 Stefan Nicolau
Stefan Nicolau Margherita Milone
Margherita Milone- Department of Neurology, Mayo Clinic, Rochester, MN, United States
Congenital myasthenic syndromes (CMS) are a group of inherited disorders of neuromuscular transmission most commonly presenting with early onset fatigable weakness, ptosis, and ophthalmoparesis. CMS are classified according to the localization of the causative molecular defect. CMS with presynaptic dysfunction can be caused by mutations in several different genes, including those involved in acetylcholine synthesis, its packaging into synaptic vesicles, vesicle docking, and release from the presynaptic nerve terminal and neuromuscular junction development and maintenance. Electrodiagnostic testing is key in distinguishing CMS from other neuromuscular disorders with similar clinical features as well as for revealing features pointing to a specific molecular diagnosis. A decremental response on low-frequency repetitive nerve stimulation (RNS) is present in most presynaptic CMS. In CMS with deficits in acetylcholine resynthesis however, a decrement may only appear after conditioning with exercise or high-frequency RNS and characteristically displays a slow recovery. Facilitation occurs in CMS caused by mutations in VAMP1, UNC13A, SYT2, AGRN, LAMA5. By contrast, facilitation is absent in the other presynaptic CMS described to date. An understanding of the underlying molecular mechanisms therefore assists the interpretation of electrodiagnostic findings in patients with suspected CMS.
Introduction
Congenital myasthenic syndromes (CMS) are a heterogeneous group of rare inherited disorders of neuromuscular transmission. Typical clinical features include hypotonia, fatigable weakness, ptosis, and ophthalmoparesis (1, 2). Less frequently, CMS may present with limb girdle weakness (3). Most CMS manifest in the neonatal period or in infancy (4), but adult onset is also possible (5). The pathophysiological mechanisms underlying the different CMS are diverse, including alterations of presynaptic, synaptic or postsynaptic neurotransmission as well as defects of protein glycosylation, endplate development, and maintenance. The majority of CMS result from genetic defects in postsynaptic proteins, with acetylcholine receptor subunits being the most frequently implicated. Presynaptic CMS are much less frequent (6). These are caused by defects in proteins involved in acetylcholine synthesis, its packaging into synaptic vesicles, vesicle docking, and release from the presynaptic nerve terminal (Figure 1). In addition, mutations in genes encoding synaptic space proteins or proteins implicated in neuromuscular synapse development and maintenance can result in presynaptic neuromuscular junction dysfunction (6).
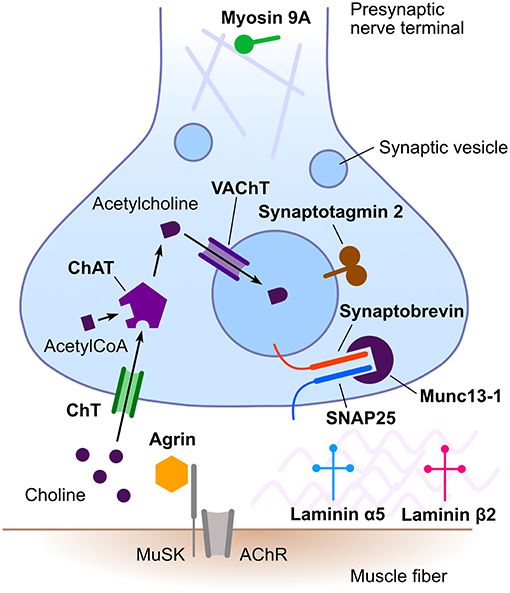
Figure 1. Schematic representation of the neuromuscular junction emphasizing proteins involved in presynaptic congenital myasthenic syndromes. Names in bold designate proteins implicated in presynaptic congenital myasthenic syndromes. AChR, acetylcholine receptor; ChAT, choline acetyltransferase; ChT, high-affinity presynaptic choline transporter; MuSK, muscle-specific kinase; SNAP25, synaptosomal-associated protein 25; VAChT, vesicular acetylcholine transporter. Rabphilin 3A is not illustrated, as its precise role in neuromuscular transmission remains to be established.
Electrodiagnostic testing is a key component of the assessment of patients with a suspected CMS and can help distinguish CMS from other neuromuscular disorders presenting with similar clinical features. Despite their clinical and molecular heterogeneity, CMS are characterized by a compromised safety margin of neuromuscular transmission. Like acquired disorders of the neuromuscular junction, CMS therefore generally demonstrate a decremental response to low-frequency (2–3 Hz) repetitive nerve stimulation (RNS) and increased jitter or blocking on single-fiber electromyography (SFEMG).
In certain presynaptic disorders of neuromuscular transmission, an increased calcium concentration in the presynaptic nerve terminal can overcome a defect in synaptic vesicle release. Such an increase in calcium concentration may be achieved via high-frequency RNS (20–50 Hz) or maximal voluntary muscle contraction and electrophysiologically results in an increase in the compound muscle action potential (CMAP) amplitude. This phenomenon is referred to as facilitation and is most prominent in the autoimmune Lambert-Eaton myasthenic syndrome (LEMS), where ≥100% increment is observed (7). While facilitation has been traditionally considered the hallmark of presynaptic defects of neuromuscular transmission, this finding is absent in many presynaptic CMS. The presence of facilitation depends on the pathophysiological mechanism compromising acetylcholine quantal release (8). The number of quanta released by a nerve impulse (m) represents the product of the number of readily releasable quanta (n) and their probability of release (p). Thus, various factors altering the number of acetylcholine molecules within a vesicle, the pool of readily releasable quanta, or their probability of release can lead to a presynaptic CMS with or without facilitation (Table 1).
