 Chiara Mandoj
Chiara Mandoj Luigi Tomao
Luigi Tomao Laura Conti
Laura Conti- 1Clinical Pathology, IRCCS Regina Elena National Cancer Institute, Rome, Italy
- 2Department of Hematology/Oncology, IRCCS Bambino Gesù Children Hospital, Rome, Italy
Cancer patients commonly show abnormal laboratory coagulation tests, indicating a subclinical hypercoagulable condition that contribute to morbidity and mortality. The hypercoagulation status not only increases the risk of thromboembolic events but also influences the tumor biology promoting its growth and progression by stimulating intracellular signaling pathways. Recent molecular studies characterized the role of oncogene and suppressor gene in activating clotting pathways, as an integral feature of the neoplastic transformation. It is now clear how haemostatic processes, activated by cancer cells harboring oncogenic mutations, rely on the molecular profile of a particular malignancy, an aspect particularly evident in the differential coagulome profiles showed by different molecular subtypes of brain tumors, such as glioblastoma and medulloblastoma. This review focuses on the biological and clinical aspects of haemostasis in cancer with particular regard on brain tumors.
Introduction
Thromboembolic events are well-recognized complication of malignant disease and are associated with significant mortality and reduced survival. Very often, at the same extent, an “idiopathic” venous thrombosis can exacerbate a latent cancer not diagnosed until then. The first evidence of an higher risk to develop a venous thromboembolism (VTE) in cancer patients was documented initially in 1865 by Armand Trousseau, who first reported the risk of migratory thrombophlebitis in occult malignancy (Trousseau's syndrome) (1). In 1878, instead, Theodor Billroth confirmed the role of VTE in the metastatic process demonstrating the presence of tumor cells in patient-derived thrombi (2).
VTE occurs in the 4–20% of cancer patients, but its frequency reach the 50% when also postmortem examinations are considered; however, approximately the 20% of all cases of VTE derives from cancer patients.
The clinical manifestations of haemostasis activation vary from a subclinical asymptomatic hypercoagulable state to manifest thrombosis of the large vessels. The pathogenesis of cancer-associated coagulopathy is complex and involves various mechanisms, however all of them are linked to abnormalities of Virchow's triad: stasis of the blood flow (prolonged hospitalization, mechanical tumor-derived compression of blood vessels); endothelial injury (tumor invasion, central venous catheters insertion, administration of chemotherapic agents, radiotherapy, and growth factors); hypercoagulability (release of tumor-associated procoagulants molecules and cytokines) (3–6).
Pathogenesis of Thrombosis in Cancer
The mechanisms bearing the activation of the haemostatic system in cancer are complex and likely multifactorial, involving some important biological and clinical factors, such as the primary cancer site and the stage or extent of the disease, but also the anticancer treatments, including chemotherapy, hormone therapy, anti-angiogenic therapy, and combination regimens; finally the practice of central venous catheter indwelling and the surgery (7–12).
Thrombotic events occur more frequently in patients with pancreas, lung and colon cancer, as well as in haematologic malignancies and, when adjusted for disease prevalence, in ovary and brain tumors (13–16).
Prothrombotic Mechanisms
Malignant cells display prothrombotic properties and promote the development of a persistent hypercoagulability status by expressing procoagulant and fibrinolytic molecules. Cancer cells also release proinflammatory and proangiogenic cytokines, such as interleukin 1β (IL-1β), tumor necrosis facror α (TNF-α) and vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), respectively. Finally, they surely interact with endothelial cells, leucocytes and platelets through the expression of several adhesion molecules (17–19).
Tumor cells directly activate haemostasis by producing tissue factor (TF), the principal activator of the blood coagulation process, promoting thrombin generation, and facilitating fibrin deposition creating a microenvironment, in solid tumors, that protects cancer cells from the immune system attack (15). Circulating TF can actually be detected in cancer patients plasma samples associated to microparticles (MPs). These are submicrometric vesicles, derived from activated or apoptotic cells, carrying both TF and phosphatidylserine on their surface, that promotes the coagulation process (20–22). Elevated levels of TF-MPs are associated with higher risk of VTE in cancer patients and correlate with D-dimer levels and other biomarkers of coagulation activation (23–27). In addition, a high rate of TF-MPs activity in patients-derived plasma correlates with the diagnostic and prognostic features of pancreatic cancer, revealing the less differentiated and more aggressive forms (28). This suggests that TF-MPs not only may contribute to development of cancer-associated thrombosis but also to tumor progression (27).
