 Niels Hansen1,2*
Niels Hansen1,2*- 1Department of Epileptology, University of Bonn Medical Center, Bonn, Germany
- 2Department of Psychiatry and Psychotherapy, University of Goettingen, Goettingen, Germany
Limbic encephalitis (LE) is an autoimmune disease defined by clinical criteria, such as seizures, psychiatric and in particular working memory abnormalities in conjunction with apparative criteria underlying structural or functional changes in the temporal lobe according to autoimmune encephalitis guidelines (1). Working memory encompasses a transient encoding of information in readiness for further processing within a time window of seconds during cognitive task operations based on neurophysiological mechanisms, such as short-term synaptic facilitation (2). On the contrary, long-term memory (LTM) serves to encode, consolidate, and finally store information for long intervals ranging from minutes to months or even life (3) through cellular mechanisms, such as long-term potentiation (LTP) (4).
In translational transfer experiments of autoimmune encephalitis from humans to the mouse, critical impairment of synaptic LTP in the hippocampus was proved by autoantibodies against the N-methyl-D-aspartate receptor (NMDAR) (5) and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunit GluA2 (6). Synaptic LTP in the hippocampus is considered to be a mechanism of synaptic consolidation (7) serving to enable LTM storage within the hippocampus. Not only antibodies against glutamatergic receptors cause LTM dysfunction probably due to altered synaptic transmission and plasticity—LGI1-LE is also assumed to be based on a synaptic mechanism, as hippocampal AMPA 2 receptors are reduced via neutralization of LGI1-ADAM22 interaction by LGI1-antibodies (8). AMPA receptor reduction is functionally partially equivalent to enhanced AMPAR endocytosis as fewer AMPARs become available for synaptic transmission. LTP might in turn be impaired (9) and LTM formation as well consequently via this suggested process.
On the contrary, the pathophysiology of accelerated long-term forgetting (ALF) is still not well-understood. It occurs often in temporal lobe epilepsy patients (10), has been reported recently in LE, and is even more predominant in LE not associated with autoantibodies (11) (Table 1A). ALF can be assessed by neuropsychological tests assessing long-term free recall entailing a 1-week time period with not derogated free recall after half an hour (11). ALF is believed to be based on a shortage of memory consolidation (23), so that ALF in LE clearly depicts LTM dysfunction. Persistent deficits in memory retrieval (19) and recognition (14), anterograde (21) and retrograde (24), autobiographic (25), visuospatial, verbal, and episodic LTM deficits have been reported in LE patients (15, 19, 20, 22, 26, 27) (for reports on LTM dysfunction in adult LE patients see Table 1A, n ≥ 6). These distinct facets of LTM deficits often occurred in LE patients in association with antibodies against membrane surface antigens, such as voltage-gated potassium channels (VGKC) with its subgroups of leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein-like 2 (CASPR2), AMPARs as well as antibodies against intracellular antigens, such as glutamic acid decarboxylase 65 (GAD65) and Ma/Ta2. The time frame of LTM decline ranged from a month to years (15, 16, 19, 22, 28) and even decades, with dynamic fluctuations in memory capacity over time (18) (Table 1A).
