int. (2017) 108:121–32. doi: 10.1016/j.neuint.2017.03.004
91. Sun GY, Xu J, Jensen MD, Simonyi A. Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. J Lipid Res. (2004) 45:205–13. doi: 10.1194/jlr.R300016-JLR200
92. Lopez-Vales R, Ghasemlou N, Redensek A, Kerr BJ, Barbayianni E, Antonopoulou G, et al. Phospholipase A2 superfamily members play divergent roles after spinal cord injury. FASEB J. (2011) 25:4240–52. doi: 10.1096/fj.11-183186
93. Palavicini JP, Wang C, Chen L, Hosang K, Wang J, Tomiyama T, et al. Oligomeric amyloid-beta induces MAPK-mediated activation of brain cytosolic and calcium-independent phospholipase A2 in a spatial-specific manner. Acta Neuropathol Commun. (2017) 5:56. doi: 10.1186/s40478-017-0460-6
94. Engel LA, Jing Z, O'brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS ONE (2010) 5:e12897. doi: 10.1371/journal.pone.0012897
95. Klein C, Löchte T, Delamonte SM, Braenne I, Hicks AA, Zschiedrich-Jansen K, et al. PLA2G6mutations and Parkinsonism: long-term follow-up of clinical features and neuropathology. Mov Disord. (2016) 31:1927–9. doi: 10.1002/mds.26814
Keywords: PLA2G6, iPLA2β, PLAN, INAD, ANAD, DP, AREP
Citation: Guo Y, Tang B and Guo J (2018) PLA2G6-Associated Neurodegeneration (PLAN): Review of Clinical Phenotypes and Genotypes. Front. Neurol. 9:1100. doi: 10.3389/fneur.2018.01100
Received: 24 August 2018; Accepted: 03 December 2018;
Published: 18 December 2018.
Edited by:
Jiyan Ma, Van Andel Institute, United StatesReviewed by:
Jian Wang, Huashan Hospital Affiliated to Fudan University, ChinaJennifer Steiner, Van Andel Institute, United States
Copyright © 2018 Guo, Tang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-feng Guo, Z3VvamlmZW5nMjAwM0AxNjMuY29t