TF release, and the following thrombin activation, not only trigger the hemostatic process but also several pathogenic events crucial for cancer progression, such as cell survival and invasion, as well as angiogenesis processes, through the activation of the protease-activated receptor (PARs) proteins, a group of membrane receptor proteins (PAR 1–4), expressed by tumor cells and vascular cells, that mediate cell activation via G protein pathways (29–32). The cytoplasmic domain of TF, in fact, participate in propagating the intracellular signaling initiated by PARs activation, affecting cancer cell behavior. In particular, the TF/factor VII (FVII) complex proteolytically activates PAR-2 and the formed TF-FVII-PAR-2 complex guides the expression of tumor metastasis by modulating genes that regulate cell survival and migration, inducing, at the same time, the release of pro-angiogenic factors, such as CXCL1, VEGF, and IL-8. Likewise, the interaction of thrombin with PAR-1, PAR-3, and PAR-4 triggers several signaling pathways in cancer cells ending with the overexpression of many angiogenesis-related genes, like bFGF, TF, VEGF, VEGF receptor (VEGFR), and metalloproteinase 2(MMP-2) (Figure 1) (33, 34). Another way of angiogenesis activation resides in the ability of cancer cells, as well as other cells of the tumor microenvironment (e.g., the pericytes), to express the NG2 proteoglycan, which promotes β-1 integrin signaling activation and the downstream FAK and PI3K/Akt pathways. Several studies, in fact, demonstrated that NG2 expression by tumor and stroma cells contributes to tumor progression and correlates with malignancy, triggering cancer aggressiveness and the generation of new tumor-associated blood vessels (35–37).
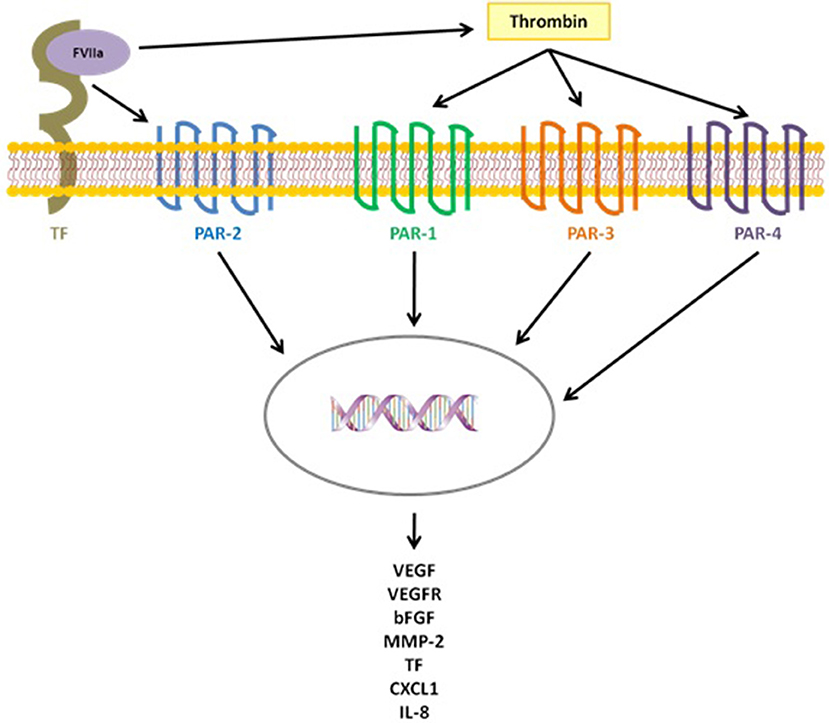
Figure 1. TF triggers several pathogenic events, crucial for cancer progression, both directly, activating PAR-2 after binding FVIIa, and indirectly activating the coagulation cascade with thrombin generation and the following activation of PAR-1,3,4.
Tumor cells, at the same time, can synthesize all of the components of the fibrinolytic system, such as urokinase-type plasminogen activator (uPA) and its receptor (uPAR), tissue-type plasminogen activator (tPA) and plasminogen activator inhibitors 1 (PAI-1) and 2 (PAI-2). Several studies demonstrated that these proteins may facilitate tumor invasion and metastasis, laying the foundation for their use as potential biomarkers, being detectable both in plasma samples and in histological sections. It is actually well known, in vivo, that high levels of uPA promote matrix degradation and cancer progression and that high levels of PAI-1 expression, in particular in breast cancer, is associated with poor prognosis. At the same time a poor prognosis was also found in patients with breast, ovarian and colorectal cancer that showed high pre-operative plasma levels of uPAR and PAI-1 (38–41).