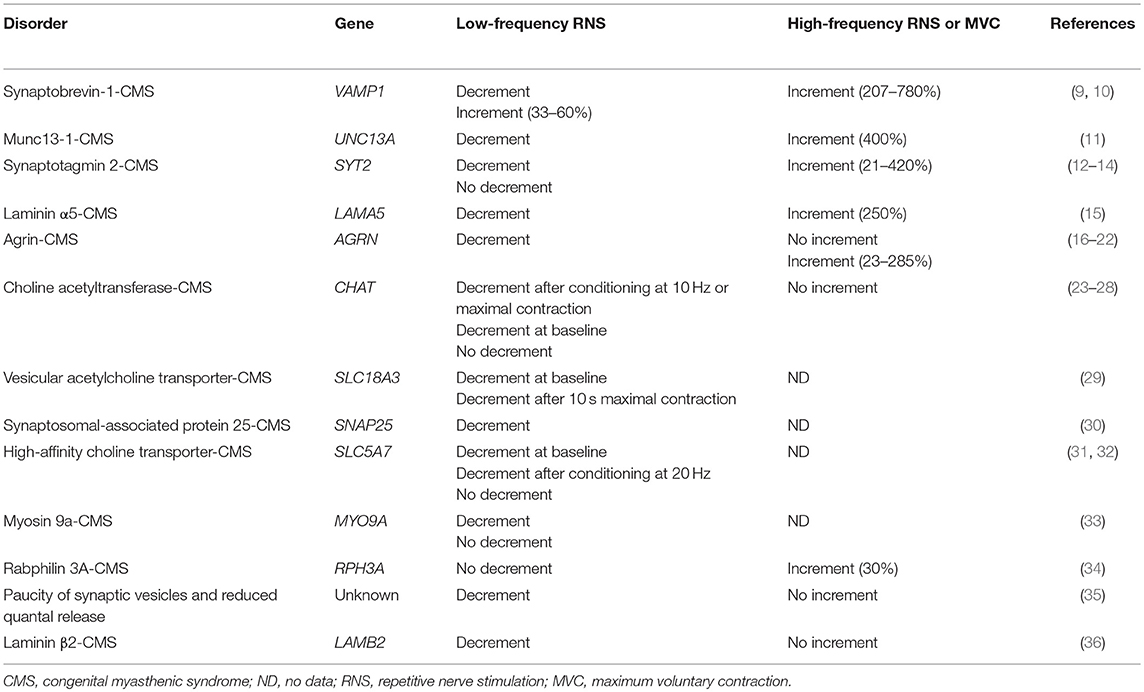
Table 1. Responses to RNS in CMS due to mutations in presynaptic proteins and other proteins affecting presynaptic function.
Herein, we focus on the electrophysiological features of presynaptic CMS and their underlying molecular mechanisms. We particularly aim to highlight how the lack of facilitation may still stem form a presynaptic defect and to increase awareness of presynaptic CMS without facilitation. It should be noted that due to the extreme rarity of many of these disorders, it is possible that new cases may emerge with mutations in the same genes, but clinical and electrophysiological phenotypes deviating from previous reports.
Presynaptic CMS With Facilitation
Synaptobrevin-1-CMS
Synaptobrevin-1, also called vescicle-associated membrane protein 1 (VAMP1), is a component of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. The SNARE complex, which additionally consists of synaptosomal-associated protein 25 (SNAP25) and syntaxin, constitutes the core of the synaptic vesicle docking, priming and fusion machinery (37). Vamp1-null mice were found to lack movements due to compromised neuromuscular transmission (38). Homozygous missense or frameshift mutations in VAMP1 were detected in 3 unrelated CMS families (9, 10). Patients presented in the neonatal period with hypotonia, severe weakness, feeding and respiratory difficulties, delayed motor development and contractures. One patient showed joint hyperlaxity. Affected individuals had low CMAP amplitudes and an incremental response (up to 780%) to 20 Hz RNS. One individual had a decremental response to low-frequency RNS, while two others had incremental responses of 33 and 60%. Myopathic abnormalities on needle examination were also reported in several patients. Parameters of quantal release were not studied in these patients. An incremental response to high-frequency RNS was also identified in Vamp1-null mice, supporting the presence of a presynaptic defect of neuromuscular transmission (10). Transfection of mutant synaptobrevin-1 into bovine chromaffin cells significantly decreased depolarization-elicited exocytosis (9). Pyridostigmine was beneficial in most patients.
Munc 13-1-CMS
Munc13-1, encoded by UNC13A, is expressed at the neuromuscular junction, where it facilitates assembly of the SNARE complex (39, 40). Loss of Munc13 in mice prevents docking of synaptic vesicles to active zones (40). Munc13 is also expressed at glutamatergic synapses in the central nervous system (39). A single patient with a CMS due to a homozygous UNC13A non-sense mutation has been reported to date (11). This patient had severe hypotonia at birth and required ventilatory support. In addition, she had cortical hyper-excitability, microcephaly, a thin corpus callosum, contractures, and dysmorphism. She died of respiratory failure at age 50 months (11). Baseline CMAP amplitudes were reduced and a decremental response (20–40%) was observed on low-frequency RNS, while 50 Hz RNS produced an incremental response of up to 400%. In vitro electrophysiological studies demonstrated that the defect of neuromuscular transmission was attributable to reduced quantal content resulting from the low number of readily releasable quanta, while the probability of quantal release was preserved (11). This finding differentiates Munc13-1-CMS from LEMS, in which there is a reduced probability of quantal release (41). Therapy with pyridostigmine and 3,4-diaminopyridine (3,4-DAP) increased CMAP amplitudes, but only the latter improved the patient's symptoms.
