 Siobhan E. Kirk
Siobhan E. Kirk Timothy J. Tracey
Timothy J. Tracey Frederik J. Steyn
Frederik J. Steyn Shyuan T. Ngo
Shyuan T. Ngo- 1The Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD, Australia
- 2Centre for Clinical Research, The University of Queensland, Brisbane, QLD, Australia
- 3Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the deterioration of motor neurons. However, this complex disease extends beyond the boundaries of the central nervous system, with metabolic alterations being observed at the systemic and cellular level. While the number of studies that assess the role and impact of metabolic perturbations in ALS is rapidly increasing, the use of metabolism biomarkers in ALS remains largely underinvestigated. In this review, we discuss current and potential metabolism biomarkers in the context of ALS. Of those for which data does exist, there is limited insight provided by individual markers, with specificity for disease, and lack of reproducibility and efficacy in informing prognosis being the largest drawbacks. However, given the array of metabolic markers available, the potential exists for a panel of metabolism biomarkers, which may complement other current biomarkers (including neurophysiology, imaging, as well as CSF, blood and urine markers) to overturn these limitations and give rise to new diagnostic and prognostic indicators.
Overview
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease caused by the death of motor neurons in the brain and spinal cord. The loss of neuronal input leads to progressive paralysis and patient mortality within 2–5 years from diagnosis (1). ALS likely arises from a combination of genetic susceptibility and environmental exposures (2, 3), although it is recognized that ALS is a complex, multi-system disease (4, 5).
Given the complex and heterogeneous nature of ALS, diagnosis and tracking of prognosis remains difficult. Current diagnostic criteria typically follow tests to rule out other pathological causes of symptoms and include: indicators of upper and lower motor neuron involvement, nerve conduction tests, electromyography and “watchful waiting” (4). As a result, researchers have attempted to utilize a wide range of biomarkers—observable biological measurements that confirm the presence or progression of a change in body status, as a means of diagnosing and following disease progression. While the current range of biomarkers in ALS offer some diagnostic and prognostic benefit, there is a need to identify a biomarker that satisfies the following six attributes: specificity to disease; reproducibility; appearance early in the disease; stability across the diurnal period; independence of dietary status and behavior; and a notable change during disease progression. By meeting these criteria, a biomarker can be used to reliably identify and track disease progression, in a manner that can easily be reproduced in a clinical setting.
Metabolic perturbations occur in ALS patients and in mouse models of the disease; both at the systemic and cellular level (6, 7). Clinically, an increase in resting energy expenditure (REE) and decline in body mass index (BMI) is linked to worse outcome (8–10), suggesting prognostic potential in metabolic biomarkers. Given that changes in metabolic status are generally reflected in overall body weight, body composition, and tissue/cellular metabolic function, metabolic changes at the anthropometric, tissue and cellular levels may represent appreciable metabolism biomarkers of ALS onset, progression, and/or severity (Figure 1). A list of the potential biomarkers of metabolism in ALS, and their quality relative to the aforementioned identifying attributes are summarized in Table 1.
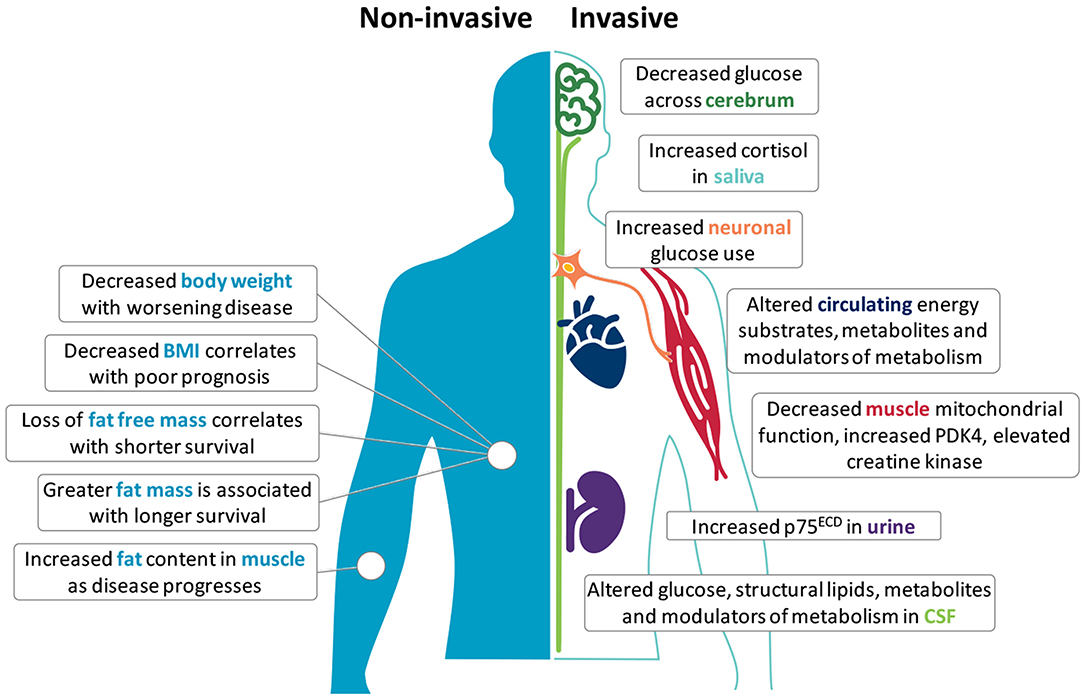
Figure 1. Potential metabolism biomarkers in amyotrophic lateral sclerosis (ALS). Metabolic alterations in ALS offer opportunities to use metabolism biomarkers for the diagnosis, categorization, and tracking of disease. Non-invasive anthropometric measures include body weight, body mass index (BMI), fat free mass, fat mass, and fat distribution. Invasive measures include the use of F18-PET to assess glucose metabolism in the central nervous system, or require the sampling of saliva, blood, cerebrospinal fluid (CSF), muscle tissue, and urine. Although few independent markers are specific, reproducible or able to track disease in ALS, used together with complementary biomarkers (including neurophysiology and imaging), these markers may provide deeper insights into metabolic perturbations that are potentially involved in the onset and progression of disease.