Tumor cells can also drive platelet activation (inducing an overexpression of CD62P) and aggregation, directly by cell-cell adhesion, indirectly by releasing activation factors (such as ADP and protease) and by inducing thrombin generation via coagulation activation. Circulating platelets contain several angiogenesis-regulating agents (such as VEGF, PDGF, bFGF, EGF, HGF, IGF1, TGFβ) that are released upon their activation. The interaction between tumor cells and platelets, as well as leukocytes and endothelial cells, during the metastatic progression occur thanks to the expression of different integrins (i.e., E, P, and L) that facilitate cancer cell adhesion, diapedesis, and the formation of the metastatic lesions (42–44).
Both tumor cells, through the activation of prothrombotic pathways, and the host cell, through their ability to trigger the inflammatory response, take part in the pathogenesis of cancer-related thrombosis, revealing the tight interrelation between inflammation and haemostasis, two overlapping processes that notably affect each other (45, 46).
Oncogenic Pathways and the Coagulant Phenotype in Cancer Cells
The role of cancer-derived hemostatic system deregulation in tumor progression and metastasization is well-characterized. Cancer cells, with their altered gene profile, promotes a selective advantage for their own survival in the tumor niche, as, thanks to a procoagulant activity, they are able to induce the formation of fibrin scaffolds in which the potential to anchor and invade is promoted. At the same time, coagulation proteins recognized from cancer cell receptors can induce the propagation of intracellular signals favoring tumor growth and progression. Until now coagulopathy thrombosis and Trousseau syndrome have been considered as “unspecific” consequences of cancer related disruption of tissue anatomy and vascular continuity, representing the very first element of tissue response to injury.
Recent molecular studies demonstrated that neoplastic transformation, induced by oncogene and suppressor gene mutations, may activate clotting pathways, both in vitro and in vivo (47–51). For example it has been reported, in colorectal cancer, that mutations of K-ras and p53 (associated to p53 loss of function) are associated with a high TF expression. Moreover, an association between circulating MP-TF activity levels and the mutational status of cancer cells was found (47, 52). In the same way a TF upregulation was found in squamous cell carcinoma (SCC) and glioblastoma multiforme (GBM), especially when mutations of the epidermal growth factor receptor (EGFR) and loss of E-cadherin occur (48, 53). It was then demonstrated that in cancer cells a higher EGFR expression, together with the overexpression of the EGFR variant III (EGFRvIII), trigger the TF expression. On the other side, when phosphatase and tensin homolog (PTEN) is restored in these cells, causing the inhibition of the phosphatidylinositol 3-phosphate kinase (PI3PK) and mitogen-activated protein kinase (MAPK) pathways, a downregulation of the EGFR-dependent TF expression was found (48, 54). In a mouse model of sporadic tumorigenesis, instead, the activation of the oncogene MET brought to the generation of spontaneous multifocal hepatocellular carcinoma (HCC), together with a lethal thrombohemorrhagic syndrome as a consequence of cyclooxygenase-2 (COX-2) and PAI-1 up-regulation, since some clinical symptoms got milder when treatment with their inhibitors was performed (49, 55).
These findings suggested that specific cancer cell phenotypes may affect the coagulation system, that the deregulation of haemostasis in tumors microenvironment is not “unspecific” and that the activation of oncogenes (such as EGFR, MET, or RAS) and the inactivation of tumor suppressor genes (such as PTEN or p53) directly affect the expression of hemostasis-controlling genes (50, 51). Other studies pointed out how oncogenic mutations and non-coding RNAs (e.g., microRNAs) can cooperate with hypoxia and cellular differentiation to control the expression of several proteins of the coagulation system, such as TF, PAR-1 and PAR-2, FII and FVII, as well as molecules of the fibrinolytic system and platelet activation (56) (Figure 2).
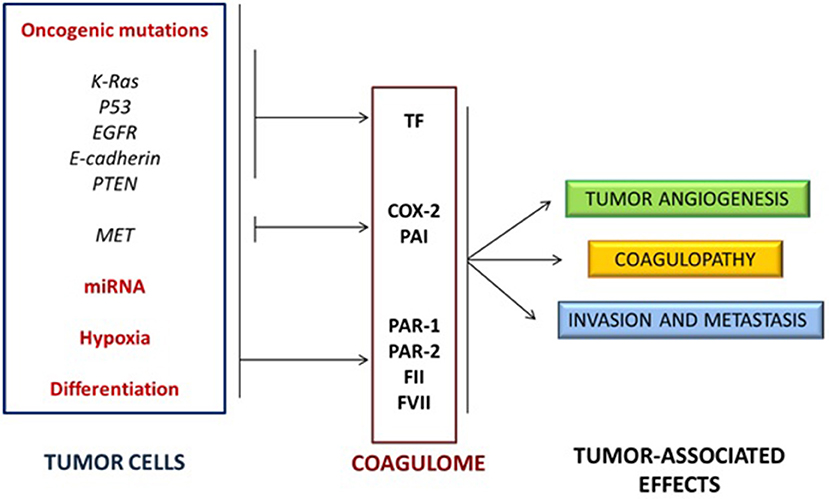
Figure 2. Activation of oncogenes and inactivation of tumor suppressor genes cooperate with non-coding RNA expression, hypoxia and cellular differentiation to control the expression of several proteins of the coagulation/fibrinolytic system and platelet activation.
