 Bruno Stuhlmüller
Bruno Stuhlmüller Udo Schneider
Udo Schneider José-B. González-González
José-B. González-González Eugen Feist
Eugen Feist- 1Department of Rheumatology and Clinical Immunology, Charité-Universitätsmedizin, Berlin, Germany
- 2Labor Berlin-Charité Vivantes GmbH, Berlin, Germany
Idiopathic inflammatory myopathies represent still a diagnostic and therapeutic challenge in different disciplines including neurology, rheumatology, and dermatology. In recent years, the spectrum of idiopathic inflammatory myopathies has been significantly extended and the different manifestations were described in more detail leading to new classification criteria. A major breakthrough has also occurred with respect to new biomarkers especially with the characterization of new autoantibody-antigen systems, which can be separated in myositis specific antibodies and myositis associated antibodies. These markers are detectable in approximately 80% of patients and facilitate not only the diagnostic procedures, but provide also important information on stratification of patients with respect to organ involvement, risk of cancer and overall prognosis of disease. Therefore, it is not only of importance to know the significance of these markers and to be familiar with the optimal diagnostic tests, but also with potential limitations in detection. This article focuses mainly on antibodies which are specific for myositis providing an overview on the targeted antigens, the available detection procedures and clinical association. As major tasks for the near future, the need of an international standardization is discussed for detection methods of autoantibodies in idiopathic inflammatory myopathies. Furthermore, additional investigations are required to improve stratification of patients with idiopathic inflammatory myopathies according to their antibody profile with respect to response to different treatment options.
Introduction
Idiopathic inflammatory myopathies (IIM) represent a heterogeneous group of acquired muscle diseases with so far unclear etiology. The different entities are associated with diverse clinical symptoms ranging from amyopathic to necrotic inflammatory muscle involvement, typical skin and internal organ involvement. In addition to the clinical picture, histological and serological findings are especially supportive in differentiation and stratification of disease (1). Based on this characteristics, IIM can be classified into different major sub-types: (i) polymyositis (PM), (ii) sporadic inclusion body myositis (sIBM), (iii) dermatomyositis (DM), (iv) immune-mediated necrotizing myopathy (IMNM), and (v) overlap syndromes with myositis (2, 3). Within the spectrum of antibodies two classes have been proposed (4), designated as myositis specific antibodies (MSAs) due to their exclusive association with IIM or as myositis-associated antibodies (MAAs) due to their prevalence in different other connective tissue disorders. This article focuses on an overview on so far identified specific autoantibody-antigen systems in IIM. Furthermore, we discuss the available methods, strategies and pitfalls of autoantibody detection in IIM as well as their diagnostic performance and clinical association.
Characterization of Disease Specific Antigen-Antibody Systems in IIM
As with many other systemic autoimmune diseases, it is unclear so far, whether and how the observed autoantibody formation is directly associated with the pathogenesis of disease or is just an epiphenomenon. However, the striking association between certain autoantibodies with a distinct clinical phenotype, their high disease specificity and their value for stratification and prognosis of disease suggests that they may play a role in disease induction and propagation (5). This section describes in detail the nature and function of so far identified IIM specific antigens. A complete overview on so far identified antigens and the corresponding autoantibodies in adult and in juvenile patients with IIM is given in Table 1.
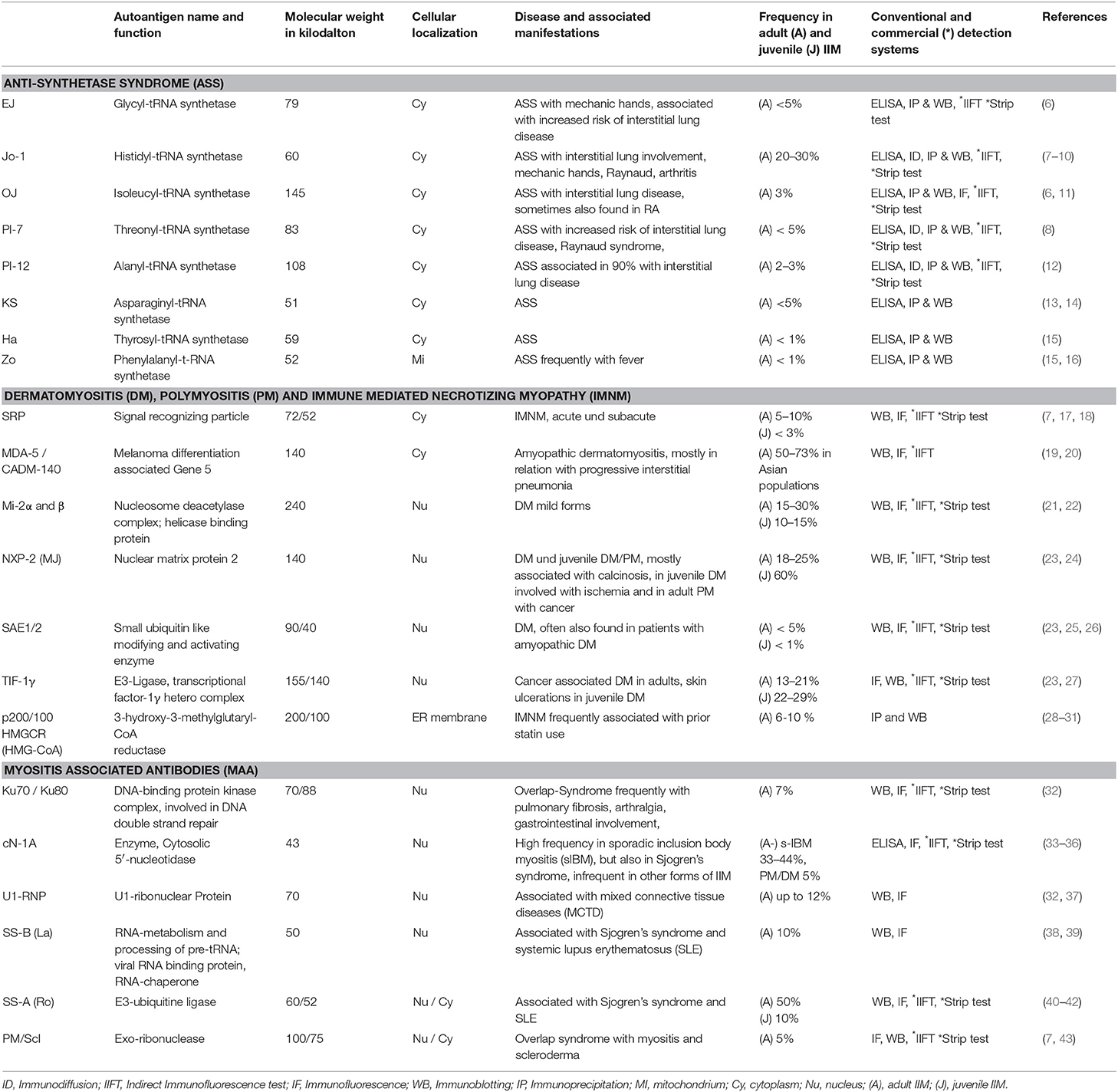
Table 1. Characterization of myositis specific and myositis associated autoantibodies and their respective antigens.
The best-known autoantigen-autoantibody system in IIM is directed against transport ribonucleoacid (t-RNA) synthetases and represents a specific finding in patients with so named anti-synthetase syndrome (ASS). The targeted synthetases catalyze the binding of a specific aminoacid to their t-RNA in the cytoplasm of each eukaryotic cell for transportation to the ribosome and subsequent protein synthesis. At least eight t-RNA synthetases have been identified as autoantigens. Anti-Jo1 antibodies are the most common ones and received their designation after the initials of the index patients (44). It is unclear, why not all t-RNA synthetases are targeted by the immune system in ASS, but only the tRNA synthetase for threonyl (PL-7), alanyl (PL-12), isoleucyl (OJ), glycyl (EJ), asparaginyl (KS), phenylalanyl (Zo), tyrosil (Ha), and finally the histidyl synthetase (Jo-1). Even more of interest is the question, whether the antibodies can interfere with the function of the respective t-RNA synthetase as it has been shown for anti-Jo1 antibodies by in-vitro experiments (45, 46). This particular antibody specificity belongs mainly to the IgG1 isotype and binds to common epitopes (47).
For the anti-Jo1 antibodies, it was shown that the formation of the major autoepitope is strongly dependent on proper folding of the molecule (46). As a shared risk factor for anti–Jo-1 autoantibody positivity, the HLA–DRB1*0301 allele was identified in European as well as African Americans (48). In the Japanese population, HLA–DRB1*0405 was associated with the formation of an anti-tRNA antibody response (49).
Another specific antigen is the transcription intermediary factor-1 gamma (TIF-1γ). This multi-functional protein with a molecular weight (MW) of 140/155 kilo-Dalton (kDa) is mainly involved in gene transcription (27, 50–53). The TIF-1 family is composed of tripartite motif-containing (TRIM) proteins, which are all implicated in cell proliferation, development, apoptosis, and innate immunity (54). All TIF1 proteins share a C-terminal chromatin reading unit consisting of a plant homeodomain finger and a bromo-domain (BROMO) that is highly conserved among TIF1 family members, but which is not present in any of the other TRIM proteins (55, 56). Of note, while the most common target in anti-TIF1-positive CAM (cancer-associated myositis) is TIF1γ, other proteins of the TIF1 family (TIF1α and β) may also be simultaneously targeted by the immune system (53).