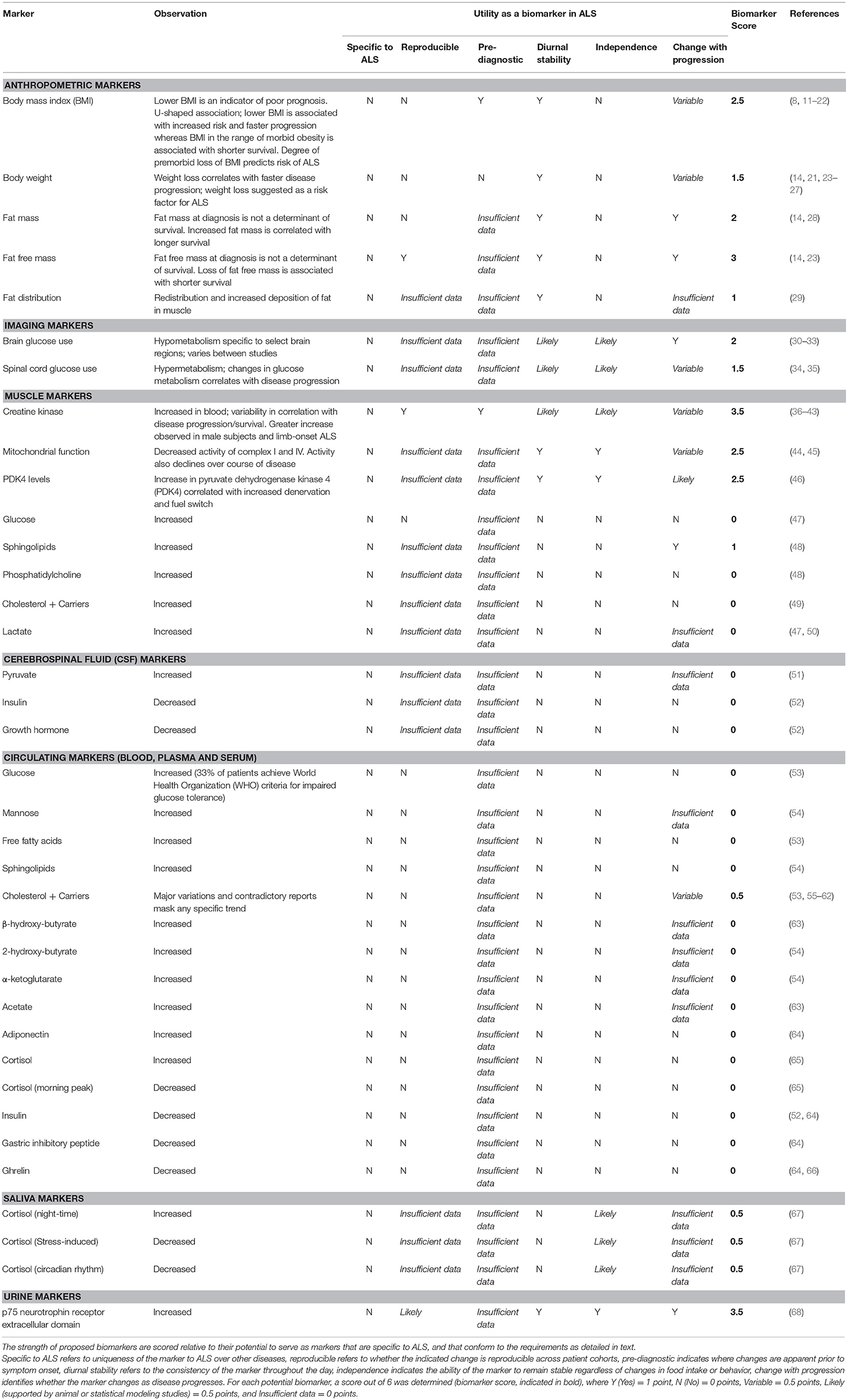
Table 1. Classification of potential biomarkers of metabolism in amyotrophic lateral sclerosis (ALS).
Anthropometric Body Measures
Lower premorbid BMI is associated with increased risk for ALS (11–13), and the degree of decline in premorbid BMI predicts ALS risk and survival (14, 15). Lower BMI, or a decline in BMI following diagnosis correlates with worse survival (16, 17), although this association is not always observed (18, 19, 23, 24). Rather, the mortality risk for ALS relative to BMI exists as a U-shaped curve, in which mortality decreases with increasing BMI, until BMI levels indicate premorbid obesity. Thereafter, mortality risk increases again (8, 20). This seemingly complex association could be explained by changes in body composition throughout disease progression.
BMI is often used as an indirect measure of fatness. However, conventional anthropometric measures of BMI and body adiposity index (BAI) do not always accurately reflect changes in fat and/or fat free mass (FFM) in ALS (69). In this regard, fat mass (FM) and FFM at diagnosis are not associated with survival risk (14), yet redistribution of adipose tissue does occur in ALS (29), and visceral fat is correlated with functional status and survival (28). Moreover, serial assessment of body FM indicates that increases in FM are associated with longer survival (14). While a decrease in FFM serves as an independent prognostic factor for shorter survival in ALS (23), we did not identify any studies that document progressive changes in muscle mass as a potential marker of disease progression in ALS. As a hallmark of ALS, however, there is potential to use the loss of FFM as a marker of disease progression. Such measures must consider the technical difficulties associated with assessing FFM in patients who experience significant and progressive disability, while also accounting for whole body and regional changes in FFM, which differ greatly between patients.