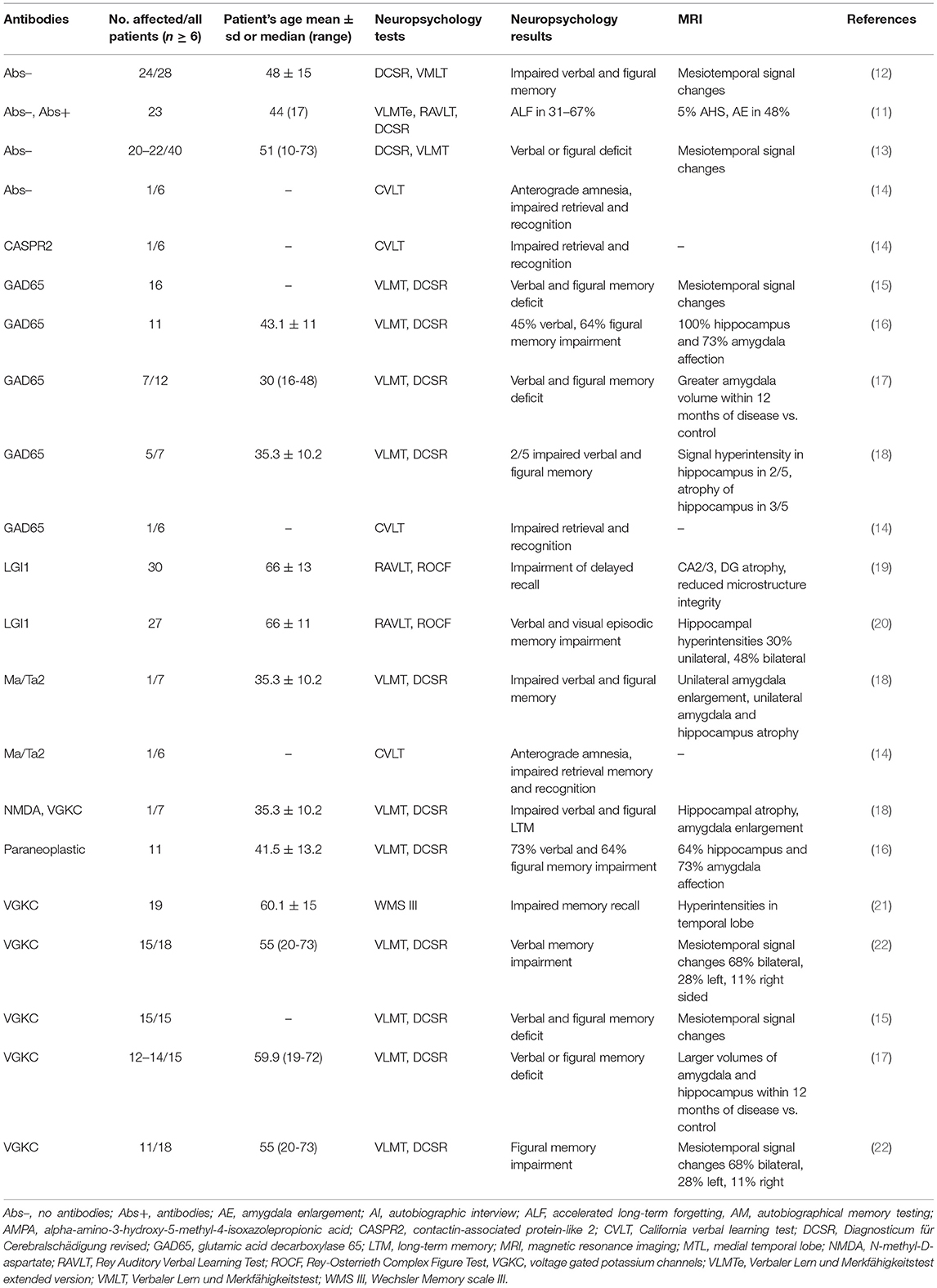
Table 1A. Reports in limbic encephalitis patients on memory including LTM dysfunction.
Functional memory impairment seems to be based on the structural integrity of mesiotemporal brain structures, as memory function is known to correlate with reduced hippocampal subfield volumes, e.g., the CA1-4, dentate gyrus or subiculum in VGKC or paraneoplastic antibody positive-encephalitis (16, 29). LE involves frontal lobe structures (20) also, but the main underlying brain pathology on the macroscopic (17) and microscopic level (involving infiltrating lymphocytes) affects the amygdalohippocampal complex (30) indicating that dysfunctional LTM is more probable than impaired working-memory pathways. In particular, some LE forms are susceptible to LTM dysfunction as their disease-mediating antibodies against membrane receptors, such as AMPA- (31), NMDA- (32), metabotropic glutamate receptor 5 (mGluR5) receptors (33) are critically involved in hippocampal synaptic long-term plasticity and LTM formation (9, 34, 35). It is thus not surprising that some AMPAR-antibodies associated LE patients present with an amnestic syndrome, such as the unique clinical manifestation of autoimmunity (36). Antibody-mediated immunopathology involving distinct memory phenotypes fluctuates (18), but its pathogenic antigen-antibody interaction of glutamatergic receptors often take days to develop functional changes in receptor electrophysiology [neuronal incubation with antibody serum requires days: Ohkawa et al. (8)] and antibody-directed epitopes undergo post-translational changes in protein expression (37), indicating time preconditions to worsen LTM function. LTM dysfunction is not a unique feature of limbic dysfunction induced by autoimmunity, but can also be caused by viral encephalitis, such as herpes simplex encephalitis. The clinical features of viral encephalitis affecting the temporal lobe can resemble those of autoimmune-mediated limbic encephalitis (LE), but frequently start with a more fulminant onset, often with fever or aphasia. The diagnosis of viral encephalitis must be ascertained by detecting viral DNA in the cerebrospinal fluid via a polymerase chain reaction. The type of LTM impairment in herpes simplex encephalitis affecting either verbal memory (pattern a) and/or memory of names (pattern b) (38), and/or memory of living things (pattern c) (39) depends on structural lesions in the temporal lobe [involving the hippocampus (a) (38) or the lateral temporal lobe (b) (38) or antero-medial temporal cortex (c) (39)].