One of the most fascinating theory in this field focuses on the differential coagulome profiles expressed by different tumor subtypes, such as medulloblastoma (MB), GBM, and other tumors, trying to reveal the possible linkage between tumorigenesis and specific procoagulant phenotypes expressed by cancer cells (57). GBM is the most lethal type of primary brain tumor and is associated with florid angiogenesis, thrombotic complications and up-regulation of TF. In the last few years emerged that different molecular subtypes of GBM (i.e., proneural, neural, classical, and mesenchymal) also showed specific coagulomes features. In the classical GBM, for example, cancer cells overexpress the TF, showing an important procoagulant phenotype, hypothetically driven by the expression of the oncogenic EGFR and its mutant form EGFRvIII. Classical GBM cells, actually, do not show only TF overexpression, but also higher levels of PAR-1 and PAR-2, as well as an ectopic synthesis of FVII. A particular study, on the other side, elegantly demonstrated that the overexpression of TF and the procoagulant activity of GBM cells, after the inactivation of PTEN, are triggered only under hypoxic conditions or together with the EGFRvIII expression, demonstrating how the activation of specific oncogenic pathways, rather than individual mutations, may drive tumor cells to express a particular procoagulant phenotype (48, 53, 54).
The proneural subtype of GBM, a tumor often bearing isocitrate dehydrogenase 1 (IDH1) mutations, is instead associated with lower TF expression. IDH1-mutated cancer cells produce high levels of D-2-hydroxyglutarate (D-2-HG), a molecule that rapidly inhibit platelet aggregation and the related blood clotting events, in a calcium-dependent way (58). It is interesting to notice, in this case, that patients with mutated IDH1 do not suffer from thrombotic events, while the 26–30% of wild-type patients develop a VTE, indicating that the IDH1 mutated form give an antithrombotic potential to GBM cells (59). In addition to this, mutant IDH1 is associated with lower PI3K signaling and podoplanin (PDPN) expression (60). PDPN is a sialomucin-like glycoprotein that binds C-type lectin receptor type-2 (CLEC-2) and causes platelet aggregation. A recent study showed that PDPN expression in GBM is associated with risk of VTE and mortality (61).
Finally, in the mesenchymal form of GBM, in which a pronounced inflammatory status was found, TF is downregulated, while high levels of PAI-1, uPA, uPAR, EPCR, and Thrombomodulin were observed. All these data indicate that GBM cancer cells, characterized by a particular mutational status, may induce the activation of the coagulation system, influencing angiogenic processes and impacting tumor progression (62–64).
Similar studies have been conducted on other types of brain tumors. Pediatric brain malignancies, for example, are not procoagulant tumors, with the MB representing the most common and aggressive one. Molecular classification of MB distinguishes four disease subgroups, including those showing abnormalities in the wingless pathway (WNT) or in the sonic hedgehog pathway (SHH), along with the group 3 (G3) and 4 (G4). Among them, G3 accounts for patients with the poorest prognosis, especially when the amplification of the proto-oncogene MYC is found. Analyzing the coagulation status in MB, a TF overexpression was found only in the SHH subgroup, in association with a higher expression of MET and with altered responsiveness to thrombin and changes in angiogenic and inflammatory factors (63, 65, 66). SSH tumors are highly vascular but do not show an association with systemic thrombosis, confirming that the coagulant phenothype can contribute and influence the tumor biology in different ways.
The sequence of oncogenic mutations related to the different stages of the disease (i.e., pre-malignant, indolent and aggressive) seems to be also correlated with the vascular abnormalities that induce the neoangiogenesis process (62, 64). Oncogenic pathways, in fact, influence the recruitment of inflammatory cells which may exhibit proangiogenic and procoagulant phenotypes, inducing the expression of TF not only in malignant cells but also ectopically in tissue that normally do not produce coagulation factors. These factors modulate the biogenesis of procoagulant extracellular vesicles (EVs) and TF-MPs that, entering the general circulation, promote the metastatic spread (25, 67, 68).