Despite BMI and BAI being poor predictors of body composition in ALS, changes in BMI may offer reliable measures for progressive changes in the overall nutritional status of the patient, and by proxy, disease progression. As documented by Kasarskis et al. a progressive decline in body weight is commonly observed in ALS patients in the months prior to death, and this reduction in body weight or BMI likely reflects a state of undernutrition (25). In recent years, lower BMI has been found to be associated with lower ALSFRS-R scores (70), and a loss of body weight (14, 21, 23, 24, 26, 27, 71) and BMI (14, 17, 22, 24) throughout disease course is consistently associated with shorter survival. Not surprisingly, these observations, while serving as markers for disease progression, have resulted in the adoption of interventions aimed at slowing weight loss in ALS (72).
Skeletal Muscle Pathology
With findings suggesting that FFM is a prognostic factor in ALS (23), analysis of skeletal muscle, the primary component of FFM, may offer insights into tissue-specific metabolism biomarkers. Assessment of cellular metabolic changes in skeletal muscle can be challenging, especially when weighing the clinical benefit against that of an invasive procedure on a patient undergoing significant muscle wasting. Furthermore, heterogeneity in site of disease onset leads to variable muscle pathophysiology between patients (73).
Despite these limitations, creatine kinase, an enzyme that is linked with muscle damage and deterioration, has been studied intensely in ALS. While not strictly a metabolic marker, creatine kinase can be considered as an important modulator of body composition (74). As such, it may indirectly influence systemic metabolic processes. Numerous reports of increased creatine kinase in ALS (36–43), and particularly in limb-onset patients (38, 43), highlight the potential for its use as a marker of disease. However, contradictory observations of associations between creatine kinase and clinical parameters of disease, and disease progression and survival attest to the need for further investigations into determining the utility of creatine kinase as a biomarker in ALS.
Mitochondrial Dysfunction
In human ALS muscle, mitochondrial defects including dysregulation of respiratory complex I (44), decreased respiratory complex I and IV activity (45, 75), decreased muscle mitochondrial protein expression (75) and upregulation of muscular mitochondrial uncoupling protein 3 (76) indicate that impairments in mitochondrial function could serve as a metabolic marker of ALS. It should be noted, however, that these studies were unable to correlate mitochondrial defects with functional parameters of disease progression, despite studies in animal models reporting a strong relationship between the two (77–79). Therefore, while there is clear evidence of mitochondrial defects in ALS, mitochondrial defects per se cannot currently be used as a biomarker due to the difficulty in both easily observing these defects in a clinical setting, and linking such defects to a marker of disease progression and/or survival. Instead, emphasis could be placed on the assessment of the more easily detectable metabolites that drive mitochondrial function.
Glucose Metabolism
Glucose use in the brain of ALS patients has been evaluated using fluorodeoxyglucose F18 positron emission tomography (F18-PET) (30–33). These studies have identified decreased glucose use in the primary motor cortex of ALS patients, suggesting that this brain region is hypometabolic (32). Other studies have reported a decrease in the use of glucose across other brain regions (31, 33); although this may reflect the differences in experimental cohorts. In this regard, Claassen et al. investigated a cohort of patients with primary lateral sclerosis, while the study by Ludolph et al. evaluated ALS patients with both upper and lower motor symptoms. Given that the degree of cerebral hypometabolism in ALS is correlated with the duration of clinically-identified symptoms (30), the ability of the motor cortex to utilize glucose may allow for monitoring of disease progression. However, since brain glucose hypometabolism is not specific to ALS (80), its use as a diagnostic/prognostic marker is limited.
F18-PET has also been used to assess the uptake and utilization of glucose in the cervical spinal cords of ALS patients (34, 35, 81). Overall, observations of spinal cord glucose hypermetabolism (34, 35, 81) is congruent with increased levels of glucose in the CSF of ALS patients (47). In a study by Yamashita et al. glucose hypermetabolism on the ipsilateral side to the patient's symptoms was found to be positively correlated with ALSFRS-R, suggesting that changes in spinal cord glucose metabolism are specific to the affected corticospinal tract and the degree of disease severity (35). By contrast, the study by Marini et al. reported spinal cord glucose hypermetabolism independent of disease duration and functional impairment (34). As such, the degree of glucose use in the spinal cord may present some use for diagnostic testing, but provides limited insights for evaluation of disease progression and prognosis. Indeed, glucose hypermetabolism in the spinal cord extends to other neurological conditions (82, 83), thereby limiting its use as a specific biomarker for ALS. Finally, as the reproducibility of F18-PET in both the brain and spinal cord is low (84), more rigorous testing is required to determine if results are consistent across a heterogeneous ALS population.
Alterations in glucose metabolism in ALS extend beyond the central nervous system (CNS). Glucose tolerance tests conducted by Pradat et al. indicate that ALS patients have a significant increase in blood glucose levels following the provision of a glucose load when compared to age- and sex-matched controls. Within ALS patients, a degree of heterogeneity was observed, with 33% of participants meeting World Health Organization criteria for impaired glucose tolerance (53). Impaired glucose tolerance is in line with reports of insulin resistance in ALS (85), and could explain observations of increased expression of pyruvate dehydrogenase kinase 4 (PDK4) in skeletal muscle of ALS patients (46). Similarly, mannose, an epimer of glucose that has recently been shown to be a predictor of insulin resistance (86), has been reported to be significantly increased in the plasma of ALS patients (54). While the assessment of glucose tolerance and insulin resistance is relatively straightforward, these tests lack reproducibility and specificity to ALS (87–89). Therefore, although glucose metabolism is altered in ALS, it cannot be used as an independent biomarker for ALS diagnosis and prognosis.