Synaptotagmin 2-CMS
Synaptotagmin 2 (SYT2) is expressed on the synaptic vesicle membrane, where it acts as a calcium sensor, interacting with SNAP25 to promote calcium-triggered synaptic vesicle release (42). The role of synaptotagmin-2 at the neuromuscular junction was demonstrated by the reduced evoked neurotransmitter release observed in synaptotagmin 2-deficient mice (42). To date, heterozygous SYT2 missense mutations have been reported to cause CMS in 3 families (12–14). Affected patients had pes cavus, hammertoes, and distal lower limb weakness and atrophy since childhood. In addition, some had proximal weakness and extraocular muscle involvement. Deep tendon reflexes were diminished in all patients at baseline but elicitable after exercise in some. Electrophysiological studies revealed reduced CMAP amplitudes, a decremental response to low frequency RNS in all but one patient, and an incremental response of up to 420% following 10 s of maximum voluntary contraction. The duration of the incremental response was prolonged, lasting up to 60 min (12). No in vitro electrophysiological studies of neuromuscular transmission are available in SYT2-CMS. A drosophila model revealed that the synaptotagmin 2 mutation exerted a dominant negative effect on synaptic transmission by abolishing calcium-triggered neurotransmitter release (13). It was speculated that the facilitation could result from increased calcium concentrations overcoming the decreased calcium-binding affinity of mutated synaptotagmin. The reason for the prolonged post-tetanic facilitation, longer than observed in LEMS and botulism, remains poorly understood. In the reported patients, pyridostigmine provided no clinical benefit. 3,4-DAP resulted in improved motor function but had no impact on CMAP amplitude or the degree of post-exercise facilitation. SFEMG however showed improvement of jitter and blocking during treatment with pyridostigmine and further improvement during treatment with 3,4-DAP (12).
Laminin α5-CMS
Laminins are heterotrimeric extracellular matrix proteins composed of combinations of α, β, and γ subunits. They interact with receptors on the sarcolemmal membrane and serve both a structural and signaling role in axon growth and repair (43). Laminin α5 (LAMA5) is one of the subunits selectively expressed at the neuromuscular junction (44). Mice deficient in laminin α5 displayed delayed neuromuscular junction maturation (45). One patient has been reported with a homozygous LAMA5 mutation leading to a CMS associated with myopia and facial tics. A significant decrement was seen on low-frequency RNS, while 30 s of maximum muscle contraction resulted in a 250% increment of CMAP amplitude (15). In vitro microelectrode recordings revealed a severe reduction of mean endplate potential (EPP) quantal content, which was attributable to a failure of nerve stimulation to elicit EPPs at a majority of neuromuscular junctions. This defect was partially corrected with 3,4-DAP. Ultrastructural studies showed nerve terminals that were smaller than their respective post-synaptic endplates, entirely absent, or encased by Schwann cell projections, similar to the ultrastructural findings observed in acetylcholinesterase deficiency. There was also a moderate reduction of the density of synaptic vesicles. The partial apposition of the nerve terminal to the post-synaptic endplate was reminiscent of the changes seen in early development in laminin α5-deficient mice. While not strictly a presynaptic CMS, laminin α5 deficiency is thus included here due to the occurrence of post-exercise facilitation in this disorder.
Agrin-CMS
Agrin (AGRN) is an extracellular matrix protein crucial for neuromuscular junction development and maintenance (46). It is secreted by the presynaptic nerve terminal and interacts with the postsynaptic LDL Receptor Related Protein 4 (LRP4) receptor. LRP4 subsequently activates muscle-specific kinase (MuSK), inducing aggregation of acetylcholine receptors at the neuromuscular junction (46). Mutations in AGRN have been reported to cause a recessive CMS (16–22). Symptoms manifested in the first or second decade of life. Most patients displayed distal muscle weakness and atrophy, with or without facial weakness, and developed proximal weakness later in the course of the disease (19). Other patients presented with generalized limb weakness (21). All patients showed a decremental response to low-frequency RNS. Post-exercise increment up to 285% was observed in some patients (19) but not in others (17). Intracellular microelectrode studies of neuromuscular transmission were not performed. The mutated agrin exhibited reduced acetylcholine receptor clustering activity and impaired neuromuscular junction maintenance (17, 19, 21). Some patients responded positively to ephedrine and others to salbutamol. Cholinergic drugs were ineffective. Similar to laminin α5, agrin-CMS is included here due to the occurrence of post-exercise facilitation in some patients.