In 1984, instead, a particular study focused on the association between head injuries and the incidence of GBM, concluding that some traumas may increase the risk to develop brain tumors (69). Karpatkin was the first that postulated that thrombin may trigger growth in dormant cancer cells awaken by tissue injury (e.g., head trauma) or the following repair processes (i.e., clotting and inflammation) (70). Certain human GBM cell lines, in fact, exhibit a permanent dormant phenotype, when injected in mice, expressing low TF levels and procoagulant activity. When a concomitant expression of EGFRvIII occur, however, a higher expression of TF is found, together with a highly procoagulant and more aggressive phenotype. Interestingly, inducing high TF levels by transfection, in absence of EGFRvIII, also interrupted the dormant state but in a delayed and less aggressive manner. These observations suggested that TF could play a role in regulating cellular properties inducing changes in the tumor microenvironment, linked to an epigenetic evolution of tumor cells that switch from a “dormant” to a malignant phenotype, not only expressing a procoagulant behavior (51).
Conclusions
Malignant disease is characterized by a hypercoagulable state and several data have improved our knowledge about the relationship between tumor and thromboembolic risk, intimately linked to the processes of tumor growth, progression and metastasis. Haemostatic mechanisms are activated by cancer cells harboring oncogenic mutations in a way that seems to be dependent from the tumor molecular subtype. Markers of coagulation activation, such as cross-linked fibrin degradation products (D-dimer), prothrombin fragment 1+2 (PF1+2), Thombin-antithrombin complex (TAT), von Willebrand factor (vWF), circulating procoagulant MPs, and the manifestation of thrombosic events are associated with tumor progression and prognosis (71–73). Genetic strategies targeting TF, thrombin, FVII and PARs have been successfully used as modulators of tumor pathogenesis and progression, suggesting that deregulation of the hemostatic system may influence tumor microenvironment in several ways. In this scenario it would be of great importance to develop risk assessment models to predict thrombosis events in cancer, in order to identify high-risk patients and lead to the definition of target-specific treatment for both cancer and thrombosis.
Author Contributions
LC and CM elaboration of the review. LT analysis of the literature and revision of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Trousseau A. Phlegmasia alba dolens. Clinique Medicale de l'Hotel-Dieu de Paris. Vol. 3. Paris: JB Ballie‘re et Fils (1865). p. 654–712.
2. Billroth T. Lectures on Surgical Pathology and Therapeutics: A Handbook for Students and Practictioners, 8th ed. London: The New Sydenham Society (1878).
3. Rickles FR, Levine MN. Epidemiology of thrombosis in cancer. Acta Haematol. (2001) 106:6–12. doi: 10.1159/000046583
4. Rickles FR. Mechanisms of cancer-induced thrombosis in cancer. Pathophysiol Haemost Thromb. (2006) 35:103–10. doi: 10.1159/000093551
5. Beleva E, Grudeva-Popova J. From Virchow's triad to metastasis: circulating hemostatic factors as predictors of risk for metastasis in solid tumors. J BUON. (2013) 18:25–33.
6. Byrnes JR, Wolberg AS. New findings on venous thrombogenesis. Hamostaseologie. (2017) 37:25–35. doi: 10.5482/HAMO-16-09-0034
7. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol. (2005) 6:401–10. doi: 10.1016/S1470-2045(05)70207-2
8. Rickles FR, Falanga A Molecular basis for the relationship between thrombosis and cancer. Thromb Res. (2001) 102:V215–24. doi: 10.1016/S0049-3848(01)00285-7
9. Levine MN, Lee AY, Kakkar AK. From Trousseau to targeted therapy: new insights and innovations in thrombosis and cancer. J Thromb Haemost. (2003) 1:1456–63. doi: 10.1046/j.1538-7836.2003.00275.x
10. Gaddh M, Antun A, Yamada K, Gupta P, Tran H, El Rassi F, et al. Venous access catheter-related thrombosis in patients with cancer. Leuk Lymphoma. (2014) 55:501–8. doi: 10.3109/10428194.2013.813503
11. Heidegger I, Porres D, Veek N, Heidenreich A, Pfister D. Predictive factors for developing venous thrombosis during cisplatin-based chemotherapy in testicular cancer. Urol Int. (2017) 99:104–9. doi: 10.1159/000471888
12. Lin HF, Liao KF, Chang CM, Lin CL, Lai SW, Hsu CY. Correlation of the tamoxifen use with the increased risk of deep vein thrombosis and pulmonary embolism in elderly women with breast cancer: a case-control study. Medicine. (2018) 97:e12842. doi: 10.1097/MD.0000000000012842
13. Lee AY, Levine MN. Venous thromboembolism and cancer: risks and outcomes. Circulation. (2003) 107:I17–21. doi: 10.1161/01.CIR.0000078466.72504.AC
14. Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost. (2013) 11:223–33. doi: 10.1111/jth.12075
15. Falanga A, Marchetti M, Russo L. The mechanisms of cancer-associated thrombosis. Thromb Res. (2015) 135:S8–11. doi: 10.1016/S0049-3848(15)50432-5
16. Ikushima S, Ono R, Fukuda K, Sakayori M, Awano N, Kondo K. Trousseau's syndrome: cancer-associated thrombosis. JPN J Clin Oncol. (2016) 46:204–8. doi: 10.1093/jjco/hyv165
17. Winter PC. The Pathogenesis of venous thromboembolism in cancer: emerging links with tumour biology. Hematol Oncol. (2006) 24:126–33. doi: 10.1002/hon.785
18. Wang LM, Duan QL, Yi XH, Zeng Y, Gong Z, Yang F. Venous thromboembolism is a product in proliferation of cancer cells. Int J Clin Exp Med. (2014) 7:1319–23. eCollection 2014.