Fatty Acids and Ketones
In patients with ALS, the resting level of circulating free fatty acids (FFAs) is significantly increased (53). While higher levels of FFAs has been linked to impaired glucose tolerance in ALS, it has not been shown to be correlated with any markers of disease progression or severity. Ketones, including β-hydroxy-butyrate (63) and 2-hydroxy-butyrate and α-ketoglutarate (54), which are produced through fatty acid metabolism under fasting conditions, are also significantly increased in ALS. Similar to FFAs, no correlations have been observed between disease status and the expression of ketones. Thus, FFAs and ketones cannot currently be considered as reliable biomarkers for ALS, and the lack of specificity for ALS-centric pathology indicate that they may not present as particularly valuable diagnostic markers individually.
Downstream Metabolites
Metabolites, the downstream indicators of metabolic function, are also impacted in ALS. While not specific to ALS, altered expression of metabolites may offer a potential avenue for biomarker discovery. In line with disease heterogeneity, reported levels of metabolites in the blood and CSF are variable. Notably, the levels of lactate (47, 50) and pyruvate (51) in the CNS are increased, potentially reflecting an increase in metabolic output, or increased release of metabolites into the CSF following neuronal deterioration. Given that mitochondrial dysfunction is observed in ALS, further evaluation of the ratio between these metabolites may hold significant informative value in ALS due to the diagnostic value of this test for mitochondrial disorders (90).
Blood levels of acetate are increased in ALS (63), although this is not readily observed in the CSF (47, 51). Acetate is a key metabolite in the oxidation of fatty acids. As acetate synthesis precedes the formation of citric acid in the Krebs cycle, changes in circulating acetate may occur due to excess production via an increase in fatty acid oxidation, increased release from deteriorating muscle cells, or other disruptions to mitochondrial membrane integrity (e.g., due to the presence of free radicals). Such potential mechanisms align with ALS pathology. As a whole, downstream metabolites hold promise as potential biomarkers, and further work that can interrogate relationships between metabolites and clinical parameters of disease would add merit to their use as metabolic biomarkers of disease.
Endocrine Modulators of Metabolism
Insulin is an anabolic hormone that has been reported to be decreased in the blood (64) and CSF (52) of ALS patients. By contrast, other studies have reported no significant differences in plasma insulin levels in ALS patients (91, 92). Other anabolic hormones that have been found to be decreased in ALS include growth hormone (in CSF and blood) (52, 92–94) and gastric inhibitory peptide in blood (64). Conversely, hormones that promote catabolism, such as cortisol (65, 67), and adiponectin (64) are increased or dysregulated in saliva and blood of patients with ALS. Furthermore, ghrelin, an important modulator of appetite, is also reduced in the plasma/blood of ALS patients (64, 66). Given that alterations in these hormones are likely to be symbolic of a change in metabolic function/homeostasis, studies that confirm a link between endocrine markers of metabolism and clinical markers of disease offer potential for their development as prognostic biomarkers.
Metabolism of Structural Lipids
While fatty acids and their derivatives serve as energy substrates through mitochondrial respiration, they also play an essential role in maintaining cellular integrity. Phospholipids, particularly phosphatidylcholine, are significantly increased in the CSF of ALS patients (48). Sphingolipids, such as stearoyl sphingomyelin and ceramide, are also increased in patient blood (48, 54). Interestingly, in the study by Blasco et al. predictions of clinical measurements, such as ALSFRS-R, were found to be correlated to CSF sphingomyelins and triglycerides with long-chain fatty acids (48). Such findings are favorable for the development of biomarker assays, but further tests are required to confirm the reliability of predictive models, before use as a prognostic biomarker.
An increase in cholesterol esters has been observed in ALS patient spinal cord (95). However, cholesterol and its carriers prove to be more difficult to characterize, with variable levels of HDL and LDL cholesterol being reported in ALS. In a population-based longitudinal study, a positive association was found between LDL cholesterol and ALS risk (55), however, there was no indication of the impact of LDL on disease progression or mortality. Nonetheless, this could serve as a diagnostic biomarker for ALS risk. Previously, higher levels of cholesterol, LDL, as well as an elevated LDL/HDL ratio in ALS patient blood have been correlated with increased survival (56–58). Conversely, similar increases in total cholesterol, LDL, and HDL cholesterol in ALS patient blood (59, 60) and CSF (49) have not been found to be correlated with disease progression. Furthermore, a small number of studies contradict these findings, reporting that cholesterol, LDL, and HDL levels do not vary between ALS patients and controls (53, 61, 62), although lower levels of serum lipids may correlate with worse respiratory function (61). Based on these contradictory observations, the validity of cholesterol as a biomarker remains uncertain. Further studies that address these disparate data are required.
Novel Metabolism-associated Biomarkers
p75 neurotrophin receptor (NTR) belongs to the tumor necrosis factor family of receptors. It is a transmembrane receptor which binds neurotrophins and pro-neurotrophins (96). p75NTR has been implicated in processes of energy expenditure (97), glucose uptake, and insulin sensitivity (98). In ALS, the secretion of the extracellular domain of p75NTR (p75ECD) in urine was recently established as a biomarker for disease progression and prognosis (68, 99). Urinary p75ECD increases as disease progresses, and an elevation of urinary p75ECD is observed alongside a decrease in ALSFRS-R scores (68). While it is not clear if increases in urinary p75ECD in ALS match metabolic derangements that accompany disease progression (such as changes in energy metabolism, glucose uptake and insulin sensitivity), the introduction of p75ECD as a fluid biomarker in ALS provides an opportunity for the evaluation and possible co-development of metabolism-associated biomarkers.
Conclusion
The complexity and heterogeneity of disease between patients limits the scope for the use of a single reliable biomarker of ALS. Significant changes in metabolism seen in ALS may represent a potential avenue for biomarker development. As documented in this review, a range of markers might be relevant (Figure 1). However, as investigations into the cause for metabolic derangements in ALS are ongoing, and little emphasis has been placed on the development of metabolism biomarkers as diagnostic or prognostic indicators, few reliable metabolism biomarkers exist (Table 1). Moreover, because metabolic alterations in ALS likely arise from the dysregulation of a number of processes, the utility of biomarkers for assessing early or progressive changes in the metabolic state of ALS patients would necessitate the development of a panel that captures the spectrum of metabolic changes that occur at the systemic and cellular level.