Presynaptic CMS Without Documented Facilitation
Choline Acetyltransferase-CMS
Choline acetyltransferase (CHAT) is the enzyme catalyzing the synthesis of acetylcholine from choline and acetyl-CoA in the presynaptic nerve terminal. Recessive mutations in CHAT cause a CMS with episodic apneas and intermittent exacerbations. This is the most common presynaptic CMS, accounting for 5% of all CMS (6). It was also the first presynaptic CMS characterized at molecular level (23). The apneic spells manifest in infancy or childhood and patients can have minimal or no myasthenic symptoms between spells (24). Different CHAT mutations reduce enzyme activity by reduced expression, altered kinetics, or impaired thermal stability. Some CHAT mutations have an allosteric effect (25). Patients with CHAT-CMS may have a decrement on low frequency RNS. Between episodic exacerbations however, most patients display no decrement at baseline because the nerve terminal contains stores of acetylcholine. In such cases, SFEMG can unmask the defect of neuromuscular transmission. Conditioning with 10 Hz RNS for 5 min can also trigger a decremental response on low-frequency RNS and a drop in CMAP amplitude. While the latter can also be observed in other CMS, including postsynaptic CMS, it is the subsequent slow post-subtetanic recovery of CMAP amplitude over up to 30 min that suggests a defect in acetylcholine resynthesis (26, 47). CHAT-CMS patients display no facilitation on high-frequency RNS. In vitro electrophysiological studies demonstrated miniature endplate potentials (MEPP) of essentially normal amplitude and normal EPP quantal content in rested muscle. Ten Hz RNS for 5 min however reduced the MEPP amplitude by ~50% and resulted in a rapid decrease of EPP amplitude, which returned to baseline over more than 10 min, consistent with insufficient acetylcholine resynthesis (2, 23). In regard to treatment, cholinesterase inhibitors can control crises (27, 28) and some patients may respond to salbutamol or 3,4-DAP as adjunctive therapy, while others do not (27). Monitoring for life-threatening apneas is essential, especially early in life.
Vesicular Acetylcholine Transporter-CMS
The vesicular acetylcholine transporter, encoded by the SLC18A3 gene, is responsible for transporting acetylcholine into synaptic vesicles. Of note, SLC18A3 is located within the first intron of the CHAT gene. The crucial role of the vesicular acetylcholine transporter was demonstrated in Slc18a3 knockout and knockdown mice, which display neuromuscular junction dysfunction and die of respiratory failure or develop weakness responsive to cholinesterase inhibitors (48, 49). Recessive mutations in SLC18A3 were detected in two unrelated individuals with CMS manifesting with apneic crises, ptosis, opthalmoplegia and fatigable weakness (29). One of the two patients had learning difficulties. Low-frequency RNS resulted in a severe decremental response in one patient, while in the other patient, a decremental response was only seen after 10 s of isometric contraction. No facilitation was described. In vitro electrophysiological studies of neuromuscular transmission were not performed. Pyridostigmine partially improved the myasthenic symptoms and addition of 3,4-DAP and ephedrine resulted in further clinical improvement.
Synaptosomal-Associated Protein 25-CMS
The plasma membrane-associated SNAP25 exists in two different isoforms, designated SNAP25A and SNAP25B, each characterized by distinct patterns of cellular and anatomical localization and developmental expression. The more abundant isoform, SNAP25B, is expressed at both central and peripheral nervous system synapses (50, 51). In vitro studies showed that SNAP25 deficiency abolishes vesicle priming and fast calcium-triggered exocytosis (52). A missense mutation in SNAP25B was identified in a patient with a CMS associated with cortical hyperexcitability, ataxia and intellectual disability (30). The patient's symptoms manifested in utero; she was born with contractures and subsequently exhibited motor developmental delay. She displayed a decremental response to low-frequency RNS. In vitro analysis of neuromuscular transmission showed that the quantal content of EPP and the number of readily releasable quanta had an almost bimodal distribution, with some being reduced while others were normal or increased. In addition, the probability of quantal release was reduced to 63% of normal. MEPP frequency was decreased as well, suggesting that the mutation also affects spontaneous vesicle exocytosis. The in vitro electrophysiological finding of a reduced probability of quantal release would predict the presence of facilitation upon tetanic stimulation, but responses to high-frequency RNS or maximum voluntary muscle contraction were not reported. The pathogenic role of the SNAP25 mutation was demonstrated by the compromised calcium-driven SNARE complex assembly observed in the presence of the mutated SNAP25 and by reduced depolarization-evoked exocytosis in bovine chromaffin cells transfected with the mutant SNAP25B. 3,4-DAP improved patient's muscle strength, while pyridostigmine was not beneficial.
High-Affinity Choline Transporter-CMS
Cholinergic neurotransmission is ended by cleavage of acetylcholine by acetylcholinesterase within the synaptic cleft. Choline is then taken up into the nerve terminal by the sodium-dependent high affinity choline transporter solute carrier family 5 member 7 (SLC5A7), also known as choline transporter 1. Choline is used for resynthesis of acetylcholine. The crucial role of the choline transporter in neuromuscular transmission was demonstrated in choline transporter knockout mice, which have breathing abnormalities and die shortly after birth. These mice have loss of spontaneous and evoked responses at the neuromuscular junction as well as developmental abnormalities of the neuromuscular synapse (53). Recessive mutations in SLC5A7 were found to cause a CMS with episodic apneas in 8 patients from 7 unrelated families (31, 32). Most affected patients presented at birth or in infancy with apneas, ophthalmoparesis, ptosis, bulbar weakness, and subsequent motor developmental delay. Two siblings were more severely affected, presenting antenatally with polyhydramnios and arthrogryposis. The more mildly affected patients with episodic apneas had a favorable prognosis. The phenotype observed in this disorder is similar to that caused by mutations in CHAT, which is also involved in acetylcholine resynthesis. Decremental responses to low-frequency RNS were found in 5 of 6 patients, including one in whom a decremental response was only seen after 10 s of conditioning with 20 Hz RNS. No in vitro microelectrode studies of the neuromuscular junction were performed. Expression studies demonstrated that the mutations lead to near-total loss of choline transporter activity. In addition to causing a CMS, dominant mutations in SLC5A7 have been reported to cause distal hereditary motor neuropathy type VII A (DHMN-VII A), which is characterized by vocal cord paresis (54). While RNS in DHMN-VII patients did not reveal a decrement, increased jitter was reported on SFEMG, although this could result from the presence of immature neuromuscular junctions in the setting of reinnervation (55). Meanwhile, no neurogenic changes were described in SLC5A7-CMS patients (31). Most patients with SLC5A7-CMS responded favorably to cholinesterase inhibitors.