The tripartite containing motif (TRIM) allows these proteins also to function as E3-ligases in the ubiquitination pathway to control protein degradation, localization, and function. In this context, it is interesting to mention that TIF1-γ is involved in the regulation of TGF-β signaling via mono-ubiquitination of SMAD-4 leading to suppression of TGF- β. Thus, by stimulating cell growth and differentiation, TIF1-γ could play a pivotal role in promoting or suppressing malignant cell growth and differentiation (57). The known association of anti-TIF1-γ antibodies with a high risk of cancer development in DM suggest that this link could be not random. Of note, anti-TIF antibodies were only rarely detectable in patients with solid cancer (3.1%) or paraneoplastic rheumatic syndrome (3.3%) without DM (58).
Recently, it was recognized that tumors from paraneoplastic anti-TIF1-γ positive patients showed an increased number of genetic alterations, such as mutations and loss of heterozygosity (LOH) in TIF1 genes (59). Compared with type-matched control tumors from non-myositis patients, TIF1-γ staining was also significantly more intense in tumors as well as muscle tissue from anti-TIF1-γ positive patients. This finding could indicate that the co-occurrence of mutations in peptide regions of TIF1 with high affinity for HLA class I and tumors with high-level TIF1 protein expression may initiate a strong adaptive immune response against neoplastic cells with the mutation. Interestingly, LOH is the most frequent way to lose a mutant allele in human cancer and this is key to tumor immune-editing, since tumor cells with mutations producing a neo-antigen may be eliminated by the immune system and replaced by tumor cells with LOH in that region (without the antigenic mutation) (60, 61). Thus, these modifications can induce an immune response, but also cause an escape of the tumor cell from clearance.
It has been described that DM disease increases toward the equator and strongly associate with latitude. Recently, this observation was confirmed by another study showing that relative prevalence of DM and frequency of anti-TIF1-γ autoantibodies were found to be significantly negatively associated with latitude in adult myositis. Furthermore, HLA alleles HLA-DRB1* 07:01 and HLA-DQB1*02 were strongly associated with the DM-specific autoantibodies anti-Mi-2 as well as anti-TIF1-γ (62).
The component of the nucleosome remodeling deacetylase complex, Mi-2, is an autoantigen of 240 kDa MW and exists in two isoforms Mi-2α (CHD3) and Mi-2β (CHD4). Mi-2 is responsible for the remodeling of chromatin by de-acetylating histones and plays a role as transcription repressor (63). The role of anti-Mi-2 autoantibodies in the pathogenesis of DM is unclear. However, the autoantigen Mi-2 is found to be up-regulated in the muscle tissue of DM patients. Of note, exposure of keratinocytes to UV radiation has been shown to increase the expression of Mi-2 protein supporting the hypothesis that UV radiation may be associated with the induction of anti-Mi-2 autoantibodies (64). However, a reported increase in the presence of anti-Mi-2 autoantibodies toward the equator was not confirmed by a current study (62, 65). A preferential expression was described in the nucleus of myofibers within fascicles affected by perifascicular atrophy, particularly in the centralized nuclei of small perifascicular muscle fibers expressing markers of regeneration (66). In a mouse model of muscle injury and repair, Mi-2 levels were dramatically and persistently up-regulated during muscle regeneration in-vivo. Of note, premature silencing of Mi-2 with RNA interference in vitro resulted in accelerated myoblast differentiation. In summary, these results indicate that this protein may play a role in modulating the kinetics of myoblast differentiation. European and American anti–Mi-2 antibody positive DM patients have a common genetic risk factor DRB1*0701 (67). Furthermore, HLA-DRB1*0302 was identified to be associated with anti–Mi-2 autoantibody positive African American patients (48). Of note, all HLA molecules were found to share a 4–amino-acid sequence motif, which was predicted by comparative homology analyses to have identical 3-dimensional orientations within the peptide-binding groove.
The target antigen of antibodies against small ubiquitin-like modifier activating enzyme (SAE1/2) is the SUMO-1 activating enzyme heterodimer with a MW of 40 and 90 kDa, respectively. This antigen is involved in the posttranslational modification of proteins, the so called “sumoylation” (68, 69). Of note, a strong association with the HLA-DRB1*04-DQA1*03-DQB1*03 haplotype has been reported (25). This is another example in IIM, where the association between genotype, serotype and clinical picture suggest a link to the pathogenesis of disease.
Immune reactivity against the signal recognizing protein hetero-complex (SRP) is associated with immune-mediated necrotizing myopathy (IMNM). This 72/52 kDa antigen is expressed in the cytoplasm and responsible for transport proteins to the endoplasmic reticulum. SRP consists of six polypeptides (including SRP19 and MIM 182175) as well as seven SL-RNA molecules with partial homology to Alu-DNA (70, 71). Although antibodies against SRP can target each of the different SRP components, a signal peptide-binding 54 kDa subunit (SRP54) represents a major epitope recognized by almost every sera and is, therefore, preferentially used in immunoassays (72–76). Although it is not clear, how the antibodies can interact with the SRP1/2 autoantigen, it was shown that anti-SRP antibodies can inhibit the translocation of secretory proteins into the endoplasmic reticulum in-vitro and that a passive transfer of IgG from anti-SRP+ patients with IMNM provoked muscle deficiency through a complement-mediated mechanism in mice model. Interestingly, also active immunization with SRP was able to induce an immune response and provoked disease (76, 77). A correlation between anti-SRP54 antibody titers and disease activity was also shown in a longitudinal follow-up study suggesting a pathogenic role of this antibody entity (78). As a genetic risk factor, HLA- DQA1*0102 was identified in anti–SRP autoantibody positive African American patients (48).
The melanoma differentiation antigen 5 (MDA-5, synonym CADM-140) with a MW of 140 kDa belongs to the family of RIG-I-like receptors of adhesion molecules and represents a resistance factor against double stranded RNA viruses (49, 79). The MDA-5 molecule plays an important role in the regulation of the immune response by the innate system. In this context, it was shown that MDA-5 bind virus particles, e.g., picornaviruses such as coxsackievirus, and induce an antiviral responses by producing type-I interferons and tumor necrosis factor (80). It was also reported that hyperferritinemia could be a marker for rapidly progressive ILD in anti–MDA-5 antibody positive DM patients (81, 82). In this context many cytokines regulate the ferritin synthesis, including IL-1β, IL-18, TNF, IFN-γ, and IL-6 and several of these cytokines are considered to be involved in IIM pathogenesis. HLA–DRB1*0101/*0405 was found to be associated with susceptibility to anti–MDA-5 antibody positive DM in the Japanese population (49). Interestingly the same alleles are well-known to play a role in the susceptibility to autoantibody induction in rheumatoid arthritis (83).
Autoantibodies against the nuclear protein 2 (NXP-2), also known as MJ or p140 antibodies, are directed against a nuclear matrix protein complex named NXP2/MORC3, which is involved in regulation of p53-induced cell senescence in the context of oncogenic signals (23). NXP-2 is associated with the small ubiquitin modifier SUMO-2 and represses its expression (84).
The enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) was recently identified as a target of autoantibodies induced under treatment with statins. The antigen is expressed in the ER membrane and has a MW of 200/100 kDa. HMGCR catalyzes the conversion of HMG-CoA to mevalonic acid, which is an important step in cholesterol biosynthesis. So far, reports about HMGCR in myositis are rare, however in animal studies it was demonstrated that loss of HMGCR function disrupts vascular stability during developmental processes (4).
Diagnostic Testing for Autoantibodies in IIM
To assess disease activity and as indicators for muscle injury in IIM, basic laboratory diagnostics are used, such as measurements of levels of inflammatory markers as well as serum activity and/or concentration of muscle specific proteins including creatine phosphokinase (CPK) or myoglobin. However, these markers are unspecific and not helpful to distinguish between the different forms of inflammatory muscle damage. In contrast, the reactivity of the described autoantibodies does not sufficiently correlate with disease activity in IIM, but can rather serve for stratification of the disease process and outcome (85). This section focuses on helpful information with respect to routine procedures of autoantibody detection and the recommended diagnostic approaches, since it is of enormous importance to know these basics for appropriate interpretation of results.
Serum is usually used for the detection of autoantibodies, and test procedures are performed according to the manufacturer's validation. From a preanalytical point of view, time point of venepuncture and fasting status of the patient are not relevant. However, hemolytic, lipemic or contaminated blood samples should not be used, since the released proteins and proteinases can interfere with the immunologic method of detection. Especially in case of a prolonged transportation of the blood sample, serum should be separated in advance by centrifugation at 1,300 g. Subsequently, the serum samples can be stored at 2–8°C for up-to 2 weeks before analysis. A longer period requires freezing at minus 20°C, which presumably allows conservation of the autoantibody reactivity for years (86, 87). Furthermore, some treatment procedures including administration of strong immunosuppressive drugs such as B-cell directed therapies as well as plasmapheresis or administration of intravenous immunoglobulins can influence the result of detection by decreasing the concentration of the autoantibodies.
From the methodical point of few, detection of autoantibodies in IIM is not well-standardized and no international reference samples are available so far. In general, immunoprecipitation of radio-labeled proteins or RNA molecules is still considered to represent the “gold standard” (68). However, due to the time-consuming procedure, high amount of required antigens and low sensitivity, this method is not a routine procedure in most clinical laboratories. Therefore, for the detection of IIM specific and/or associated autoantibodies several, more efficient approaches such as immunofluorescence, ELISA and western blotting are widely used (88–90). In general, immunofluorescence requires large experience to interpret patterns and may not be sensitive enough to detect all MSAs/MAA. On the other hand, ELISA methods using recombinant antigens or immunoblotting with denaturated antigens can probably not detect antibodies to certain conformational epitopes. Line-blot assays allow a qualitative detection of many antibodies in one run. In contrast, and in accordance to the manufacturer specification only a limited number of MSAs/MAAs can be detected by commercial available ELISAs so far.