As there is no single biomarker for ALS that sufficiently meets the six major attributes of a biomarker, it is clear that the assessment of biomarkers that cover multiple dimensions of the disease is needed in order to generate a comprehensive view of the state of disease. The complementary assessment of metabolism markers alongside other biomarkers including neurophysiology, imaging, as well as CSF, blood, and urine markers may form a more convincing and reliable diagnostic/prognostic platform, while providing insights into the multifactorial nature of disease.
Author Contributions
SEK, TJT, FJS, and STN conducted the literature search and wrote the manuscript. FJS produced all artwork. STN critically revised the manuscript.
Funding
TJT is supported by an Australian Postgraduate Award from the University of Queensland, and a Postgraduate top-up grant from the Motor Neurone Disease Research Institute of Australia. STN is supported by the Scott Sullivan MND Research Fellowship (MND and Me Foundation, The Royal Brisbane and Women's Hospital Foundation, and the Queensland Brain Institute) and the Australian Institute for Bioengineering and Nanotechnology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. (2007) 369:2031–41. doi: 10.1016/S0140-6736(07)60944-1
2. Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. (2014) 13:1108–13. doi: 10.1016/S1474-4422(14)70219-4
3. Chio A, Mazzini L, D'Alfonso S, Corrado L, Canosa A, Moglia C, et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology. (2018) 91:e635–42. doi: 10.1212/WNL.0000000000005996
4. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. (2011) 377:942–55. doi: 10.1016/S0140-6736(10)61156-7
5. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017) 377:162–72. doi: 10.1056/NEJMra1603471
6. Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. (2004) 101:11159–64. doi: 10.1073/pnas.0402026101
7. Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. (2011) 10:75–82. doi: 10.1016/S1474-4422(10)70224-6
8. Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. (2011) 44:20–4. doi: 10.1002/mus.22114
9. Jesus P, Fayemendy P, Nicol M, Lautrette G, Sourisseau H, Preux PM, et al. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur J Neurol. (2018) 25:97–104. doi: 10.1111/ene.13468
10. Steyn FJ, Ioannides ZA, van Eijk RPA, Heggie S, Thorpe KA, Ceslis A, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry. (2018) 89:1016–23. doi: 10.1136/jnnp-2017-317887
11. Scarmeas N, Shih T, Stern Y, Ottman R, Rowland LP. Premorbid weight, body mass, and varsity athletics in ALS. Neurology. (2002) 59:773–5. doi: 10.1212/WNL.59.5.773
12. Doyle P, Brown A, Beral V, Reeves G, Green J. Incidence of and risk factors for Motor Neurone Disease in UK women: a prospective study. BMC Neurol. (2012) 12:25. doi: 10.1186/1471-2377-12-25
13. O'Reilly EJ, Wang H, Weisskopf MG, Fitzgerald KC, Falcone G, McCullough ML, et al. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2013) 14:205–11. doi: 10.3109/21678421.2012.735240
14. Marin B, Desport JC, Kajeu P, Jesus P, Nicolaud B, Nicol M, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. (2011) 82:628–34. doi: 10.1136/jnnp.2010.211474
15. Mariosa D, Beard JD, Umbach DM, Bellocco R, Keller J, Peters TL, et al. Body mass index and amyotrophic lateral sclerosis: a study of US military veterans. Am J Epidemiol. (2017) 185:362–71. doi: 10.1093/aje/kww140
16. Desport JC, Preux PM, Truong CT, Courat L, Vallat JM, Couratier P. Nutritional assessment and survival in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:91–6. doi: 10.1080/14660820050515386
17. Jawaid A, Murthy SB, Wilson AM, Qureshi SU, Amro MJ, Wheaton M, et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lateral Scler. (2010) 11:542–8. doi: 10.3109/17482968.2010.482592
18. Wolf J, Safer A, Wohrle JC, Palm F, Nix WA, Maschke M, et al. Factors predicting one-year mortality in amyotrophic lateral sclerosis patients—data from a population-based registry. BMC Neurol. (2014) 14:197. doi: 10.1186/s12883-014-0197-9
19. Wolf J, Safer A, Wohrle JC, Palm F, Nix WA, Maschke M, et al. Factors predicting survival in ALS patients—data from a population-based registry in Rhineland-Palatinate, Germany. Neuroepidemiology. (2015) 44:149–55. doi: 10.1159/000381625
20. Reich-Slotky R, Andrews J, Cheng B, Buchsbaum R, Levy D, Kaufmann P, et al. Body mass index (BMI) as predictor of ALSFRS-R score decline in ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. (2013) 14:212–6. doi: 10.3109/21678421.2013.770028
21. Peter RS, Rosenbohm A, Dupuis L, Brehme T, Kassubek J, Rothenbacher D, et al. Life course body mass index and risk and prognosis of amyotrophic lateral sclerosis: results from the ALS registry Swabia. Eur J Epidemiol. (2017) 32:901–8. doi: 10.1007/s10654-017-0318-z
22. Shimizu T, Nagaoka U, Nakayama Y, Kawata A, Kugimoto C, Kuroiwa Y, et al. Reduction rate of body mass index predicts prognosis for survival in amyotrophic lateral sclerosis: a multicenter study in Japan. Amyotroph Lateral Scler. (2012) 13:363–6. doi: 10.3109/17482968.2012.678366