Myosin 9A-CMS
Myosin 9A (MYO9A) is an unconventional myosin expressed in the axons of motor neurons and at the neuromuscular junction. Unlike previously-discussed proteins causing presynaptic CMS, myosin 9A is not directly involved in neuromuscular transmission. Unconventional myosins are molecular motors that bind to and move along the actin cytoskeleton, serving functions such as cargo transport, actin organization, and cell motility (56). Three patients have been described with a CMS caused by recessive mutations in MYO9A. All patients had prenatal or neonatal onset of symptoms and presented with hypotonia, ptosis, ophthalmoparesis, bulbar weakness, motor developmental delay, and recurrent respiratory crises, often triggered by infections. Within one affected family, several of the proband's siblings had died of respiratory failure within the first year of life. Cognitive impairment was also present in some patients. The presence of an impairment of neuromuscular transmission was demonstrated by a decremental response to low-frequency RNS in one patient, while the other two were found to have increased jitter on SFEMG. In vitro experiments found that MYO9A knockdown alters neurite extension and branching in cultured neurons, while knockdown in zebrafish leads to aberrant growth of motor neuron axons during neuromuscular junction formation (33). More recent studies in a mouse motor neuron-derived cell line showed that myosin 9A exerts a role in neuronal cytoskeleton maintenance and protein secretion. Myosin 9A mutations were found to result in impaired agrin secretion (57). Pyridostigmine improved symptoms in all 3 patients. In addition, one patient benefited from 3,4-DAP, while 3,4-DAP and fluoxetine precipitated respiratory crises in another.
Rabphilin 3A-CMS
Rabphilin 3A (RPH3A) is a membrane trafficking protein that plays a role in synaptic vesicle docking, re-priming and endocytosis. It has been suggested that rabphilin 3A acts as a regulator of synaptic vesicle release, but its precise functions remain incompletely understood (58, 59). While rabphilin knock-out mice lack any overt phenotype, cultures of hippocampal neurons from these animals demonstrated an accelerated recovery from synaptic depression (60). Compound heterozygous variants in RPH3A were recently identified in one patient with a neurological phenotype of childhood-onset fatigable weakness without extraocular muscle involvement as well as learning disabilities, tremor and ataxia (34). The patient did not demonstrate a decremental response to low-frequency RNS but did have increased jitter on SFEMG and a 30% incremental response to 30 Hz RNS. In vitro intracellular microelectrode studies of neuromuscular transmission were not performed. Ultrastructural studies of the neuromuscular junction showed a decreased density of synaptic vesicles, which had a variable shape. There was also an increase in non-vesicular membranes and degenerative lamellar bodies in the nerve terminals. Expression studies showed that only one of the detected variants impairs the interaction between rabphilin 3A and 14-3-3, a regulator of synaptic transmission. The patient's response to cholinergic drugs was not described. The role of rabphilin 3A in causing CMS therefore requires further investigation.
Paucity of Synaptic Vesicles and Reduced Quantal Release
Walls et al. described a 23-year-old patient with a CMS characterized by infantile onset of fatigue, weakness, ptosis, ophthalmoparesis and recurrent episodes of bulbar dysfunction. The patient responded to cholinesterase inhibitors. Electrodiagnostic testing revealed a decremental response to low-frequency RNS, while high-frequency RNS did not result in an incremental response. In vitro electrophysiological testing demonstrated a reduced EPP quantal content attributable to a reduction in the number of readily releasable quanta to 20% of normal, with a preserved probability of release (35). Electron microscopy confirmed an 80% reduction in synaptic vesicle density. Although the molecular basis of the syndrome was not identified, the ultrastructural and electrophysiological findings showed a paucity of synaptic vesicles, explaining the lack of facilitation on high frequency RNS. It was suggested that the paucity of synaptic vesicles could result from impairment of synaptic vesicle formation, axonal transport or membrane recycling.