The occurrence of episodic LTM deficits in LE are often not followed by working memory disturbances (14), so that there may be patients suffering from LE who are not registered due to application of the Graus et al. criteria (1). Redefining the memory criteria in LE has been proposed to consider episodic LTM function in LE patients (14). We suggest an even more amplified and elaborated LTM-dysfunction criterion in addition to working memory performance to adapt LE criteria to include several aspects of episodic, semantic and visuospatial LTM and ALF. Thus, to diagnose limbic encephalitis, we suggest incorporating this aforementioned novel memory criterion within the existing criteria from Graus et al. (1) (Table 1B). Furthermore, we recommend utilizing specific neuropsychological tests (Table 1C) to detect subtle LTM deficits in LE patients.

Table 1B. Proposal for revised criteria for limbic encephalitis.

Table 1C. Neuropsychological test battery for assessing LTM function in limbic encephalitis.
This suggested framework provides a more realistic imprint of memory impairment in LE and might help us identify and treat LE patients with LTM disabilities. This is particularly important, as early immunosuppressive or other treatment options (e.g., tumor resection) are essential to improve or recover memory performance in LE patients.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. (2016) 15:391–404. doi: 10.1016/S1474-4422(15)00401-9
2. Barak O, Tsodyks M. Working models of working memory. Curr Opin Neurobiol. (2014) 25:20–4. doi: 10.1016/j.conb.2013.10.008
3. Dudai Y, Karni A, Born J. The consolidation and transformation of memory. Neuron. (2015) 88:20–32. doi: 10.1016/j.neuron.2015.09.004
4. Lynch G, Browning M, Bennett WF. Biochemical and physiological studies of long-term synaptic plasticity. Fed Proc. (1979) 38:2117–22.
5. Planagumà J, Haselmann H, Mannara F, Petit-Pedrol M, Grünewald B, Aguilar E, et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann Neurol. (2016) 80:388–400. doi: 10.1002/ana.24721
6. Haselmann H, Mannara F, Werner C, Planagumà J, Miguez-Cabello F, Schmidl L, et al. Human autoantibodies against the AMPA receptor subunit GluA2 induce receptor reorganization and memory dysfunction. Neuron. (2018) 100:91–105. doi: 10.1016/j.neuron.2018.07.048
7. Bramham CR. Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications. Prog Brain Res. (2007) 163:453–71. doi: 10.1016/S0079-6123(07)63025-8
8. Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. (2013) 33:18161–74. doi: 10.1523/JNEUROSCI.3506-13.2013
9. Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, et al. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest. (2015) 125:234–47. doi: 10.1172/JCI77888
10. Butler CR, Zeman AZ. Recent insights into the impairment of memory in epilepsy: transient epileptic amnesia, accelerated long-term forgetting and remote memory impairment. Brain. (2008) 131:2243–63. doi: 10.1093/brain/awn127
11. Helmstaedter C, Winter B, Melzer N, Lohmann H, Witt JA. Accelerated long-term forgetting in focal epilepsies with special consideration given to patients with diagnosed and suspected limbic encephalitis. Cortex. (2019) 110:58–68. doi: 10.1016/j.cortex.2018.01.003
12. von Rhein B, Wagner J, Widman G, Malter MP, Elger CE, Helmstaedter C. Suspected antibody negative autoimmune limbic encephalitis: outcome of immunotherapy. Acta Neurol Scand. (2017) 135:134–41. doi: 10.1111/ane.12575