23. Roubeau V, Blasco H, Maillot F, Corcia P, Praline J. Nutritional assessment of amyotrophic lateral sclerosis in routine practice: value of weighing and bioelectrical impedance analysis. Muscle Nerve. (2015) 51:479–84. doi: 10.1002/mus.24419
24. Fasano A, Fini N, Ferraro D, Ferri L, Vinceti M, Errals, et al. Percutaneous endoscopic gastrostomy, body weight loss and survival in amyotrophic lateral sclerosis: a population-based registry study. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:233–42. doi: 10.1080/21678421.2016.1270325
25. Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ. Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am J Clin Nutr. (1996) 63:130–7. doi: 10.1093/ajcn/63.1.130
26. Stambler N, Charatan M, Cedarbaum JM. Prognostic indicators of survival in ALS. ALS CNTF Treatment Study Group Neurology. (1998) 50:66–72. doi: 10.1212/WNL.50.1.66
27. Clavelou P, Blanquet M, Peyrol F, Ouchchane L, Gerbaud L. Rates of progression of weight and forced vital capacity as relevant measurement to adapt Amyotrophic Lateral Sclerosis management for patient result of a French multicentre cohort survey. J Neurol Sci. (2013) 331:126–31. doi: 10.1016/j.jns.2013.06.002
28. Lindauer E, Dupuis L, Muller HP, Neumann H, Ludolph AC, Kassubek J. Adipose tissue distribution predicts survival in amyotrophic lateral sclerosis. PLoS ONE. (2013) 8:e67783. doi: 10.1371/journal.pone.0067783
29. Gerevini S, Agosta F, Riva N, Spinelli EG, Pagani E, Caliendo G, et al. MR imaging of brachial plexus and limb-girdle muscles in patients with amyotrophic lateral sclerosis. Radiology. (2016) 279:553–61. doi: 10.1148/radiol.2015150559
30. Dalakas MC, Hatazawa J, Brooks RA, Di Chiro G. Lowered cerebral glucose utilization in amyotrophic lateral sclerosis. Ann Neurol. (1987) 22:580–6. doi: 10.1002/ana.410220504
31. Ludolph AC, Langen KJ, Regard M, Herzog H, Kemper B, Kuwert T, et al. Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. (1992) 85:81–9. doi: 10.1111/j.1600-0404.1992.tb04003.x
32. Claassen DO, Josephs KA, Peller PJ. The stripe of primary lateral sclerosis: focal primary motor cortex hypometabolism seen on fluorodeoxyglucose F18 positron emission tomography. Arch Neurol. (2010) 67:122–5. doi: 10.1001/archneurol.2009.298
33. Pagani M, Chio A, Valentini MC, Oberg J, Nobili F, Calvo A, et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology. (2014) 83:1067–74. doi: 10.1212/WNL.0000000000000792
34. Marini C, Cistaro A, Campi C, Calvo A, Caponnetto C, Nobili FM, et al. A PET/CT approach to spinal cord metabolism in amyotrophic lateral sclerosis. Eur J Nucl Med Mol Imaging. (2016) 43:2061–71. doi: 10.1007/s00259-016-3440-3
35. Yamashita T, Hatakeyama T, Sato K, Fukui Y, Hishikawa N, Ohta Y, et al. Flow-metabolism uncoupling in the cervical spinal cord of ALS patients. Neurol Sci. (2017) 38:659–65. doi: 10.1007/s10072-017-2823-y
36. Williams ER, Bruford A. Creatine phosphokinase in motor neurone disease. Clin Chim Acta. (1970) 27:53–6. doi: 10.1016/0009-8981(70)90373-6
37. Sinaki M, Mulder DW. Amyotrophic-lateral-sclerosis—relationship between serum creatine-kinase level and patient survival. Arch Phys Med Rehabil. (1986) 67:169–71. doi: 10.1016/0003-9993(86)90064-X
38. Felice KJ, North WA. Creatine kinase values in amyotrophic lateral sclerosis. J Neurol Sci. (1998) 160:S30–2. doi: 10.1016/S0022-510X(98)00195-6
39. Ilzecka J, Stelmasiak Z. Creatine kinase activity in amyotrophic lateral sclerosis patients. Neurol Sci. (2003) 24:286–7. doi: 10.1007/s10072-003-0158-3
40. Lima AF, Evangelista T, de Carvalho M. Increased creatine kinase and spontaneous activity on electromyography, in amyotrophic lateral sclerosis. Electromyogr Clin Neurophysiol. (2003) 43:189–92.
41. Gibson SB, Kasarskis EJ, Hu N, Pulst SM, Mendiondo MS, Matthews DE, et al. Relationship of creatine kinase to body composition, disease state, and longevity in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:473–7. doi: 10.3109/21678421.2015.1062516
42. Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Creatine kinase enzyme level correlates positively with serum creatinine and lean body mass, and is a prognostic factor for survival in amyotrophic lateral sclerosis. Eur J Neurol. (2016) 23:1071–8. doi: 10.1111/ene.12995
43. Tai HF, Cui LY, Guan YZ, Liu MS, Li XG, Shen DC, et al. Correlation of creatine kinase levels with clinical features and survival in amyotrophic lateral sclerosis. Front Neurol. (2017) 8:322. doi: 10.3389/fneur.2017.00322
44. Wiedemann FR, Winkler K, Kuznetsov AV, Bartels C, Vielhaber S, Feistner H, et al. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. (1998) 156:65–72. doi: 10.1016/S0022-510X(98)00008-2
45. Echaniz-Laguna A, Zoll J, Ponsot E, N'Guessan B, Tranchant C, Loeffler JP, et al. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp Neurol. (2006) 198:25–30. doi: 10.1016/j.expneurol.2005.07.020
46. Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Grosch S, Henriques A, et al. A metabolic switch towards lipid use in glycolytic muscle is an early pathologic event in a mouse model of Amyotrophic Lateral Sclerosis. EMBO Mol Med. (2015) 7:526–46. doi: 10.15252/emmm.201404433