Laminin β2-CMS
Laminin β2 (LAMB2) is another laminin subunit expressed at the neuromuscular junction as well as in the glomerular basement membrane and ocular structures. Biallelic loss of function mutations in LAMB2 cause Pierson syndrome, which is characterized by neonatal onset of nephrotic syndrome and ocular abnormalities, often along with hypotonia and developmental delay. The classical phenotype is severe, resulting in death in infancy or early childhood (61), which may limit assessment of neuromuscular involvement. Long-term survival however has been reported in some patients, one of whom displayed a CMS phenotype (36). This patient presented in the neonatal period with nephrosis and episodes of respiratory distress. She displayed motor developmental delay, severe proximal weakness, ptosis and ophthalmoparesis. Electrodiagnostic testing revealed a 24% decrement on low-frequency RNS. Unlike LAMA2-CMS, facilitation was absent and high-frequency RNS worsened the decrement. In vitro microelectrode recordings demonstrated an 82% reduction of EPP quantal content as well as a reduction of MEPP amplitude and frequency. Ultrastructural examination demonstrated a reduced size of presynaptic nerve terminals, which were encased by Schwann cells. Synaptic clefts were widened and invaded by Schwann cell processes. Treatment with cholinesterase inhibitors resulted in a severe exacerbation of weakness requiring ventilatory support, but ephedrine was beneficial. Although laminin β2 is a synaptic space protein, it is listed here because the reduced quantal content suggests the presence of presynaptic dysfunction.
Conclusions
Presynaptic CMS can be caused by mutations in a number of genes with different functions, leading to different electrophysiological findings. Of these, CHAT deficiency is the most common and displays a distinctive electrophysiological phenotype, with a decremental response often absent at baseline but uncovered by conditioning with 10 Hz RNS for 5 min, followed by slow recovery of the CMAP amplitude. This underscores the importance of performing provocative testing to unmask the defect of neuromuscular transmission in patients with a suspected CMS. Other presynaptic CMS are caused by mutations in other proteins involved in acetylcholine resynthesis and packing, or in the SNARE complex and related proteins. In these conditions, the presence of facilitation depends on the mechanism responsible for the compromised quantal release. The absence of facilitation is thus not limited to post-synaptic disorders of neuromuscular transmission. A sound understanding of the pathophysiology of these disorders is therefore key in the interpretation of electrodiagnostic findings in CMS patients.
Author Contributions
SN and MM jointly reviewed the literature and drafted the manuscript.
Funding
MM receives research support through generous gifts from Mayo Clinic benefactors.
Conflict of Interest Statement
MM receives an honorarium as associate editor of Neurology Genetics and an MDA care center grant.
The remaining author declares that the research was conducted in the absence of any competing relationships, whether personal, professional, commercial, financial or otherwise, that could be construed as a potential conflict of interest.
References
1. McMacken G, Abicht A, Evangelista T, Spendiff S, Lochmuller H. The increasing genetic and phenotypical diversity of congenital myasthenic syndromes. Neuropediatrics. (2017) 48:294–308. doi: 10.1055/s-0037-1602832
2. Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. (2015) 14:420–34. doi: 10.1016/S1474-4422(14)70201-7
3. O'Connor E, Topf A, Zahedi RP, Spendiff S, Cox D, Roos A, et al. Clinical and research strategies for limb-girdle congenital myasthenic syndromes. Ann N Y Acad Sci. (2018) 1412:102–12. doi: 10.1111/nyas.13520
4. Shieh PB, Oh SJ. Congenital myasthenic syndromes. Neurol Clin. (2018) 36:367–78. doi: 10.1016/j.ncl.2018.01.007
5. Kao JC, Milone M, Selcen D, Shen XM, Engel AG, Liewluck T. Congenital myasthenic syndromes in adult neurology clinic: a long road to diagnosis and therapy. Neurology. (2018) 91:e1770–e7. doi: 10.1212/WNL.0000000000006478
6. Engel AG. Congenital myasthenic syndromes in 2018. Curr Neurol Neurosci Rep. (2018) 18:46. doi: 10.1007/s11910-018-0852-4
7. Kesner VG, Oh SJ, Dimachkie MM, Barohn RJ. Lambert-eaton myasthenic syndrome. Neurol Clin. (2018) 36:379–94. doi: 10.1016/j.ncl.2018.01.008
8. Lorenzoni PJ, Scola RH, Kay CSK, Werneck LC, Horvath R, Lochmuller H. How to spot congenital myasthenic syndromes resembling the lambert-eaton myasthenic syndrome? a brief review of clinical, electrophysiological, and genetics features. Neuromol Med. (2018) 20:205–14. doi: 10.1007/s12017-018-8490-1
9. Shen XM, Scola RH, Lorenzoni PJ, Kay CS, Werneck LC, Brengman J, et al. Novel synaptobrevin-1 mutation causes fatal congenital myasthenic syndrome. Ann Clin Transl Neurol. (2017) 4:130–8. doi: 10.1002/acn3.387
10. Salpietro V, Lin W, Delle Vedove A, Storbeck M, Liu Y, Efthymiou S, et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann Neurol. (2017) 81:597–603. doi: 10.1002/ana.24905