19. Prodger A, Saha P, Smith A, Evans CE. Cancer-associated thrombosis: regulatory mechanisms and emerging directions. Adv Exp Med Biol. (2017) 906:115–22. doi: 10.1007/5584_2016_110
20. Falanga A, Tartari CJ, Marchetti M. Microparticles in tumor progression. Thromb Res. (2012) 129:S132–6. doi: 10.1016/S0049-3848(12)70033-6
21. Nomura S, Niki M, Nisizawa T, Tamaki T, Shimizu M. Microparticles as biomarkers of blood coagulation in cancer. Biomark Cancer. (2015) 7:51–6. doi: 10.4137/BIC.S30347
22. Date K, Ettelaie C, Maraveyas A. Tissue factor-bearing microparticles and inflammation: a potential mechanism for the development of venous thromboembolism in cancer. J Thromb Haemost. (2017)15:2289–99. doi: 10.1111/jth.13871
23. Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S. Microparticle associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost. (2007) 5:520–7. doi: 10.1111/j.1538-7836.2007.02369.x
24. Sartori MT, Della Puppa A, Ballin A, Saggiorato G, Bernardi D, Padoan A, et al. Prothrombotic state in glioblastoma multiforme: an evaluation of the procoagulant activity of circulating microparticles. J Neurooncol. (2011) 104:225–31. doi: 10.1007/s11060-010-0462-8
25. Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, et al. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. (2009) 15:6830–40. doi: 10.1158/1078-0432.CCR-09-0371
26. Thaler J, Ay C, Mackman N, Bertina RM, Kaider A, Marosi C, et al. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastic, colorectal and brain cancer patients. J. Thromb Haemost. (2012) 10:1363–70. doi: 10.1111/j.1538-7836.2012.04754.x
27. Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood. (2013) 122:1873–80. doi: 10.1182/blood-2013-04-460139
28. Bharthuar A, Khorana AA, Hutson A, Wang JG, Key NS, Mackman N, et al. Circulating microparticle tissue factor, thromboembolism and survival in pancreaticobiliary cancers. Thromb Res. (2013) 132:180–4. doi: 10.1016/j.thromres.2013.06.026
29. Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F. Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost. (2011) 9:306–15. doi: 10.1111/j.1538-7836.2011.04318.x
30. Lima LG, Monteiro RQ. Activation of blood coagulation in cancer: implications for tumour progression. Biosci Rep. (2013) 33:e00064. doi: 10.1042/BSR20130057
31. Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med. (2017) 9:eaaf5294. doi: 10.1126/scitranslmed.aaf5294
32. Rickles FR, Patierno S, Fernandez PM. Tissue factor, thrombin, and cancer. Chest. (2003) 124:58S−68. doi: 10.1378/chest.124.3_suppl.58S
33. Garnier D, Milsom C, Magnus N, Meehan B, Weitz J, Yu J, et al. Role of the tissue factor pathway in the biology of tumor initiating cells. Thromb Res. (2010) 125(Suppl. 2):S44–50. doi: 10.1016/S0049-3848(10)70012-8
34. Chanakira A, Westmark PR, Ong IM, Sheehan JP. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol Oncol. (2017) 145:167–75. doi: 10.1016/j.ygyno.2017.01.022
35. Wang J, Svendsen A, Kmiecik J, Immervoll H, Skaftnesmo KO, Planagumà J, et al. Targeting the NG2/CSPG4 proteoglycan retards tumour growth and angiogenesis in preclinical models of GBM and melanoma. PLoS ONE. (2011) 6:e23062. doi: 10.1371/journal.pone.0023062
36. You WK, Yotsumoto F, Sakimura K, Adams RH, Stallcup WB. NG2 proteoglycan promotes tumor vascularization via integrin-dependent effects on pericyte function. Angiogenesis. (2014) 17:61–76. doi: 10.1007/s10456-013-9378-1
37. Stallcup WB, You WK, Kucharova K, Cejudo-Martin P, Yotsumoto F. NG2 proteoglycan-dependent contributions of pericytes and macrophages to brain tumor vascularization and progression. Microcirculation. (2016) 23:122–33. doi: 10.1111/micc.12251
38. Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. (2003) 22:205–22. doi: 10.1023/A:1023099415940
39. McMahon GA, Petitclerc E, Stefansson S, Smith E, Wong MK, Westrick RJ, et al. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. J Biol Chem. (2001) 276:33964–8. doi: 10.1074/jbc.M105980200
40. Kwaan HC, Mazar AP, McMahon BJ. The apparent uPA/PAI-1 paradox in cancer: more than meets the eye. Semin Thromb Hemost. (2013) 39:382–91. doi: 10.1055/s-0033-1338127
41. Madunić J. The urokinase plasminogen activator system in human cancers: an overview of its prognostic and predictive role. Thromb Haemost. (2018) 118:2020–36. doi: 10.1055/s-0038-1675399
42. Mannori G, Crottet P, Cecconi O, Hanasaki K, Aruffo A, Nelson RM, et al. Differential colon cancer cell adhesion to E-, P-, and L-selectin: role of mucin-type glycoproteins. Cancer Res. (1995) 55:4425–31.
43. Hamidi H, Pietilä M, Ivaska J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br J Cancer. (2016) 115:1017–23. doi: 10.1038/bjc.2016.312
44. Meikle CK, Kelly CA, Garg P, Wuescher LM, Ali RA, Worth RG. Cancer and thrombosis: the platelet perspective. Front Cell Dev Biol. (2017) 4:147. doi: 10.3389/fcell.2016.00147
45. Margetic S. Inflammation and haemostasis. Biochem Med. (2012) 22:49–62. doi: 10.11613/BM.2012.006
46. Abdol Razak NB, Jones G, Bhandari M, Berndt MC, Metharom P. Cancer-associated thrombosis: an overview of mechanisms, risk factors, and treatment. Cancers. (2018) 10:E380. doi: 10.3390/cancers10100380
47. Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, et al. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. (2005) 105:1734–41. doi: 10.1182/blood-2004-05-2042
48. Rong Y, Post DE, Pieper RO, Durden DL, Van Meir EG, Brat DJ. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. (2005) 65:1406–13. doi: 10.1158/0008-5472.CAN-04-3376
49. Boccaccio C, Sabatino G, Medico E, Girolami F, Follenzi A, Reato G, et al. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature. (2005) 434:396–400. doi: 10.1038/nature03357
50. Rak J, Yu JL, Luyendyk J, Mackman N. Oncogenes, trousseau syndrome, and cancer-related changes in the coagulome of mice and humans. Cancer Res. (2006) 66:10643–6. doi: 10.1158/0008-5472.CAN-06-2350
51. Magnus N, D'Asti E, Meehan B, Garnier D, Rak J. Oncogenes and the coagulation system–forces that modulate dormant and aggressive states in cancer. Thromb Res. (2014) 133(Suppl.2):S1–9. doi: 10.1016/S0049-3848(14)50001-1
52. Magnus N, Garnier D, Meehan B, McGraw S, Lee TH, Caron M, et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci USA. (2014) 111:3544–9. doi: 10.1073/pnas.1314118111
53. Monteiro RQ, Lima LG, Gonçalves NP, DE Souza MR, Leal AC, Demasi MA, et al. Hypoxia regulates the expression of tissue factor pathway signaling elements in a rat glioma model. Oncol Lett. (2016) 12:315–22. doi: 10.3892/ol.2016.4593
54. Magnus N, Meehan B, Garnier D, Hashemi M, Montermini L, Lee TH, et al. The contribution of tumor and host tissue factor expression to oncogene-driven gliomagenesis. Biochem Biophys Res Commun. (2014) 454:262–8. doi: 10.1016/j.bbrc.2014.10.041
55. Boccaccio C, Comoglio PM. Genetic link between cancer and thrombosis. J Clin Oncol. (2009) 27:4827–33. doi: 10.1200/JCO.2009.22.7199
56. D'Asti E, Rak J. Biological basis of personalized anticoagulation in cancer: oncogene and oncomir networks as putative regulators of coagulopathy. Thromb Res. (2016) 140(Suppl.1):S37–43. doi: 10.1016/S0049-3848(16)30096-2
57. D'Asti E, Fang Y, Rak J. Brain neoplasms and coagulation-lessons from heterogeneity. Rambam Maimonides Med J. (2014) 5:e0030. doi: 10.5041/RMMJ.10164