13. Malter MP, Widman G, Galldiks N, Stoecker W, Helmstaedter C, Elger CE, et al. Suspected new-onset autoimmune temporal lobe epilepsy with amygdala enlargement. Epilepsia. (2016) 57:1485–94. doi: 10.1111/epi.13471
14. Nascimento Alves P, Maruta C, Albuquerque L, Martins IP. Dissociation findings between short-term and long-term memory in autoimmune limbic encephalitis. J Neurol Sci. (2017) 381:126–7. doi: 10.1016/j.jns.2017.08.016
15. Frisch C, Malter MP, Elger CE, Helmstaedter C. Neuropsychological course of voltage-gated potassium channel and glutamic acid decarboxylase antibody related limbic encephalitis. Eur J Neurol. (2013) 20:1297–304. doi: 10.1111/ene.12186
16. Hansen N, Widman G, Witt JA, Wagner J, Becker AJ, Elger CE, et al. Seizure control and cognitive improvement via immunotherapy in late onset epilepsy patients with paraneoplastic versus GAD65 autoantibody-associated limbic encephalitis. Epilepsy Behav. (2016) 65:18–24. doi: 10.1016/j.yebeh.2016.10.016
17. Wagner J, Weber B, Elger CE. Early and chronic gray matter volume changes in limbic encephalitis revealed by voxel-based morphometry. Epilepsia. (2015) 56:754–61. doi: 10.1111/epi.12968
18. Hansen N, Ernst L, Rüber T, Widman G, Becker AJ, Elger CE, et al. Pre- and long-term postoperative courses of hippocampus-associated memory impairment in epilepsy patients with antibody-associated limbic encephalitis and selective amygdalohippocampectomy. Epilepsy Behav. (2018) 79:93–9. doi: 10.1016/j.yebeh.2017.10.033
19. Finke C, Prüss H, Heine J, Reuter S, Kopp UA, Wegner F, et al. Evaluation of cognitive deficits and structural hippocampal damage in encephalitis with leucine-rich, glioma-inactivated 1 antibodies. JAMA Neurol. (2017) 74:50–9. doi: 10.1001/jamaneurol.2016.4226
20. Heine J, Prüss H, Kopp UA, Wegner F, Then Bergh F, Münte T, et al. Beyond the limbic system: disruption and functional compensation of large-scale brain networks in patients with anti-LGI1 encephalitis. J Neurol Neurosurg Psychiatry. (2018) 89:1191–9. doi: 10.1136/jnnp-2017-317780
21. Butler CR, Miller TD, Kaur MS, Baker IW, Boothroyd GD, Illman NA, et al. Persistent anterograde amnesia following limbic encephalitis associated with antibodies to the voltage-gated potassium channel complex. J Neurol Neurosurg Psychiatry. (2014) 85:387–91. doi: 10.1136/jnnp-2013-306724
22. Malter MP, Frisch C, Schoene-Bake JC, Helmstaedter C, Wandinger KP, Stoecker W, et al. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol. (2014) 261:1695–705. doi: 10.1007/s00415-014-7408-6
23. Blake RV, Wroe SJ, Breen EK, McCarthy RA. Accelerated forgetting in patients with epilepsy: evidence for an impairment in memory consolidation. Brain. (2000) 3:472–83. doi: 10.1093/brain/123.3.472
24. Bataller L, Galiano R, García-Escrig M, Martínez B, Sevilla T, Blasco R, et al. Reversible paraneoplastic limbic encephalitis associated with antibodies to the AMPA receptor. Neurology. (2010) 74:265–7. doi: 10.1212/WNL.0b013e3181cb3e52
25. Joubert B, Saint-Martin M, Noraz N, Picard G, Rogemond V, Ducray F, et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. (2016) 73:1115–24. doi: 10.1001/jamaneurol.2016.1585
26. Kanazawa K, Matsumoto R, Shimotake A, Kinoshita M, Otsuka A, Watanabe O, et al. Persistent frequent subclinical seizures and memory impairment after clinical remission in smoldering limbic encephalitis. Epileptic Disord. (2014) 16:312–7. doi: 10.1684/epd.2014.0664