47. Toczylowska B, Jamrozik Z, Liebert A, Kwiecinski H. NMR-based metabonomics of cerebrospinal fluid applied to amyotrophic lateral sclerosis. Biocybernet Biomed Eng. (2013) 33:21–32. doi: 10.1016/S0208-5216(13)70053-6
48. Blasco H, Veyrat-Durebex C, Bocca C, Patin F, Vourc'h P, Nzoughet JK, et al. Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci Rep. (2017) 7:17652. doi: 10.1038/s41598-017-17389-9
49. Abdel-Khalik J, Yutuc E, Crick PJ, Gustafsson JA, Warner M, Roman G, et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J Lipid Res. (2017) 58:267–78. doi: 10.1194/jlr.P071639
50. Gray E, Larkin JR, Claridge TDW, Talbot K, Sibson NR, Turner MR. The longitudinal cerebrospinal fluid metabolomic profile of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:456–63. doi: 10.3109/21678421.2015.1053490
51. Blasco H, Corcia P, Moreau C, Veau S, Fournier C, Vourc'h P, et al. H-1-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE. (2010) 5:e13223. doi: 10.1371/journal.pone.0013223
52. Bilic E, Rudan I, Kusec V, Zurak N, Delimar D, Zagar M. Comparison of the growth hormone, IGF-1 and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls. Eur J Neurol. (2006) 13:1340–5. doi: 10.1111/j.1468-1331.2006.01503.x
53. Pradat PF, Bruneteau G, Gordon PH, Dupuis L, Bonnefont-Rousselot D, Simon D, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2010) 11:166–71. doi: 10.3109/17482960902822960
54. Lawton KA, Cudkowicz ME, Brown MV, Alexander D, Caffrey R, Wulff JE, et al. Biochemical alterations associated with ALS. Amyotroph Lateral Scler. (2012) 13:110–8. doi: 10.3109/17482968.2011.619197
55. Mariosa D, Hammar N, Malmstrom H, Ingre C, Jungner I, Ye WM, et al. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: a more than 20-year follow-up of the Swedish AMORIS cohort. Ann Neurol. (2017) 81:718–28. doi: 10.1002/ana.24936
56. Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. (2008) 70:1004–9. doi: 10.1212/01.wnl.0000285080.70324.27
57. Dorst J, Kuhnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol. (2011) 258:613–7. doi: 10.1007/s00415-010-5805-z
58. Ikeda K, Hirayama T, Takazawa T, Kawabe K, Iwasaki Y. Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in japanese patients with amyotrophic lateral sclerosis: a cross-sectional study. Intern Med. (2012) 51:1501–8. doi: 10.2169/internalmedicine.51.7465
59. Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: insights from the olesoxime clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:478–84. 10.3109/21678421.2015.1062517
60. Delaye JB, Patin F, Piver E, Bruno C, Vasse M, Vourc'h P, et al. Low IDL-B and high LDL-1 subfraction levels in serum of ALS patients. J Neurol Sci. (2017) 380:124–7. doi: 10.1016/j.jns.2017.07.019
61. Chio A, Calvo A, Ilardi A, Cavallo E, Moglia C, Mutani R, et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology. (2009) 73:1681–5. doi: 10.1212/WNL.0b013e3181c1df1e
62. Huang R, Guo X, Chen X, Zheng Z, Wei Q, Cao B, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: a study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:359–65. doi: 10.3109/21678421.2015.1047454
63. Kumar A, Bala L, Kalita J, Misra UK, Singh RL, Khetrapal CL, et al. Metabolomic analysis of serum by 1H NMR spectroscopy in amyotrophic lateral sclerosis. Clin Chim Acta. (2010) 411:563–7. doi: 10.1016/j.cca.2010.01.016
64. Ngo ST, Steyn FJ, Huang L, Mantovani S, Pfluger CMM, Woodruff TM, et al. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J Neurol Sci. (2015) 357:22–7. doi: 10.1016/j.jns.2015.06.053
65. Spataro R, Volanti P, Vitale F, Meli F, Colletti T, Di Natale A, et al. Plasma cortisol level in amyotrophic lateral sclerosis. J Neurol Sci. (2015) 358:282–6. doi: 10.1016/j.jns.2015.09.011
66. Czell D, Baldinger R, Schneider U, Neuwirth C, Weber M. The role of the SenseWear device and ghrelin for metabolism in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:295–6. doi: 10.3109/21678421.2015.1113299
67. Patacchioli FR, Monnazzi P, Scontrini A, Tremante E, Caridi I, Brunetti E, et al. Adrenal dysregulation in amyotrophic lateral sclerosis. J Endocrinol Invest. (2003) 26:Rc23–5. doi: 10.1007/BF03349149
68. Shepheard SR, Wuu J, Cardoso M, Wiklendt L, Dinning PG, Chataway T, et al. Urinary p75(ECD) A prognostic, disease progression, and pharmacodynamic biomarker in ALS. Neurology. (2017) 88:1137–43. doi: 10.1212/WNL.0000000000003741
69. Ioannides ZA, Steyn FJ, Henderson RD, McCombe PA, Ngo ST. Anthropometric measures are not accurate predictors of fat mass in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:486–91. doi: 10.1080/21678421.2017.1317811
70. Park Y, Park J, Kim Y, Baek H, Kim SH. Association between nutritional status and disease severity using the amyotrophic lateral sclerosis (ALS) functional rating scale in ALS patients. Nutrition. (2015) 31:1362–7. doi: 10.1016/j.nut.2015.05.025
71. Moglia C, Calvo A, Grassano M, Canosa A, Manera U, D'Ovidio F, et al. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry. (2019). doi: 10.1136/jnnp-2018-319611. [Epub ahead of print].