Indirect immunofluorescence (IIF) on HEp-2 cells, a human epithelioma cell line, is commonly be used for detection of anti-nuclear antibodies (ANA), but also enables detection of antibodies against cytoplasmatic antigens. This method allows a screening for a wide range of autoantibodies especially in connective tissue disorders by describing the staining pattern (e.g., nuclear, nucleolar, cytoplamatic), as well as the reactivity titer starting with a serum dilution of usually 1:80. Although no agreement has still been reached on the interpretation and reporting of the ANA titres, results can be considered as weak positive in antibody titer of ≥1:160 and as strong positive in a titres of ≥1:640. In this context, it is important to mention that measured titers often do not correspond with the significance of the results. In other words, a low reactivity should not be considered as irrelevant. Furthermore, it is also important to pay attention to the described pattern of immunofluorescence in the nucleus, but also the cytoplasm. It is helpful to know that IIF on HEp-2 cells shows characteristic patterns of some MSA or MAA antibodies such as those against PM/Scl with prominent homogeneous staining of the nucleous, U1-RNP with coarse speckled staining of the nucleus, Jo-1 or SRP with fine speckled stainings of the cytoplasm (Figure 1), but is not sufficiently accurate to be used as the only screening tool for myositis antibodies.
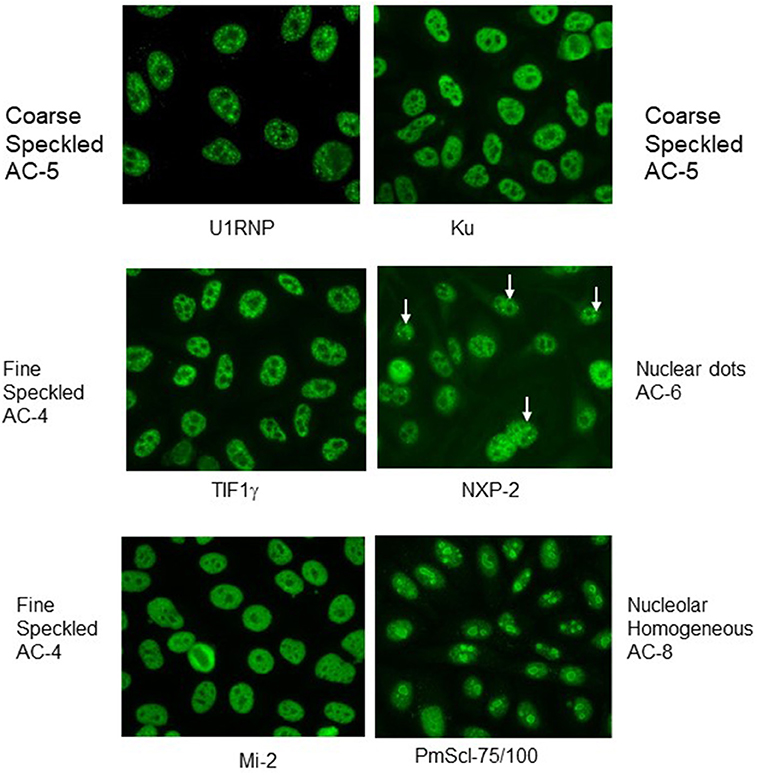
Figure 1. Indirect immunofluorescence with typical nuclear patterns of MSA and MAA. Immunofluorescence patterns are indicated with the terminology of the International Consensus on ANA Patterns (ICAP).
Although most of the targeted antigens in IMM are expressed within HEp-2 cells, their detection by IIF is clearly limited. The reasons for frequent false negative results is diverse including e.g., low expression level of the antigen or low affinity of the antibodies causing a weak staining signal. Furthermore, the intracellular distribution of relevant antigens in IIM is usually diffuse generating an unspecific staining pattern. Thus, week positive results are often not recognized, as it frequently occurs even with anti-tRNA synthetase antibodies. Nevertheless, IIF on HEp-2 cells is an important diagnostic procedure. Table 1 summarizes frequent staining patterns in conjunction with the respective antibody and, Figures 1, 2 show representative IIF staining patterns indicating the respective nomenclature for the most frequent MSAs and MAAs by International Consensus on Autoantibody Patterns (ICAP).
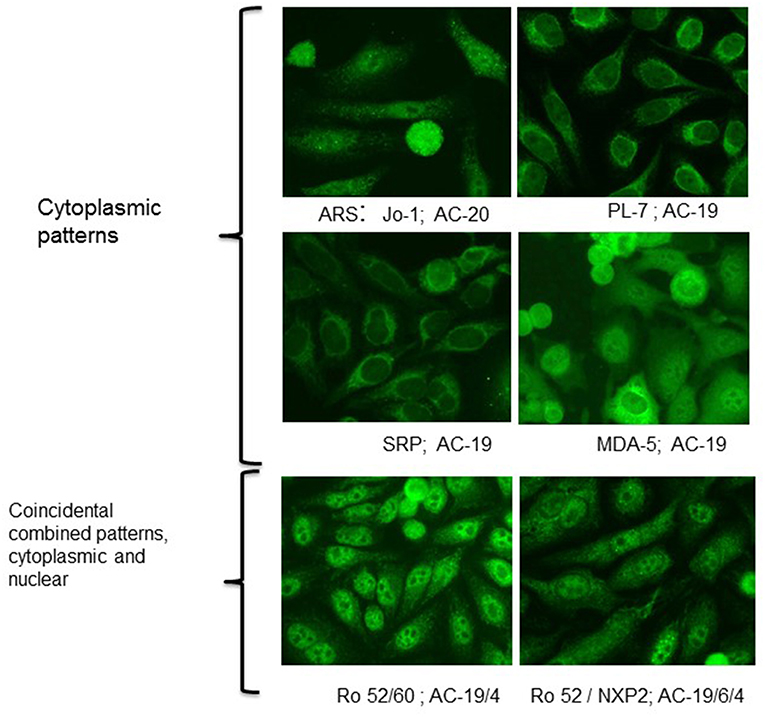
Figure 2. Indirect immunofluorescence with typical cytoplasmic patterns of MSA and MAA. Immunofluorescence patterns are indicated with the terminology of the International Consensus on ANA Patterns (ICAP). AC-19-cytoplasmic dense fine speckled, AC-20-cytoplasmic fine speckled.
For the detection of classical and new myositis antibodies, multi-analyte line blot assays and ELISA are the current routine methods of choice. In contrast to IIF, commercial ELISAs and line-blot assays use purified or recombinant expressed antigens for detection of antibodies. These methods allow the targeted detection or confirmation of respective antibodies in serum samples, which are usually diluted 1:100. In contrast to semi-quantitative results provided by line-blot assays (with negative, weak or strong positive signals, see Figure 3), ELISA based methods allow a better quantification of antibody reactivity, especially if international standards will be available. However, the majority of ELISA methods published hitherto to detect novel myositis antibodies are in-house made and most of the commercial ELISAs only report negative or positive results by using a cut-off level of reactivity defined by the providing manufacturer. A multiplex-approach for detection of several antibodies in one immunoassay has also limitations and pitfalls due to the difficult optimization of the cut-off for all investigated antibodies. Therefore, it is always important to check the plausibility of the obtained results, not only in cases of weak or borderline reactivity, but also in case of discordance between the different immunoassays (e.g., negative ANA staining pattern but positive anti-PM/Scl antibodies in ELISA or line-asays). On the solid phase of an immunoassay, the antigen structure can be altered yielding a false positive antibody reactivity, which is not directed against the native antigen. Therefore, it is important, that involved diagnostic laboratories are be informed about the clinical suspected diagnosis and provide high quality assays proven by regular internal and external quality controls.
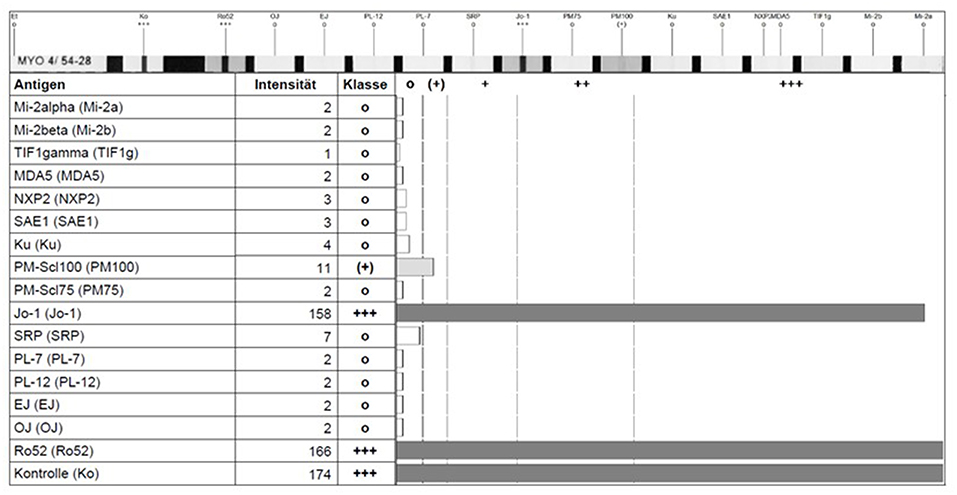
Figure 3. Detection of anti-Jo-1 and anti Ro-52 antibody reactivity in a line-blot assay showing the reaction intensity by a scan-software of the fabricant (EUROLINE, Euroimmune, Germany).