27. Dodich A, Cerami C, Iannaccone S, Marcone A, Alongi P, Crespi C, et al. Neuropsychological and FDG-PET profiles in VGKC autoimmune limbic encephalitis. Brain Cogn. (2016) 108:81–7. doi: 10.1016/j.bandc.2016.07.010
28. Witt JA, Vogt VL, Widman G, Langen KJ, Elger CE, Helmstaedter C. Loss of Autonoetic awareness of recent autobiographical episodes and accelerated long-term forgetting in a patient with previously unrecognized glutamic acid decarboxylase antibody related limbic encephalitis. Front Neurol. (2015) 6:130. doi: 10.3389/fneur.2015.00130
29. Finke C, Kopp UA, Pajkert A, Behrens JR, Leypoldt F, Wuerfel JT, et al. Structural hippocampal damage following anti-N-methyl-D-asparate receptor encephalitis. Biol Psychiatry. (2016) 79: 727–34. doi: 10.1016/j.biopsych.2015.02.024
30. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain. (2012) 135:1622–38. doi: 10.1093/brain/aws082
31. Dogan Onugoren M, Deuretzbacher D, Haensch CA, Hagedorn HJ, Halve S, Isenmann S, et al. Limbic encephalitis due to GABAB and AMPA receptor antibodies: a case series. J Neurol Neurosurg Psychiatry. (2015) 86: 965–72. doi: 10.1136/jnnp-2014-308814
32. Finke C, Kopp UA, Prüss H, Dalmau J, Wandinger KP, Ploner CJ. Cognitive deficits following anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. (2012) 83:195–8. doi: 10.1136/jnnp-2011-300411
33. Mat A, Adler H, Merwick A, Chadwick G, Gullo G, Dalmau JO, et al. Ophelia syndrome with metabotropic glutamate receptor 5 antibodies in CSF. Neurology. (2013) 80:1349–50. doi: 10.1212/WNL.0b013e31828ab325
34. Volianskis A, France G, Jensen MS, Bortolotto ZA, Jane DE, Collingridge GL. Long-term potentiation and the role of N-methyl-D-aspartate receptors. Brain Res. (2015) 1621:5–16. doi: 10.1016/j.brainres.2015.01.016
35. Hagena H, Manahan-Vaughan D. mGlu5 acts as a switch for opposing forms of synaptic plasticity at mossy fiber-CA3 and commissural associational-CA3 synapses. J Neurosci. (2015) 35:4999–5006. doi: 10.1523/JNEUROSCI.3417-14.2015
36. Joubert B, Kerschen P, Zekeridou A, Desestret V, Rogemond V, Chaffois MO, et al. Clinical spectrum of encephalitis associated with antibodies against the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor: case series and review of the literature. JAMA Neurol. (2015) 72:1163–9. doi: 10.1001/jamaneurol.2015.1715
37. van Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PA. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. (2014) 13:299–312. doi: 10.1016/j.autrev.2013.10.016
38. Frisch S, Thiel F, Marschhauser A, Villringer A, Horstmann A, Schroeter ML. Identifying neural correlates of memory and language disturbances in herpes simplex encephalitis: a voxel-based morphometry (VBM) study. J Neurol. (2015) 262:563–9. doi: 10.1007/s00415-014-7604-4
Keywords: autoimmunity, limbic encephalitis, long-term memory, autoantibodies, synaptic consolidation
Citation: Hansen N (2019) Long-Term Memory Dysfunction in Limbic Encephalitis. Front. Neurol. 10:330. doi: 10.3389/fneur.2019.00330
Received: 13 September 2018; Accepted: 18 March 2019;
Published: 26 April 2019.
Edited by:
Tjalf Ziemssen, Zentrum für Klinische Neurowissenschaften (ZKN), GermanyReviewed by:
Sonja Hochmeister, Medical University of Graz, AustriaYoshiro Ohara, Kanazawa Medical University, Japan
Copyright © 2019 Hansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niels Hansen, bmllbHMuaGFuc2VuQG1lZC51bmktZ29ldHRpbmdlbi5kZQ==