11. Engel AG, Selcen D, Shen XM, Milone M, Harper CM. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet. (2016) 2:e105. doi: 10.1212/NXG.0000000000000105
12. Whittaker RG, Herrmann DN, Bansagi B, Hasan BA, Lofra RM, Logigian EL, et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology. (2015) 85:1964–71. doi: 10.1212/WNL.0000000000002185
13. Montes-Chinea NI, Guan Z, Coutts M, Vidal C, Courel S, Rebelo AP, et al. Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype. (2018) 4:e282. doi: 10.1212/NXG.0000000000000282
14. Herrmann DN, Horvath R, Sowden JE, Gonzalez M, Sanchez-Mejias A, Guan Z, et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet. (2014) 95:332–9. doi: 10.1016/j.ajhg.2014.09.012
15. Maselli RA, Arredondo J, Vazquez J, Chong JX, University of Washington Center for Mendelian G, Bamshad MJ, et al. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. Am J Med Genet A. (2017) 173:2240–5. doi: 10.1002/ajmg.a.38291
16. Maselli RA, Fernandez JM, Arredondo J, Navarro C, Ngo M, Beeson D, et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. (2012) 131:1123–35. doi: 10.1007/s00439-011-1132-4
17. Huze C, Bauche S, Richard P, Chevessier F, Goillot E, Gaudon K, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. (2009) 85:155–67. doi: 10.1016/j.ajhg.2009.09.010
18. Karakaya M, Ceyhan-Birsoy O, Beggs AH, Topaloglu H. A novel missense mutation in the AGRN gene causing congenital myasthenic syndrome mimicking neck myopathy. Neuromus Disord. (2014) 24:843. doi: 10.1016/j.nmd.2014.06.170
19. Nicole S, Chaouch A, Torbergsen T, Bauche S, de Bruyckere E, Fontenille MJ, et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. (2014) 137:2429–43. doi: 10.1093/brain/awu160
20. Karakaya M, Ceyhan-Birsoy O, Beggs AH, Topaloglu H. A novel missense variant in the AGRN gene; congenital myasthenic syndrome presenting with head drop. J Clin Neuromuscul Dis. (2017) 18:147–51. doi: 10.1097/CND.0000000000000132
21. Xi J, Yan C, Liu WW, Qiao K, Lin J, Tian X, et al. Novel SEA and LG2 Agrin mutations causing congenital myasthenic syndrome. Orphanet J Rare Dis. (2017) 12:182. doi: 10.1186/s13023-017-0732-z
22. Zhang Y, Dai Y, Han JN, Chen ZH, Ling L, Pu CQ, et al. A novel AGRN mutation leads to congenital myasthenic syndrome only affecting limb-girdle muscle. Chin Med J. (2017) 130:2279–82. doi: 10.4103/0366-6999.215332
23. Ohno K, Tsujino A, Brengman JM, Harper CM, Bajzer Z, Udd B, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. (2001) 98:2017–22. doi: 10.1073/pnas.98.4.2017
24. Arredondo J, Lara M, Gospe SM Jr, Mazia CG, Vaccarezza M, Garcia-Erro M, et al. Choline acetyltransferase mutations causing congenital myasthenic syndrome: molecular findings and genotype-phenotype correlations. Hum Mutat. (2015) 36:881–93. doi: 10.1002/humu.22823
25. Shen XM, Crawford TO, Brengman J, Acsadi G, Iannaconne S, Karaca E, et al. Functional consequences and structural interpretation of mutations of human choline acetyltransferase. Hum Mutat. (2011) 32:1259–67. doi: 10.1002/humu.21560
26. Byring RF, Pihko H, Tsujino A, Shen XM, Gustafsson B, Hackman P, et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord. (2002) 12:548–53. doi: 10.1016/S0960-8966(01)00336-4
27. McMacken G, Whittaker RG, Evangelista T, Abicht A, Dusl M, Lochmuller H. Congenital myasthenic syndrome with episodic apnoea: clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J Neurol. (2018) 265:194–203. doi: 10.1007/s00415-017-8689-3
28. Schara U, Christen HJ, Durmus H, Hietala M, Krabetz K, Rodolico C, et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur J Paediatr Neurol. (2010) 14:326–33. doi: 10.1016/j.ejpn.2009.09.009
29. O'Grady GL, Verschuuren C, Yuen M, Webster R, Menezes M, Fock JM, et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology. (2016) 87:1442–8. doi: 10.1212/WNL.0000000000003179
30. Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. (2014) 83:2247–55. doi: 10.1212/WNL.0000000000001079
31. Bauche S, O'Regan S, Azuma Y, Laffargue F, McMacken G, Sternberg D, et al. Impaired presynaptic high-affinity choline transporter causes a congenital myasthenic syndrome with episodic apnea. Am J Hum Genet. (2016) 99:753–61. doi: 10.1016/j.ajhg.2016.06.033
32. Pardal-Fernandez JM, Carrascosa-Romero MC, Alvarez S, Medina-Monzon MC, Caamano MB, de Cabo C. A new severe mutation in the SLC5A7 gene related to congenital myasthenic syndrome type 20. Neuromuscul Disord. (2018) 28:881–4. doi: 10.1016/j.nmd.2018.06.020
33. O'Connor E, Topf A, Muller JS, Cox D, Evangelista T, Colomer J, et al. Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome. Brain. (2016) 139:2143–53. doi: 10.1093/brain/aww130
34. Maselli RA, Vazquez J, Schrumpf L, Arredondo J, Lara M, Strober JB, et al. Presynaptic congenital myasthenic syndrome with altered synaptic vesicle homeostasis linked to compound heterozygous sequence variants in RPH3A. Mol Genet Genomic Med. (2018) 6:434–40. doi: 10.1002/mgg3.370