The major advantage of ELISA and line blot test assays is the option of automation test fastness. In this context, the development of commercial quantitative immunoassays including ELISA, chemiluminescence and immunofluorometry has facilitated the use at large scale in routine laboratories and provided interesting information on correlation of clinical and serological findings in IIM.
Correlation of MSA With Clinical Findings
In addition to inflammatory lesions of skeletal muscles, involvement of other organs such as skin, joint, lung and heart is frequent in IIM. Extra-muscular involvement, especially interstitial lung disease (ILD) and underlying malignancy in cancer-associated myositis (CAM) are the two dominating factors contributing to increased mortality in IIM-patients (91, 92). This section gives an overview on the association of MSA with clinical findings and their diagnostic performance. The incidence of most of the so far identified autoantibody activities of European patients is in agreement with similar studies of Japanese and American patients (7).
Antibodies Against t-RNA Synthetases
Patients with antibodies against t-RNA synthetases are prone to develop the so called anti-synthetase syndrome (ASS) characterized by myopathy, interstitial lung disease (ILD), non-erosive arthritis, fever, Raynaud's phenomenon and mechanic's hands. Since not all symptoms are present at disease onset, ASS should be carefully considered in patients presenting with isolated arthritis, even in those with erosive manifestation and RF as well as ACPA-positivity (93, 94). The presence of anti-synthetase antibodies can be suspected if a characteristic cytoplasmic pattern on HEp2 cells is evident (Figure 1). However, confirmation is needed using ELISA, immunoblot or line-assays with the isolated antigens. Depending on the diagnostic method anti-Jo-1 antibodies are the most frequent autoantibodies in IIM, they can be detected in 20–30% of patients (7, 95). Titers of Jo-1 antibodies were shown to correlate with disease activity in adults (95). In muscle biopsies of anti-Jo-1 positive myositis patients a specific histologic pattern with peri-fascicular necrosis has been described (96).
The CT- and histomorphologic pattern of ILD in anti-synthetase syndrome can vary between non-specific interstitial and organizing pneumonia (97). Antibodies against PL-7 and PL-12 are positive in up to 5% of IIM patients. Clinically they are associated with less muscle involvement but with a higher proportion of ILD, which might have an acute onset (98). Furthermore, pericarditis was observed in up to 50% of anti-PL7 positive patients (99). Antibodies against OJ, EJ, KS, Zo, and Ha are rare and only present in 1–3% of patients with ARS. The coincidence of anti-Ro52/TRIM21with anti-ASS antibodies was described to be associated with severe myositis and arthropathy as well as with an increased risk of cancer (7, 99, 100). The onset of ILD and myositis, as leading symptoms in ASS, can be subsequently or in parallel, while the course of ILD must not necessarily be progressive.
DM-Specific Antibodies
Anti-Mi-2 antibodies have a high specificity for both adult and juvenile DM (JDM). Mi-2-antibodies produce a fine speckled nuclear pattern in IIF on HEp-2 cells (AC-4 of the ICAP nomenclature) and are detectable in approximately 30% in adult and 10% in juvenile DM (JDM) patients, respectively. Patients with positive anti-Mi-2 antibodies have usually a milder myopathy, lower risk of interstitial lung disease (ILD) and malignancy (101). Skin manifestations, such as Gottron's sign and heliotrope rash, as well, rashes in neck (V rash) and upperback (shawl rash) or cuticular overgrowth are typical. Patients respond well to steroid therapy and have a good prognosis. Furthermore, levels of Mi-2 antibodies were shown to correlate with clinical response to B-cell depletion therapy (102). Although DM with antibodies against Mi2-β can be associated with neoplasia (e.g., colon or mama-carcinoma), anti-Mi-2 antibodies are in general associated with a lower risk of paraneoplastic myositis and hence considered to be a good prognostic factor.
Anti-TIF1γ (anti-155/140) antibodies are detectable in 13–21% of patients with tumor associated adult DM and in approximately 30% of severe juvenile DM patients (23, 27). In fact, these antibodies are the most frequent marker in juvenile IIM (JIIM) and are primarily associated with JDM. They were originally found almost exclusively in JDM, in about of 23–29% of cases using immunoprecipitation and immunoblotting. Interestingly, in recent studies including a total of 374 cases with JIIM, 131 cases (35%) were found to be positive for these antibodies. In detail, the antibodies were detectable in 38% of patients with JDM (123/320) and in 26% of patients with an overlap syndrome with myositis (8/31) (103, 104). TIF-γ-antibodies are known to be very frequently associated with malignancy in adults with a specificity of 89%, sensitivity of 78%, and positive and negative predictive values of 58 and 95%, respectively (105). In contrast to adults, there is no paraneoplastic association in JIIM (106). Skin manifestations are characterized by a usually slowly progressive onset, but are more extensive than in other JDM groups. In fact, skin ulcerations and lipodystrophy were reported to be particularly associated with these antibodies. However, this was not reported uniformly, since recent studies did not observe ulceration or V-rashes significantly more frequent in this group than in others (104). Of note, it has been shown that levels of TIF-γ-antibodies correlate with response to B-cell depletion therapy in pediatric patients (102).
Anti-NXP2 antibodies (MJ or p140) cause a fine speckled ANA pattern on IIF, but this pattern is often misinterpreted as variable nuclear dots (Figure 1). These antibodies have been originally described in children with JDM in about 25% of cases (107, 108). In a cohort of 436 patients with JIIM, their prevalence was reported to be approximately 21% in JDM, 9% in JPM and 15% in overlap syndromes (juvenile connective tissue myositis, JCTM) (104). Subsequently, anti-NXP2 antibodies have been also detected in approximately 30% of patients with DM and 8% with PM in a cohort of 58 adult Italian patients (109), but in only 1.6% of 507 adult Japanese patients (110). In the Italian cohort, a good response to therapy was reported for muscle involvement (109). In juvenile DM, the autoantibodies directed against NXP-2 are frequently associated with calcinosis and ischemic muscle involvement in up to 60% of cases (111). In adults, a possible association was reported with malignancies such as mamma-, uterus- and pancreas-carcinomas.
The ANA pattern of anti-MDA5 antibodies on HEp-2 cells in IIF is usually negative. These antibodies were identified for the first time in East-Asian populations. They are more frequently observed in adult DM and were reported in a prevalence of 19–35% (19, 81). In a study with 285 patients with JDM (112), anti-MDA5 antibodies were detectable in 7.4% of patients (21/285) and a recent review reports a prevalence of 0–13% in Europe and USA (113). The typical clinical manifestations of adult IIM patients with anti-MDA5 antibodies were amyopathic myositis with rapidly progressive interstitial lung disease (ILD), which in turn determines a high mortality rate in these patients (Figure 4). However, ILD must not be rapidly progressive in every case. In contrast to Asian patients, Caucasian patients often show skin ulcerations and painful palmar papules (114, 115). Furthermore, differences were also reported for Asian compared to Caucasian JDM associated with MDA-5 antibodies. In this context, Japanese patients showed a pulmonary involvement in nearly 50% of patients, whereas the incidence of ILD was much lower in Caucasians. JDM patients with anti-MDA5 antibodies show frequent skin as well as oral ulceration, but no calcinosis and had less severe muscle weakness compared other JDM subtypes. Of note, ILD was found in 4 of 12 cases (19%) on chest X-ray. None of these anti-MDA5 antibodies positive ILD patients was positive for anti-synthetase antibodies suggesting that anti-MDA5 antibodies represent an own subtype of IIM. This study also revealed that more patients with anti-MDA5-antibodies were in remission after 2 years compared to patients without these antibodies. In the follow-up, patients in remission showed a decline in the titer of MDA5 antibodies. These results resemble those of prior studies in which antibodies were shown to disappear during disease remission (20).
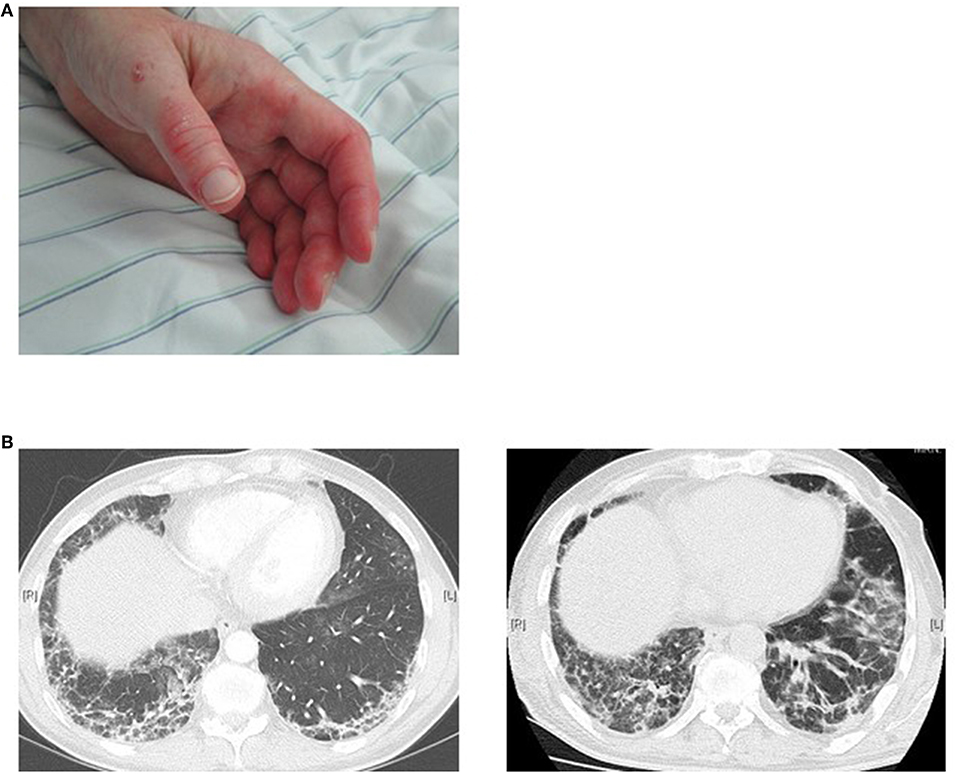
Figure 4. MDA5 positive patient with an amyopathic dermatomyositis (A) maculopapular palmar rash and hyperkeratosis, (B) rapid progression of ILD manifestations within 5 weeks.