58. Unruh D, Schwarze SR, Khoury L, Thomas C, Wu M, Chen L, et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol. (2016) 132:917–30. doi: 10.1007/s00401-016-1620-7
59. Cote DJ, Smith TR. Venous thromboembolism in brain tumor patients. J Clin Neurosci. (2016) 25:13–8. doi: 10.1016/j.jocn.2015.05.053
60. Birner P, Pusch S, Christov C, Mihaylova S, Toumangelova-Uzeir K, Natchev S, et al. Mutant IDH1 inhibits PI3K/Akt signaling in human glioma. Cancer. (2014) 120:2440–7. doi: 10.1002/cncr.28732
61. Riedl J, Preusser M, Nazari PMS, Posch F, Panzer S, Marosi C, et al. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. (2017) 129:1831–9. doi: 10.1182/blood-2016-06-720714
62. Rak J, Klement G. Impact of oncogenes and tumor suppressor genes on deregulation of hemostasis and angiogenesis in cancer. Cancer Metastasis Rev. (2000) 19:93–6. doi: 10.1023/A:1026516920119
63. Magnus N, D'Asti E, Garnier D, Meehan B, Rak J. Brain neoplasms and coagulation. Semin Thromb Hemost. (2013) 39:881–95. doi: 10.1055/s-0033-1357483
64. Tawil N, Chennakrishnaiah S, Bassawon R, Johnson R, D'Asti E, Rak J. Single cell coagulomes as constituents of the oncogene-driven coagulant phenotype in brain tumours. Thromb Res. (2018) 164(Suppl. 1):S136–42. doi: 10.1016/j.thromres.2018.01.021
65. Rak J, Milsom C, May L, Klement P, Yu J. Tissue factor in cancer and angiogenesis: the molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin Thromb Hemost. (2006) 32:54–70. doi: 10.1055/s-2006-933341
66. D'Asti E, Magnus N, Meehan B, Garnier D, Rak J. Genetic basis of thrombosis in cancer. Semin Thromb Hemost. (2014) 40: 284–95. doi: 10.1055/s-0034-1370766
67. Garnier D, Magnus N, Lee TH, Bentley V, Meehan B, Milsom C, et al. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J Biol Chem. (2012) 287:4356–72. doi: 10.1074/jbc.M112.401760
68. Gopal SK, Greening DW, Rai A, Chen M, Xu R, Shafiq A, et al. Extracellular vesicles: their role in cancer biology and epithelial-mesenchymal transition. Biochem J. (2017) 474:21–45. doi: 10.1042/BCJ20160006
69. Hochberg F, Toniolo P, Cole P. Head trauma and seizures as risk factors of glioblastoma. Neurology. (1984) 34: 1511–4. doi: 10.1212/WNL.34.11.1511
70. Karpatkin S, Does hypercoagulability awaken dormant tumor cells in the host? J. Thromb Haemost. (2014) 2:2103–6. doi: 10.1111/j.1538-7836.2004.01003.x
71. Ay C, Vormittag R, Dunkler D, Simanek R, Chiriac AL, Drach J, et al. D-Dimer and prothrombin fragment 12+2 predict venous thromboembolism in patients with cancer: results from the Vienna Cancer and Thrombosis Study. J Clin Oncol. (2009) 27:4124–9. doi: 10.1200/JCO.2008.21.7752
72. Pace A, Mandoj C, Antenucci A, Villani V, Sperduti I, Casini B, et al. A predictive value of von Willebrand Factor for early response to Bevacizumab Therapy in recurrent glioma. J Neurooncol. (2018) 138:527–35. doi: 10.1007/s11060-018-2820-x
Keywords: thromboembolism, glioblastoma, tissue factor, oncogenes, TF microparticles
Citation: Mandoj C, Tomao L and Conti L (2019) Coagulation in Brain Tumors: Biological Basis and Clinical Implications. Front. Neurol. 10:181. doi: 10.3389/fneur.2019.00181
Received: 14 December 2018; Accepted: 13 February 2019;
Published: 18 March 2019.
Edited by:
Sandro M. Krieg, Technische Universität München, GermanyReviewed by:
Ulises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, SpainJanusz Rak, McGill University, Canada
Vance Girard Nielsen, University of Arizona, United States
Copyright © 2019 Mandoj, Tomao and Conti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Conti, bGF1cmEuY29udGlAaWZvLmdvdi5pdA==