35. Walls TJ, Engel AG, Nagel AS, Harper CM, Trastek VF. Congenital myasthenic syndrome associated with paucity of synaptic vesicles and reduced quantal release. Ann N Y Acad Sci. (1993) 681:461–8. doi: 10.1111/j.1749-6632.1993.tb22930.x
36. Maselli RA, Ng JJ, Anderson JA, Cagney O, Arredondo J, Williams C, et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet. (2009) 46:203–8. doi: 10.1136/jmg.2008.063693
37. Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. (2013) 80:675–90. doi: 10.1016/j.neuron.2013.10.022
38. Nystuen AM, Schwendinger JK, Sachs AJ, Yang AW, Haider NB. A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics. (2007) 8:1–10. doi: 10.1007/s10048-006-0068-7
39. Rizo J, Xu J. The synaptic vesicle release machinery. Annu Rev Biophys. (2015) 44:339–67. doi: 10.1146/annurev-biophys-060414-034057
40. Siksou L, Varoqueaux F, Pascual O, Triller A, Brose N, Marty S. A common molecular basis for membrane docking and functional priming of synaptic vesicles. Eur J Neurosci. (2009) 30:49–56. doi: 10.1111/j.1460-9568.2009.06811.x
42. Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, et al. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci. (2006) 26:13493–504. doi: 10.1523/JNEUROSCI.3519-06.2006
43. Rogers RS, Nishimune H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. (2017) 57–58:86–105. doi: 10.1016/j.matbio.2016.08.008
44. Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. (1997) 139:1507–21. doi: 10.1083/jcb.139.6.1507
45. Nishimune H, Valdez G, Jarad G, Moulson CL, Muller U, Miner JH, et al. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol. (2008) 182:1201–15. doi: 10.1083/jcb.200805095
46. Burden SJ, Huijbers MG, Remedio L. Fundamental molecules and mechanisms for forming and maintaining neuromuscular synapses. Int J Mol Sci. (2018) 19:e490. doi: 10.3390/ijms19020490
47. Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nat Rev Neurosci. (2003) 4:339–52. doi: 10.1038/nrn1101
48. Prado VF, Martins-Silva C, de Castro BM, Lima RF, Barros DM, Amaral E, et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron. (2006) 51:601–12. doi: 10.1016/j.neuron.2006.08.005
49. De Castro BM, De Jaeger X, Martins-Silva C, Lima RD, Amaral E, Menezes C, et al. The vesicular acetylcholine transporter is required for neuromuscular development and function. Mol Cell Biol. (2009) 29:5238–50. doi: 10.1128/MCB.00245-09
50. Prescott GR, Chamberlain LH. Regional and developmental brain expression patterns of SNAP25 splice variants. BMC Neurosci. (2011) 12:35. doi: 10.1186/1471-2202-12-35
51. Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA. (1995) 92:1510–4. doi: 10.1073/pnas.92.5.1510
52. Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, et al. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. (2003) 114:75–86. doi: 10.1016/S0092-8674(03)00477-X
53. Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci USA. (2004) 101:8762–7. doi: 10.1073/pnas.0401667101
54. Salter CG, Beijer D, Hardy H, Barwick KES, Bower M, Mademan I, et al. Truncating SLC5A7 mutations underlie a spectrum of dominant hereditary motor neuropathies. Neurol Genet. (2018) 4:e222. doi: 10.1212/NXG.0000000000000222
55. Barwick KE, Wright J, Al-Turki S, McEntagart MM, Nair A, Chioza B, et al. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am J Hum Genet. (2012) 91:1103–7. doi: 10.1016/j.ajhg.2012.09.019
56. Hartman MA, Finan D, Sivaramakrishnan S, Spudich JA. Principles of unconventional myosin function and targeting. Annu Rev Cell Dev Biol. (2011) 27:133–55. doi: 10.1146/annurev-cellbio-100809-151502
57. O'Connor E, Phan V, Cordts I, Cairns G, Hettwer S, Cox D, et al. MYO9A deficiency in motor neurons is associated with reduced neuromuscular agrin secretion. Hum Mol Genet. (2018) 27:1434–46. doi: 10.1093/hmg/ddy054
58. Montaville P, Coudevylle N, Radhakrishnan A, Leonov A, Zweckstetter M, Becker S. The PIP2 binding mode of the C2 domains of rabphilin-3A. Protein Sci. (2008) 17:1025–34. doi: 10.1110/ps.073326608
59. Ferrer-Orta C, Perez-Sanchez MD, Coronado-Parra T, Silva C, Lopez-Martinez D, Baltanas-Copado J, et al. Structural characterization of the Rabphilin-3A-SNAP25 interaction. Proc Natl Acad Sci USA. (2017) 114:E5343–E51. doi: 10.1073/pnas.1702542114
60. Deak F, Shin OH, Tang J, Hanson P, Ubach J, Jahn R, et al. Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. (2006) 25:2856–66. doi: 10.1038/sj.emboj.7601165
Keywords: congenital myasthenic syndromes, electromyography, facilitation, neuromuscular junction, repetitive nerve stimulation
Citation: Nicolau S and Milone M (2019) The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms. Front. Neurol. 10:257. doi: 10.3389/fneur.2019.00257
Received: 10 January 2019; Accepted: 26 February 2019;
Published: 19 March 2019.
Edited by:
Massimiliano Filosto, Azienda Socio Sanitaria Territoriale of the Spedali Civili of Brescia, ItalyReviewed by:
Carmelo Rodolico, University of Messina, ItalyLorenzo Maggi, Neurological Institute Foundation Carlo Besta, Italy
Copyright © 2019 Nicolau and Milone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margherita Milone, bWlsb25lLm1hcmdoZXJpdGFAbWF5by5lZHU=