The anti-SAE-antibodies were originally described in adults with DM exhibiting an amyopathic onset with skin manifestations, but who may develop myositis several months later. Lung involvement was infrequent reported. The antibodies were detected in approximately 7–8% of adult Caucasian patients with DM (25, 116). In a study with 110 Japanese patients with DM including 13 with JDM, only 2 patients with DM (1,8%) were shown to have SAE-antibodies (117). In a large study with 436 patients with JIIM, only one patient with JDM was positive for anti-SAE-antibodies (118). Therefore, the presence of SAE-antibodies in patients with JIIM seems to be extremely rare.
Immune Mediated Necrotizing Myositis (IMNM)
Anti-SRP-Antibodies were detected in about 3 to 7% of adults with IIM (73, 119). In JIIM, these antibodies are even more infrequently observed in only 1.6% of patients exclusively with juvenile polymyositis (JPM) (103, 120). The presence of SRP-antibodies can be suspected by detection of a cytoplasmic staining pattern in ANA IIF (Figure 1). Adults with anti-SRP-antibodies develop typically an acute necrotizing myopathy with prominent muscle impairment without skin manifestations. Compared to other myositis forms, a satisfactory response to medicament treatment is difficult to achieve. Pulmonary involvement, Raynaud-symptoms, arthritis or overlap syndromes are infrequent. Similar to adults, juvenile patients have very high levels of CPK as well as often cardiac involvement detectable e.g., by ECG or echocardiography. Of note, adult patients show frequently Raynaud-symptoms, dysphonia and dyspnea under exertion (107). Compared to other forms of JIIM, onset of symptoms is late in patients with anti-SRP-antibodies. However, two unusual cases have been published recently with onset in the first decade of life showing muscular dystrophy and a low degree of inflammation in muscle biopsy (107). The antibody titer against SRP seems to correlate with clinical activity as well as with levels of CPK in adults as well as in JIMM and can be used for monitoring of therapy (78, 121).
Antibodies against the enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) have been identified initially in patients with immune mediated necrotizing myositis (IMNM) in association with statin treatment. Overall, autoantibodies against HMGCR are detectable in 6–7% of patients with IIM (28–30). Myalgias, as a common side effects under statins, are not associated with this form of myositis and positive HMGCR-antibodies. In fact, only a minority of patients under statin treatment develops myopathy with anti-HMGCR-antibodies (122). Furthermore, even patients without statin treatment can develop necrotizing myositis with positive anti-HMGCR-antibodies (123). Results of a multicenter study show that the majority of patients with anti-HMGCR antibodies have IMNM. In this study, the prevalence of anti-HMGCR antibodies in different subpopulations of IMNM exposed to statins was approximately 70%, and even 75% in patients above the age of 50 years. However, approximately 45% of INMN patients had no exposure to statins and in 5% of cases did not show muscle necrosis (124). In contrast to non-immune statin myopathy, which resolves after stopping statin therapy, patients with anti-HMGCR antibodies have a persistent autoimmune response despite cancelation of treatment with the inducing agent. Similar to SRP antibodies, the titer of anti-HMGCR antibodies correlates with the clinical activity of necrotizing myositis. However, in contrast to SRP antibodies it is not known, whether antibody titres can normalize under effective therapy (125–127). Of note, these antibodies have also been detected in children after statin therapy recently, however, the experience with these cases is very limited (128, 129).
The detection of anti-HMGCR antibodies was performed by only few laboratories using immunoprecipitation so far, since no standardized commercial assay was available. An introduction of other methods such as ELISA or chemiluminescence for the detection of anti-HMGCR antibodies could make this test widely available to facilitate the diagnostic possibilities (130). However, since such immunoassays can yield false positive results due to detection of low avidity antibodies, a confirmatory analysis by immunoprecipitation can be recommended (131). Interestingly, usage of rat hepatocytes in indirect immunofluorescence was proposed (132). However, although some laboratories have adopted this strategy, there are not yet available data from an extended use of this approach (133). Of note, in a French cohort study HMGCR positive IMNM-patients were found to be at higher risk for malignancy (134).
Autoantibodies in DM and the Risk of Cancer
A high risk of cancer development is well-described and has led to the sub-categorization of cancer associated myositis (CAM) (135). The overall risk of cancer in myositis is significantly higher than in the age-matched population and approximately 10% of IIM are associated with malignancies (134–138). In DM, the risk is especially increased in the first 5 years after diagnosis (139–144). The most common cancers among the reported cases associated with DM are nasopharyngeal carcinoma and adenocarcinoma of the ovary, lung, pancreas, stomach and colon (139–145). Many studies have claimed the presence or absence of certain antibodies as markers for the risk of cancer in myositis. The autoantibody profile is considered a useful tool to identify patients at risk for CAM (32). In this context, the best established antibodies are directed against TIF1-γ with a high sensitivity and specificity for cancer associated DM in adult patients (27, 52, 53, 105, 146, 147), but not in JDM (50). Furthermore, also NXP-2 positivity in DM patients has been identified as a risk factor for malignancy (148). The association of anti-synthetase and anti-HMGCR antibodies with cancer is less clear and needs further confirmation. To exclude an associated malignancy in clinical practice repetitive examinations can be recommended in DM patients at risk, whereas clear guidelines on the frequency and extension of such diagnostic procedures are missing so far.
Summary
Autoantibodies in IIM are very important diagnostic and prognostic markers, which can help to facilitate our approach to these rare and divers diseases. They correlate closely with the clinical manifestation of disease and allow stratification of patients. However, a laboratory result provides always only a piece of the diagnostic puzzle and should always be questioned if not plausible. For this purpose, a better knowledge on the limitations of the laboratory procedures is necessary and tests should only be performed if indicated according to the clinical picture. To allow comparison and improve reproducibility, the existing assays require uniform standards on an international level, and optimized methods for a broader distribution. If these issues can be solved, antibodies in IIM will play a more prominent role in the classification of disease. Another interesting and important tasks for the future will be to investigate the treatment response in IIM patients according to their antibody profile in more detail. This includes the question, whether different antibodies correlate with disease activity, but can be also used for an individualized approach to predict the response and outcome. Finally, the interaction of the antibodies with the targeted antigens is of major interest especially for the IIM specific entities. A deeper understanding on the mechanisms behind the induction of the respective immune response as well as on the potential role of the targeted antigens in IIM can improve our insight into the pathogenesis of disease.
Author Contributions
BS reviewed published articles and contributed to the manuscript on antigen-antibody systems. US reviewed published articles and contributed to the manuscript on clinical aspects. J-BG-G reviewed published articles and contributed to the manuscript on laboratory diagnostic aspects. EF reviewed published articles and contributed to all parts of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mantegazza R, Bernasconi P, Confalonieri P, Cornelio F. Inflammatory myopathies and systemic disorders: a review of immunopathogenetic mechanisms and clinical features. J Neurol. (1997) 244:277–87. doi: 10.1007/s004150050087
2. Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. (2005) 52:1824–35. doi: 10.1002/art.21103
3. Preuße C, Goebel HH, Held J, Wengert O, Scheibe F, Irlbacher K, et al. Immune-mediated necrotizing myopathy is characterized by a specific Th1-M1 polarized immune profile. Am J Pathol. (2012) 181:2161–71. doi: 10.1016/j.ajpath.2012.08.033
4. Eisa-Beygi S, Hatch G, Noble S, Ekker M, Moon TW. The 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) pathway regulates developmental cerebral-vascular stability via prenylation-dependent signalling pathway. Dev Biol. (2013) 373:258–66. doi: 10.1016/j.ydbio.2012.11.024
5. Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology. (2009) 48:607–12. doi: 10.1093/rheumatology/kep078
6. Targoff IN. Autoantibodies to aminoacyl-transfer RNA synthetases for isoleucine and glycine. Two additional synthetases are antigenic in myositis. J Immunol. (1990) 144:1737–43.
7. Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. (2001) 60:116–23. doi: 10.1136/ard.60.2.116
8. Mathews MB, Reichlin M, Hughes GR, Bernstein RM. Anti-threonyl-tRNA synthetase, a second myositis-related autoantibody. J Exp Med. (1984) 160:420–34. doi: 10.1084/jem.160.2.420
9. Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol. (1997) 109:32–40. doi: 10.1046/j.1365-2249.1997.4081308.x
10. Mileti LM, Strek ME, Niewold TB, Curran JJ, Sweiss NJ. Clinical characteristics of patients with anti-Jo-1 antibodies: a single center experience. J Clin Rheumatol. (2009) 15:254–5. doi: 10.1097/RHU.0b013e3181b0e910
11. Targoff IN, Trieu EP, Miller FW. Reaction of anti-OJ autoantibodies with components of the multi-enzyme complex of aminoacyl-tRNA synthetases in addition to isoleucyl-tRNA synthetase. J Clin Invest. (1993) 91:2556–64. doi: 10.1172/JCI116493
12. Bunn CC, Bernstein RM, Mathews MB. Autoantibodies against alanyl-tRNA synthetase and tRNAAla coexist and are associated with myositis. J Exp Med. (1986) 163:1281–91. doi: 10.1084/jem.163.5.1281
13. Hirakata M, Suwa A, Nagai S, Kron MA, Trieu EP, Mimori T, et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol. (1999) 162:2315–20.
