 Simone Migliore
Simone Migliore Joseph Jankovic
Joseph Jankovic Ferdinando Squitieri
Ferdinando Squitieri- 1Huntington and Rare Diseases Unit, Fondazione IRCCS Casa Sollievo Della Sofferenza Research Hospital, San Giovanni Rotondo, Italy
- 2Department of Neurology, Parkinson's Disease Center and Movement Disorders Clinic, Baylor College of Medicine, Houston, TX, United States
Huntington's disease (HD) is a rare, hereditary, neurodegenerative and dominantly transmitted disorder affecting about 10 out of 100,000 people in Western Countries. The genetic cause is a CAG repeat expansion in the huntingtin gene (HTT), which is unstable and may further increase its length in subsequent generations, so called anticipation. Mutation repeat length coupled with other gene modifiers and environmental factors contribute to the age at onset in the offspring. Considering the unpredictability of age at onset and of clinical prognosis in HD, the accurate interpretation, a proper psychological support and a scientifically sound and compassionate communication of the genetic test result are crucial in the context of Good Clinical Practice and when considering further potential disease-modifying therapies. We discuss various genetic test scenarios that require a particularly careful attention in psychological and genetic counseling and expect that the counseling procedures will require a constant update.
Introduction
Huntington's disease (HD) is a rare, hereditary, dominantly transmitted, neurodegenerative disease that leads to severe motor, cognitive, and psychiatric disability at any age, usually earlier in offspring than in their affected parent (i.e., onset anticipation phenomenon) (1, 2). The initial manifestation is typically chorea, but other movement disorders such as dystonia, myoclonus and parkinsonism may also be present (3). As the disease advances, dystonia and parkinsonism (eg., rigidity, bradykinesia) worsen the patients' functional capacity and independence and overlap to choreic movements, accompanied by gradual cognitive decline and dementia and body cachexia in the final stages of the disease.
HD is caused by an expanded and unstable CAG repeat mutation in the huntingtin (HTT) gene on chromosome 4p16.3 (4). CAG length above 35 is considered a pathogenic mutation (5) but there is a low penetrance with CAG expansions in the range 36–39. In this range the disease may not be clinically manifested until advanced age (6). On the other hand, CAG repeat instability, due to the intergenerational change of expanded number of trinucleotides, may result in highly expanded mutations beyond 60 that are highly penetrant and generally associated with young age at onset starting before age 20 (7); some patients with pediatric onset HD (PHD) may manifest a more severe HD variant and have CAG repeats >80 (2). Usually, the most extended CAG repeat expansions causing PHD in young children depend on paternal genetic transmission (2) and related germline CAG repeat instability (8).
Although a genetic test for HD is commercially available and can be ordered even before the onset of symptoms, the results are often misinterpreted and inappropriately applied in genetic counseling (9). HD is dominantly transmitted from an affected parent with each offspring having 50% chance of inheriting an unstable CAG expansion of variable repeat length in the mutated gene (10). The unexpanded repeat length in the general population is genetically influenced by specific haplotypes, with an intergenerational stepwise mechanism of CAG repeat increase, which is larger in Western than in Far East populations (11). As a direct consequence of intergenerational CAG repeat increase of borderline length alleles, i.e., the so-called intermediate alleles (IA 27−35CAG), there are new HD mutations originating from apparently unaffected parents (10). Unstable CAG repeats are generally associated with specific HTT Single Nucleotide Polymorphism (SNP) haplogroups, whose frequency is higher in population of European descent and affect the prevalence of the disease (12).
Considering the unpredictability of age at onset and of clinical prognosis in HD and the many possible genetic and clinical scenarios associated with the potentially diverse inherited pre-mutational and mutational conditions, accurate interpretation and scientifically sound and compassionate communication of the genetic test result is crucial in the context of Good Clinical Practice (13). This is especially important now as new interventional trials by potentially disease-modifying therapies are on horizon (14). We review here some genetic test scenarios that, in our experience, require a particularly careful attention in psychological and genetic counseling.