72. Ngo ST, Mi JD, Henderson RD, McCombe PA, Steyn FJ. Exploring targets and therapies for amyotrophic lateral sclerosis: current insights into dietary interventions. Degener Neurol Neuromuscular Dis. (2017) 7:95–108. doi: 10.2147/DNND.S120607
73. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. (2009) 4:3. doi: 10.1186/1750-1172-4-3
74. Saks VA, Ventura-Clapier R, Aliev MK. Metabolic control and metabolic capacity: two aspects of creatine kinase functioning in the cells. Biochim Biophys Acta. (1996) 1274:81–8. doi: 10.1016/0005-2728(96)00011-4
75. Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, et al. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. (2000) 123:1339–48. doi: 10.1093/brain/123.7.1339
76. Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, et al. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J. (2003) 17:2091–3. doi: 10.1096/fj.02-1182fje
77. Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. (2008) 8:425–36. doi: 10.1016/j.cmet.2008.09.002
78. Dupuis L, Gonzalez de Aguilar JL, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE. (2009) 4:e5390. doi: 10.1371/journal.pone.0005390
79. Wong M, Martin LJ. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet. (2010) 19:2284–302. doi: 10.1093/hmg/ddq106
80. Zilberter Y, Zilberter M. The vicious circle of hypometabolism in neurodegenerative diseases: ways and mechanisms of metabolic correction. J Neurosci Res. (2017) 95:2217–35. doi: 10.1002/jnr.24064
81. Marini C, Morbelli S, Cistaro A, Campi C, Caponnetto C, Bauckneht M, et al. Interplay between spinal cord and cerebral cortex metabolism in amyotrophic lateral sclerosis. Brain. (2018) 141:2272–9. doi: 10.1093/brain/awy152
82. Dubey N, Miletich RS, Wasay M, Mechtler LL, Bakshi R. Role of fluorodeoxyglucose positron emission tomography in the diagnosis of neurosarcoidosis. J Neurol Sci. (2002) 205:77–81. doi: 10.1016/S0022-510X(02)00225-3
83. Daulatzai MA. Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer's disease. J Neurosci Res. (2017) 95:943–72. doi: 10.1002/jnr.23777
84. Phelps ME, Huang SC, Hoffman EJ, Selin C, Sokoloff L, Kuhl DE. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Ann Neurol. (1979) 6:371–88. doi: 10.1002/ana.410060502
85. Reyes ET, Perurena OH, Festoff BW, Jorgensen R, Moore WV. Insulin resistance in amyotrophic lateral sclerosis. J Neurol Sci. (1984) 63:317–24. doi: 10.1016/0022-510X(84)90154-0
86. Lee S, Zhang C, Kilicarslan M, Piening BD, Bjornson E, Hallstrom BM, et al. Integrated network analysis reveals an association between plasma mannose levels and insulin resistance. Cell Metab. (2016) 24:172–84. doi: 10.1016/j.cmet.2016.05.026
87. Norhammar A, Tenerz A, Nilsson G, Hamsten A, Efendic S, Ryden L, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet. (2002) 359:2140–4. doi: 10.1016/S0140-6736(02)09089-X
88. Nathan DM, Davidson MB, DeFronzo RA, Heine RJ, Henry RR, Pratley R, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. (2007) 30:753–9. doi: 10.2337/dc07-9920
89. de la Monte SM. Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer's disease. Drugs. (2017) 77:47–65. doi: 10.1007/s40265-016-0674-0
90. De Pinieux G, Chariot P, Ammi-saïd M, Louarn F, Lejonc JL, Astier A, et al. Lipid-lowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol. (1996) 42:333–7. doi: 10.1046/j.1365-125.1996.04178.x
91. Perurena OH, Festoff BW. Reduction in insulin-receptors in amyotrophic-lateral-sclerosis correlates with reduced insulin sensitivity. Neurology. (1987) 37:1375–9. doi: 10.1212/WNL.37.8.1375
92. Pellecchia MT, Pivonello R, Monsurro MR, Trojsi F, Longo K, Piccirillo G, et al. The GH-IGF system in amyotrophic lateral sclerosis: correlations between pituitary GH secretion capacity, insulin-like growth factors and clinical features. Eur J Neurol. (2010) 17:666–71. doi: 10.1111/j.1468-1331.2009.02896.x
93. Morselli LL, Bongioanni P, Genovesi M, Licitra R, Rossi B, Murri L, et al. Growth hormone secretion is impaired in amyotrophic lateral sclerosis. Clin Endocrinol. (2006) 65:385–8. doi: 10.1111/j.1365-2265.2006.02609.x
94. Sacca F, Quarantelli M, Rinaldi C, Tucci T, Piro R, Perrotta G, et al. A randomized controlled clinical trial of growth hormone in amyotrophic lateral sclerosis: clinical, neuroimaging, and hormonal results. J Neurol. (2011) 259:132–8. doi: 10.1007/s00415-011-6146-2
95. Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. (2002) 52:448–57. doi: 10.1002/ana.10312
96. Chao MV. The p75 neurotrophin receptor. J Neurobiol. (1994) 25:1373–85. doi: 10.1002/neu.480251106
97. Baeza-Raja B, Sachs BD, Li PP, Christian F, Vagena E, Davalos D, et al. p75 Neurotrophin receptor regulates energy balance in obesity. Cell Rep. (2016) 14:255–68. doi: 10.1016/j.celrep.2015.12.028
98. Baeza-Raja B, Li P, Le Moan N, Sachs BD, Schachtrup C, Davalos D, et al. p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proc Natl Acad Sci USA. (2012) 109:5838–43. doi: 10.1073/pnas.1103638109
Keywords: amyotrophic lateral sclerosis, ALS, metabolism, biomarker, motor neurone disease
Citation: Kirk SE, Tracey TJ, Steyn FJ and Ngo ST (2019) Biomarkers of Metabolism in Amyotrophic Lateral Sclerosis. Front. Neurol. 10:191. doi: 10.3389/fneur.2019.00191
Received: 13 November 2018; Accepted: 14 February 2019;
Published: 18 March 2019.
Edited by:
Pierre-Francois Pradat, Hôpitaux Universitaires Pitié Salpêtrière, FranceReviewed by:
Mary-Louise Rogers, Flinders University, AustraliaPatrizia Longone, Fondazione Santa Lucia (IRCCS), Italy
Jean-Philippe Loeffler, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2019 Kirk, Tracey, Steyn and Ngo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shyuan T. Ngo, cy5uZ29AdXEuZWR1LmF1