14. Hirakata M, Suwa A, Takada T, Sato S, Nagai S, Genth E, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum. (2007) 56:1295–303. doi: 10.1002/art.22506
15. Vartanian OA. Detection of autoantibodies against phenylalanyl-, tyrosyl-, and tryptophanyl-tRNA-synthetase and anti-idiotypic antibodies to it in serum from patients with autoimmune diseases. Mol Biol. (1991) 25:1033–9.
16. Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology. (2007) 46:1005–8. doi: 10.1093/rheumatology/kem045
17. Vincze M, Molnár PA, Tumpek J, Szollosi L, Gyetvai A, Kapitány A, et al. An unusual association: anti-Jo1 and anti-SRP antibodies in the serum of a patient with polymyositis. Clin Rheumatol. (2010) 29:811–4. doi: 10.1007/s10067-010-1394-6
18. Genth E, Mierau R. Diagnostic significance of scleroderma and myositis-associated autoantibodies. Z Rheumatol. (1995) 54:39–49.
19. Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Fujita T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. (2009) 60:2193–200. doi: 10.1002/art.24621
20. Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology. (2012) 51:800–4. doi: 10.1093/rheumatology/ker408
21. Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. (1985) 28:796–803. doi: 10.1002/art.1780280711
22. Hirakata M. Autoantibodies and their clinical significance in idiopathic inflammatory myopathies; polymyositis/dermatomyositis and related conditions. Nihon Rinsho Meneki Gakkai Kaishi. (2007) 30:444–54. doi: 10.2177/jsci.30.444
23. Gunawardena H, Betteridge ZE, McHugh NJ. Newly identified autoantibodies: relationship to idiopathic inflammatory myopathy subsets and pathogenesis. Curr Opin Rheumatol. (2008) 20:675–80. doi: 10.1097/BOR.0b013e328313bff4
24. Aouizerate J, De Antonio M, Bader-Meunier B, Barnerias C, Bodemer C, Isapof A, et al. Muscle ischaemia associated with NXP2 autoantibodies: a severe subtype of juvenile dermatomyositis. Rheumatology. (2018) 57:873–9. doi: 10.1093/rheumatology/kex516
25. Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WE, Cooper RG, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. (2009) 68:1621–5. doi: 10.1136/ard.2008.097162
26. Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. (2011) 13:209. doi: 10.1186/ar3275
27. Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O'Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. (2006) 54:3682–9. doi: 10.1002/art.22164
28. Mohassel P, Mammen AL. Anti-HMGCR myopathy. J Neuromuscul Dis. (2018) 5:11–20. doi: 10.3233/JND-170282
29. Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. (2011) 63:713–21. doi: 10.1002/art.30156
30. Padala S, Thompson PD. Statins as a possible cause of inflammatory and necrotizing myopathies. Atherosclerosis. (2012) 222:15–21. doi: 10.1016/j.atherosclerosis.2011.11.005
31. Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. (2010) 62:2757–66. doi: 10.1002/art.27572
32. Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis. (2007) 66:1345–9. doi: 10.1136/ard.2006.068502
33. Ghirardello A, Doria A. New insights in myositis-specific autoantibodies. Curr Opin Rheumatol. (2018) 30:614–22. doi: 10.1097/BOR.0000000000000548
34. Tawara N, Yamashita S, Zhang X, Korogi M, Zhang Z, Doki T, et al. Pathomechanisms of anti-cytosolic 5'-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann Neurol. (2017) 81:512–25. doi: 10.1002/ana.24919
35. Lilleker JB, Rietveld A, Pye SR, Mariampillai K, Benveniste O, Peeters MT, et al. Cytosolic 5'-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis. (2017) 76:862–8. doi: 10.1136/annrheumdis-2016-210282
36. Herbert MK, Stammen-Vogelzangs J, Verbeek MM, Rietveld A, Lundberg IE, Chinoy H, et al. Disease specificity of autoantibodies to cytosolic 5'-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis. (2016) 75:696–701. doi: 10.1136/annrheumdis-2014-206691
37. Mimori T, Hinterberger M, Pettersson I, Steitz JA. Autoantibodies to the U2 small nuclear ribonucleoprotein in a patient with scleroderma-polymyositis overlap syndrome. J Biol Chem. (1984) 259:560–5.
38. Hilker M, Tröster H, Grölz D, Hake U, Bachmann M. The autoantigen La/SS-B: analysis of the expression of alternatively spliced La mRNA isoforms. Cell Tissue Res. (1996) 284:383–9. doi: 10.1007/s004410050599
39. Teppo AM, Gripenberg M, Kurki P, Baklien K, Helve T, Wegelius O. Purification and characterization of a nuclear SS-B antigen. Scand J Immunol. (1982) 15:1–7. doi: 10.1111/j.1365-3083.1982.tb00615.x
40. Pakozdi A, Nihtyanova S, Moinzadeh P, Ong VH, Black CM, Denton CP. Clinical and serological hallmarks of systemic sclerosis overlap syndromes. J Rheumatol. (2011) 38:2406–9. doi: 10.3899/jrheum.101248
41. Váncsa A, Gergely L, Ponyi A, Lakos G, Németh J, Szodoray P, et al. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: Relevance for clinical classification: retrospective study of 169 patients. Joint Bone Spine. (2010) 77:125–30. doi: 10.1016/j.jbspin.2009.08.008
42. Limaye VS, Cassidy J, Scott G, Roberts-Thomson PJ, Gillis D. Anti-Ro52 antibodies, antisynthetase antibodies, and antisynthetase syndrome. Clin Rheumatol. (2008) 27:521–3. doi: 10.1007/s10067-007-0762-3
43. Ghirardello A, Zampieri S, Tarricone E, Iaccarino L, Bendo R, Briani C, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity. (2006) 39:217–21. doi: 10.1080/08916930600622645
44. Wasicek CA, Reichlin M, Montes M, Raghu G. Polymyositis and interstitial lung disease in a patient with anti-Jo1 prototype. Am J Med. (1984) 76:538–44. doi: 10.1016/0002-9343(84)90677-6
45. Yang DC, Dang CV, Arnett FC. Rat liver histidyl-tRNA synthetase. Purification and inhibition by the myositis-specific anti-Jo-1 autoantibody. Biochem Biophys Res Commun. (1984) 120:15–21. doi: 10.1016/0006-291X(84)91407-4
46. Brouwer R, Vree Egberts W, Jongen PH, van Engelen BG, van Venrooij WJ. Frequent occurrence of anti-tRNA(His) autoantibodies that recognize a conformational epitope in sera of patients with myositis. Arthritis Rheum. (1998) 41:1428–37. doi: 10.1002/1529-0131(199808)41:8<1428::AID-ART12>3.0.CO;2-J
47. Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest. (1990) 85:468–75. doi: 10.1172/JCI114461
48. O'Hanlon TP, Rider LG, Mamyrova G, Targoff IN, Arnett FC, Reveille JD, et al. HLA polymorphisms in African Americans with idiopathic inflammatory myopathy: allelic profiles distinguish patients with different clinical phenotypes and myositis autoantibodies. Arthritis Rheum. (2006) 54:3670–81. doi: 10.1002/art.22205
49. Gono T, Kawaguchi Y, Kuwana M, Sugiura T, Furuya T, Takagi K, et al. Brief report: association of HLA-DRB1*0101/*0405 with susceptibility to anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis in the Japanese population. Arthritis Rheum. (2012) 64:3736–40. doi: 10.1002/art.34657
50. Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology. (2008) 47:324–8. doi: 10.1093/rheumatology/kem359
51. Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology. (2007) 46:25–8. doi: 10.1093/rheumatology/kel161
52. Fiorentino D, Casciola-Rosen L. TIF1 autoantibodies in dermatomyositis shed insight into the cancer-myositis connection. Arthritis Rheum. (2012) 64:346–9. doi: 10.1002/art.33402
53. Fujimoto M, Hamaguchi Y, Kaji K, Matsushita T, Ichimura Y, Kodera M, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. (2012) 64:513–22. doi: 10.1002/art.33403
54. Kawai T, Akira S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med. (2011) 3:513–27. doi: 10.1002/emmm.201100160
55. Khetchoumian K, Teletin M, Mark M, Lerouge T, Cerviño M, Oulad-Abdelghani M, et al. TIF1delta, a novel HP1-interacting member of the transcriptional intermediary factor 1 (TIF1) family expressed by elongating spermatids. J Biol Chem. (2004) 279:48329–41. doi: 10.1074/jbc.M404779200
56. Venturini L, You J, Stadler M, Galien R, Lallemand V, Koken MH, et al. TIF1gamma, a novel member of the transcriptional intermediary factor 1 family. Oncogene. (1999) 18:1209–17. doi: 10.1038/sj.onc.1202655
57. Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS. Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell. (2011) 43:85–96. doi: 10.1016/j.molcel.2011.05.020
58. Venalis P, Selickaja S, Lundberg K, Rugiene R, Lundberg IE. Association of anti-transcription intermediary factor 1gamma antibodies with paraneoplastic rheumatic syndromes other than dermatomyositis. Arthritis Care Res. (2018) 70:648–51. doi: 10.1002/acr.23325
59. Pinal-Fernandez I, Ferrer-Fabregas B, Trallero-Araguas E, Balada E, Martínez MA, Milisenda JC, et al. Tumour TIF1 mutations and loss of heterozygosity related to cancer-associated myositis. Rheumatology. (2018) 57:388–96. doi: 10.1093/rheumatology/kex413
60. Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science. (2014) 343:152–7. doi: 10.1126/science.1246886
61. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
62. Parkes JE, Rothwell S, Oldroyd A, Chinoy H, Lamb JA, Myositis Genetics Consortium (MYOGEN). Genetic background may contribute to the latitude-dependent prevalence of dermatomyositis and anti-TIF1-gamma autoantibodies in adult patients with myositis. Arthritis Res Ther. (2018) 20:117. doi: 10.1186/s13075-018-1617-9
63. Wang HB, Zhang Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res. (2001) 29:2517–21. doi: 10.1093/nar/29.12.2517
64. Burd CJ, Kinyamu HK, Miller FW, Archer TK. UV radiation regulates Mi-2 through protein translation and stability. J Biol Chem. (2008) 283:34976–82. doi: 10.1074/jbc.M805383200
65. Okada S, Weatherhead E, Targoff IN, Wesley R, Miller FW. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. (2003) 48:2285–93. doi: 10.1002/art.11090
66. Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A. Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum. (2009) 60:3784–93. doi: 10.1002/art.24977
67. O'Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine. (2006) 85:111–27. doi: 10.1097/01.md.0000217525.82287.eb
68. Ghirardello A, Bassi N, Palma L, Borella E, Domeneghetti M, Punzi L, et al. Autoantibodies in polymyositis and dermatomyositis. Curr Rheumatol Rep. (2013) 15:335. doi: 10.1007/s11926-013-0335-1
69. Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum. (2007) 56:3132–7. doi: 10.1002/art.22862
70. Ullu E, Weiner AM. Human genes and pseudogenes for the 7SL RNA component of signal recognition particle. EMBO J. (1984) 3:3303–10. doi: 10.1002/j.1460-2075.1984.tb02294.x
71. Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. (1990) 33:1361–70. doi: 10.1002/art.1780330908
72. Janda CY, Li J, Oubridge C, Hernández H, Robinson CV, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. (2010) 465:507–10. doi: 10.1038/nature08870
73. Satoh T, Okano T, Matsui T, Watabe H, Ogasawara T, Kubo K, et al. Novel autoantibodies against 7SL RNA in patients with polymyositis/dermatomyositis. J Rheumatol. (2005) 32:1727–33.
74. Okada N, Mimori T, Mukai R, Kashiwagi H, Hardin JA. Characterization of human autoantibodies that selectively precipitate the 7SL RNA component of the signal recognition particle. J Immunol. (1987) 138:3219–23.
75. Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with the signal-recognition particle. Proc Natl Acad Sci USA. (1986) 83:9507–11. doi: 10.1073/pnas.83.24.9507
76. Römisch K, Miller FW, Dobberstein B, High S. Human autoantibodies against the 54 kDa protein of the signal recognition particle block function at multiple stages. Arthritis Res Ther. (2006) 8:R39. doi: 10.1186/ar1895
77. Bergua C, Chiavelli H, Allenbach Y, Arouche-Delaperche L, Arnoult C, Bourdenet G, et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis. (2018) 78:131–9. doi: 10.1136/annrheumdis-2018-213518
78. Benveniste O, Drouot L, Jouen F, Charuel JL, Bloch-Queyrat C, Behin A, et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis Rheum. (2011) 63:1961–71. doi: 10.1002/art.30344
79. Betteridge ZE, Gunawardena H, McHugh NJ. Pathogenic mechanisms of disease in myositis: autoantigens as clues. Curr Opin Rheumatol. (2009) 21:604–9. doi: 10.1097/BOR.0b013e328331638a
80. Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. (2008) 20:17–22. doi: 10.1016/j.coi.2008.01.002
81. Nakashima R, Imura Y, Kobayashi S, Yukawa N, Yoshifuji H, Nojima T, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology. (2010) 49:433–40. doi: 10.1093/rheumatology/kep375
82. Gono T, Kawaguchi Y, Satoh T, Kuwana M, Katsumata Y, Takagi K, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology. (2010) 49:1713–9. doi: 10.1093/rheumatology/keq149
83. Szodoray P, Szabó Z, Kapitány A, Gyetvai A, Lakos G, Szántó S, et al. Anti-citrullinated protein/peptide autoantibodies in association with genetic and environmental factors as indicators of disease outcome in rheumatoid arthritis. Autoimmun Rev. (2010) 9:140–3. doi: 10.1016/j.autrev.2009.04.006
84. Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, et al. NXP-2 association with SUMO-2 depends on lysines required for transcriptional repression. Proc Natl Acad Sci USA. (2006) 103:5308–13. doi: 10.1073/pnas.0601066103
85. Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. (2002) 28:859–90. doi: 10.1016/S0889-857X(02)00032-7
86. Männistö T, Surcel HM, Bloigu A, Ruokonen A, Hartikainen AL, Järvelin MR, et al. The effect of freezing, thawing, and short- and long-term storage on serum thyrotropin, thyroid hormones, and thyroid autoantibodies: implications for analyzing samples stored in serum banks. Clin Chem. (2007) 53:1986–7. doi: 10.1373/clinchem.2007.091371
87. Gislefoss RE, Grimsrud TK, Morkrid L. Stability of selected serum proteins after long-term storage in the Janus Serum Bank. Clin Chem Lab Med. (2009) 47:596–603. doi: 10.1515/CCLM.2009.121
88. Nakashima R, Hosono Y, Mimori T. Clinical significance and new detection system of autoantibodies in myositis with interstitial lung disease. Lupus. (2016) 25:925–33. doi: 10.1177/0961203316651748
89. Aggarwal R, Oddis CV, Goudeau D, Fertig N, Metes I, Stephens C, et al. Anti-transcription intermediary factor 1-gamma autoantibody ELISA development and validation. Rheumatology. (2014) 53:433–7. doi: 10.1093/rheumatology/ket383
90. Fujimoto M, Murakami A, Kurei S, Okiyama N, Kawakami A, Mishima M, et al. Enzyme-linked immunosorbent assays for detection of anti-transcriptional intermediary factor-1 gamma and anti-Mi-2 autoantibodies in dermatomyositis. J Dermatol Sci. (2016) 84:272–81. doi: 10.1016/j.jdermsci.2016.09.013
91. Johnson C, Pinal-Fernandez I, Parikh R, Paik J, Albayda J, Mammen AL, et al. Assessment of mortality in autoimmune myositis with and without associated interstitial lung disease. Lung. (2016) 194:733–7. doi: 10.1007/s00408-016-9896-x
92. Qiang JK, Kim WB, Baibergenova A, Alhusayen R. Risk of malignancy in dermatomyositis and polymyositis. J Cutan Med Surg. (2017) 21:131–6. doi: 10.1177/1203475416665601
93. Cavagna L, Nuño L, Scirè CA, Govoni M, Longo FJ, Franceschini F, et al. Serum Jo-1 autoantibody and isolated arthritis in the antisynthetase syndrome: review of the literature and report of the experience of AENEAS collaborative group. Clin Rev Allergy Immunol. (2017) 52:71–80. doi: 10.1007/s12016-016-8528-9
94. Klein M, Mann H, Pleštilová L, Betteridge Z, McHugh N, Remáková M, et al. Arthritis in idiopathic inflammatory myopathy: clinical features and autoantibody associations. J Rheumatol. (2014) 41:1133–9. doi: 10.3899/jrheum.131223
95. Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. (2007) 56:3125–31. doi: 10.1002/art.22865
96. Mescam-Mancini L, Allenbach Y, Hervier B, Devilliers H, Mariampillay K, Dubourg O, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain. (2015) 138:2485–92. doi: 10.1093/brain/awv192
97. Maturu VN, Lakshman A, Bal A, Dhir V, Sharma A, Garg M, et al. Antisynthetase syndrome: an under-recognized cause of interstitial lung disease. Lung India. (2016) 33:20–6. doi: 10.4103/0970-2113.173055
98. Shi J, Li S, Yang H, Zhang Y, Peng Q, Lu X, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol. (2017) 44:1051–7. doi: 10.3899/jrheum.161480
99. Labirua-Iturburu A, Selva-O'Callaghan A, Vincze M, Dankó K, Vencovsky J, Fisher B, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Medicine. (2012) 91:206–11. doi: 10.1097/MD.0b013e318260977c
100. Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum. (2012) 41:890–9. doi: 10.1016/j.semarthrit.2011.09.008
101. Madan V, Chinoy H, Griffiths CEM, Cooper RG. Defining cancer-risk, and assessing diagnostic usefulness of myositis serology, in dermatomyositis- Part 2. Clin Exp Dermatol. (2009) 34:561–5. doi: 10.1111/j.1365-2230.2009.03227.x
102. Aggarwal R, Oddis CV, Goudeau D, Koontz D, Qi Z, Reed AM, et al. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab. Rheumatology. (2016) 55:1710. doi: 10.1093/rheumatology/kew275
103. Rider LG, Shah M, Mamyrova G, Huber AM, Rice MM, Targoff IN, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. (2013) 92:223–43. doi: 10.1097/MD.0b013e31829d08f9
104. Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. (2013) 92:25–41. doi: 10.1097/MD.0b013e31827f264d
105. Trallero-Araguás E, Rodrigo-Pendás JÁ, Selva-O'Callaghan A, Martínez-Gómez X, Bosch X, Labrador-Horrillo M, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum. (2012) 64:523–32. doi: 10.1002/art.33379
106. Chiu YE, Co DO. Juvenile dermatomyositis: immunopathogenesis, role of myositis-specific autoantibodies, and review of rituximab use. Pediatr Dermatol. (2011) 28:357–67. doi: 10.1111/j.1525-1470.2011.01501.x
107. Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol. (2012) 24:602–8. doi: 10.1097/BOR.0b013e328358bd85
108. Espada G, Maldonado Cocco JA, Fertig N, Oddis CV. Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J Rheumatol. (2009) 36:2547–51. doi: 10.3899/jrheum.090461
109. Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Franceschini F, Quinzanini M, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther. (2012) 14:R97. doi: 10.1186/ar3822
110. Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. (2012) 71:710–3. doi: 10.1136/annrheumdis-2011-200697
111. Chance B, Younkin DP, Kelley R, Bank WJ, Berkowitz HD, Argov Z, et al. Magnetic resonance spectroscopy of normal and diseased muscles. Am J Med Genet. (1986) 25:659–79. doi: 10.1002/ajmg.1320250408
112. Tansley SL, Betteridge ZE, Gunawardena H, Jacques TS, Owens CM, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. (2014) 16:R138. doi: 10.1186/ar4600
113. Palterer B, Vitiello G, Carraresi A, Giudizi MG, Cammelli D, Parronchi P. Bench to bedside review of myositis autoantibodies. Clin Mol Allergy. (2018) 16:5. doi: 10.1186/s12948-018-0084-9
114. Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. (2011) 65:25–34. doi: 10.1016/j.jaad.2010.09.016
115. Hall JC, Casciola-Rosen L, Samedy LA, Werner J, Owoyemi K, Danoff SK, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. (2013) 65:1307–15. doi: 10.1002/acr.21992
116. Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. (2012) 384:128–34. doi: 10.1016/j.jim.2012.07.019
117. Muro Y, Sugiura K, Akiyama M. Low prevalence of anti-small ubiquitin-like modifier activating enzyme antibodies in dermatomyositis patients. Autoimmunity. (2013) 46:279–84. doi: 10.3109/08916934.2012.755958
118. Rider LG, Katz JD, Jones OY. Developments in the classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am. (2013) 39:877–904. doi: 10.1016/j.rdc.2013.06.001
119. Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. (2011) 147:391–8. doi: 10.1001/archdermatol.2011.52
120. Rouster-Stevens KA, Pachman LM. Autoantibody to signal recognition particle in African American girls with juvenile polymyositis. J Rheumatol. (2008) 35:927–9.
121. Momomura M, Miyamae T, Nozawa T, Kikuchi M, Kizawa T, Imagawa T, et al. Serum levels of anti-SRP54 antibodies reflect disease activity of necrotizing myopathy in a child treated effectively with combinatorial methylprednisolone pulses and plasma exchanges followed by intravenous cyclophosphamide. Mod Rheumatol. (2014) 24:529–31. doi: 10.3109/14397595.2013.852852
122. Mammen AL, Pak K, Williams EK, Brisson D, Coresh J, Selvin E, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res. (2012) 64:269–72. doi: 10.1002/acr.20662
123. Allenbach Y, Drouot L, Rigolet A, Charuel JL, Jouen F, Romero NB, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine. (2014) 93:150–7. doi: 10.1097/MD.0000000000000028
124. Musset L, Allenbach Y, Benveniste O, Boyer O, Bossuyt X, Bentow C, et al. Anti-HMGCR antibodies as a biomarker for immune-mediated necrotizing myopathies: a history of statins and experience from a large international multi-center study. Autoimmun Rev. (2016) 15:983–93. doi: 10.1016/j.autrev.2016.07.023
125. Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. (2012) 64:4087–93. doi: 10.1002/art.34673
126. Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve. (2013) 48:477–83. doi: 10.1002/mus.23854
127. Waters MJ, Limaye V. Clinico-serologic features of statin-induced necrotising autoimmune myopathy in a single-centre cohort. Clin Rheumatol. (2018) 37:543–7. doi: 10.1007/s10067-017-3831-2
128. Kishi T, Rider LG, Pak K, Barillas-Arias L, Henrickson M, McCarthy PL, et al. Association of Anti-3-Hydroxy-3-Methylglutaryl-Coenzyme A reductase autoantibodies with DRB1*07:01 and severe myositis in Juvenile myositis patients. Arthritis Care Res. (2017) 69:1088–94. doi: 10.1002/acr.23113
129. Liang WC, Uruha A, Suzuki S, Murakami N, Takeshita E, Chen WZ, et al. Pediatric necrotizing myopathy associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies. Rheumatology. (2017) 56:287–93. doi: 10.1093/rheumatology/kew386
130. Shovman O, Gilburd B, Chayat C, Lazar AD, Amital H, Blank M, et al. Anti-HMGCR antibodies demonstrate high diagnostic value in the diagnosis of immune-mediated necrotizing myopathy following statin exposure. Immunol Res. (2017) 65:276–81. doi: 10.1007/s12026-016-8867-x
131. Droney L, Gillis D, Wong R. New immunoassays for anti-HMG-CoA reductase antibodies may lead to incorrect diagnosis in inflammatory myositis. Pathology. (2017) 49:638–9. doi: 10.1016/j.pathol.2017.04.013
132. Alvarado-Cardenas M, Marin-Sánchez A, Martínez MA, Martínez-Martínez L, Pinal-Fernandez I, Labrador-Horrillo M, et al. Statin-associated autoimmune myopathy: a distinct new IFL pattern can increase the rate of HMGCR antibody detection by clinical laboratories. Autoimmun Rev. (2016) 15:1161–6. doi: 10.1016/j.autrev.2016.09.005
133. Palterer B, Cammelli D, Vitiello G, Giudizi MG. Anti-HMGCR and anti-DFS70 antibodies immunofluorescence patterns. Autoimmun Rev. (2017) 16:321–2. doi: 10.1016/j.autrev.2017.01.002
134. Allenbach Y, Keraen J, Bouvier AM, Jooste V, Champtiaux N, Hervier B, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. (2016) 139:2131–5. doi: 10.1093/brain/aww054
135. Zampieri S, Valente M, Adami N, Biral D, Ghirardello A, Rampudda ME, et al. Polymyositis, dermatomyositis and malignancy: a further intriguing link. Autoimmun Rev. (2010) 9:449–53. doi: 10.1016/j.autrev.2009.12.005
136. Buchbinder R, Hill CL. Malignancy in patients with inflammatory myopathy. Curr Rheumatol Rep. 4:415–26. doi: 10.1007/s11926-002-0087-9
137. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 357:96–100. doi: 10.1016/S0140-6736(00)03540-6
138. Buchbinder R, Forbes A, Hall S, Dennett X, Giles G. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based Cohort study. Ann Intern Med. (2001) 134:1087–95. doi: 10.7326/0003-4819-134-12-200106190-00008
139. Airio A, Pukkala E, Isomäki H. Elevated cancer incidence in patients with dermatomyositis: a population based study. J Rheumatol. (1995) 22:1300–3.
140. Barnes BE. Dermatomyositis and malignancya review of the literature. Ann Intern Med. (1976) 84:68–76. doi: 10.7326/0003-4819-84-1-68
141. Bonnetblanc JM, Bernard P, Fayol J. Dermatomyositis and malignancy. A multicenter cooperative study. Dermatologica. (1990) 180:212–6. doi: 10.1159/000248032
142. Chen YJ, Wu CY, Huang YL, Wang CB, Shen JL, Chang YT. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. (2010) 12:R70. doi: 10.1186/ar2987
143. Chow WH, Gridley G, Mellemkjaer L, McLaughlin JK, Olsen JH, Fraumeni JF. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. (1995) 6:9–13. doi: 10.1007/BF00051675
144. Maoz CR, Langevitz P, Livneh A, Blumstein Z, Sadeh M, Bank I, et al. High incidence of malignancies in patients with dermatomyositis and polymyositis: an 11-year analysis. Semin Arthritis Rheum. (1998) 27:319–24. doi: 10.1016/S0049-0172(98)80052-8
145. Zahr ZA, Baer AN. Malignancy in myositis. Curr Rheumatol Rep. (2011) 13:208–15. doi: 10.1007/s11926-011-0169-7
146. Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-γ antibodies have clinical significance for patients with dermatomyositis. Rheumatology. (2010) 49:1726–33. doi: 10.1093/rheumatology/keq153
147. Selva-O'Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol. (2010) 22:627–32. doi: 10.1097/BOR.0b013e32833f1075
Keywords: myositis, inflammation, autoantibodies, antigens, biomarker
Citation: Stuhlmüller B, Schneider U, González-González J-B and Feist E (2019) Disease Specific Autoantibodies in Idiopathic Inflammatory Myopathies. Front. Neurol. 10:438. doi: 10.3389/fneur.2019.00438
Received: 03 December 2018; Accepted: 10 April 2019;
Published: 08 May 2019.
Edited by:
Stefan Bittner, Johannes Gutenberg University Mainz, GermanyReviewed by:
Reinhild Klein, University of Tübingen, GermanyPatrick Joseph Waters, University of Oxford, United Kingdom
Copyright © 2019 Stuhlmüller, Schneider, González-González and Feist. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugen Feist ZXVnZW4uZmVpc3RAY2hhcml0ZS5kZQ==