Counseling in Case of Intermediate Alleles (27–35 CAG Repeats)
Although the rate of new mutations is unpredictable, some studies postulated a frequency of about 7% of new expanded mutations originating from IAs in some populations and that the frequency of IAs influence the prevalence of HD in the general population (15). There is growing body of evidence that IAs themselves may be associated with clinical manifestations of HD, although the possibility of mild phenotypes or subclinical traits, so called endophenotypes, in some of the cases should be also considered (16). Recent evidence highlighted the association between IAs and behavioral and cognitive issues (17–21), as well as the occurrence of motor features, indistinguishable from typical HD (16). In such a scenario, the communication of the genetic test result might be problematic as it has several implications not only for the individual but also for the proband's entire family. For example, siblings of a new mutation originated from a parent carrying an IA share a risk to inherit a HD mutation themselves below 50%. Therefore, even considering the potentially high frequency of IA that may be as high as 6% of all normal alleles in some populations (16), proper counseling procedures with specific competence in clinical manifestation and genetics of HD (22), coupled with knowledge of published international guidelines (23), are critical. In families without established diagnosis of HD, DNA analysis from other members of the family are often helpful. If, for example, one of the parents or adult offsprings has an IA, this would provide support for the proband's polymorphism being “pathological,” indicating increased risk for the transmission of HD and development of symptoms in the future. It is, however, difficult to predict with any certainty the degree of risk or the age at onset of symptoms, although generally individuals with low expanded repeats have a relatively late age at onset and slow progression of the disease. Individuals carrying an IA should be carefully counseled on the potential risk of genetic transmission to children, mainly if they carry an IA≥33, i.e., near to the pathological edge of repeats and the highest risk to transmit new mutations (24). Pre-test counseling should therefore contemplate the increased potentiality of a large IA to expand in offspring and the current international guidelines (23) should be further updated accordingly. Furthermore, participation in registries and observational studies, such as ENROLL-HD (25), should be encouraged to clinically monitor these conditions (Table 1, Row A).
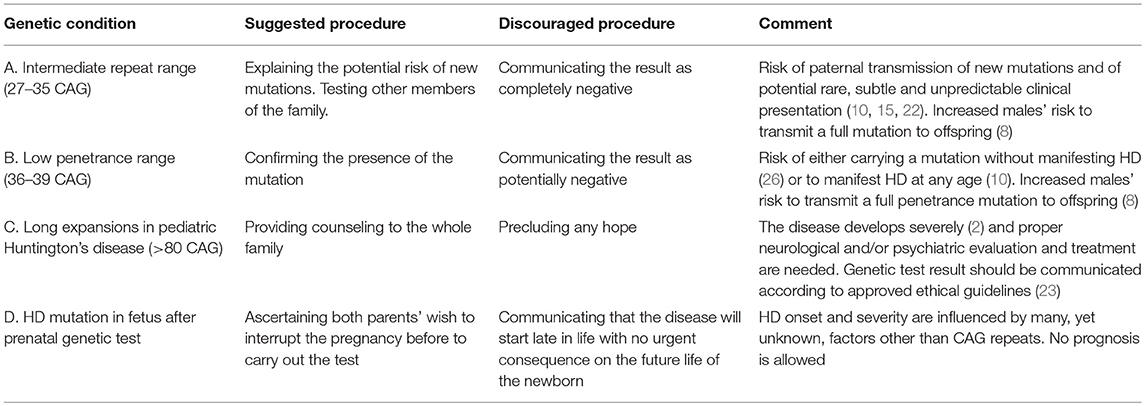
Table 1. Genetic counseling in Huntington disease.
The mechanism by which IAs result in a clinical phenotype is not well-understood but instability of CAG expansion may determine trinucleotide repeat number variability during the intergenerational parent-child mutation transmission with related somatic mosaicism, i.e., cell populations carrying different expanded repeat lengths in several body tissues (27), especially in differentiated neurons (e.g., in the striatal medium spiny neurons or cortex) (28), which may explain this phenomenon (10, 29). There is already evidence of increased mosaicism of the full CAG mutation in brain striatum neurons of transgenic mice and of symptomatic patients with large size mutations in the post-mortem neuropathological brain sample (28, 30). Therefore, we cannot exclude that CAG instability of IAs may play a key role in determining somatic triplet mosaicism, some cell populations having full mutations in the central nervous system. In other words, subjects carrying IAs in their blood cells might theoretically have a mosaic of CAG lengths, including full mutations in their brain cells and this phenomenon might contribute to symptoms of HD or endophenotypes. Future studies on peripheral, easy-to access, tissues, e.g., blood, buccal, sperm cells, might offer new chances to investigate potential markers of CAG mosaicism. In this regard, genes or their products that either facilitate somatic expansion or increase CAG mosaicism in different body tissues, may represent additional therapeutic targets (2, 31).
Counseling in Case of Low Penetrance Alleles (36–39 CAG Repeats)
When the polymorphic CAG stretch is longer than 35, the disease may develop at any age with tendency to anticipate before the affected parent's onset age. Allele repeat numbers included in 36–39 CAG length are considered low penetrance mutations (6, 10) and may randomly occur in the general population with an estimated frequency of 1/400 (26). In most of these cases, subjects do not show full HD phenotype (or will show some clinical symptoms sometime at advanced age). These conditions are difficult to detect, unless people with no evidence of positive family history show typical symptoms of HD. Considering the subtle and non-specific clinical manifestations and the possible negative or misinterpreted family history, most of these situations remain undiagnosed, thus contributing to the underestimate of the disease frequency. Communication of a genetic result when the mutation is in the low penetrance range is problematic because it may never be associated with development of a clinical manifestation of disease. (Table 1, Row B). There are many factors that must be considered during genetic counseling in these individuals, including penetrance variability determining age at onset, increased longevity in aging general population (32), parent-to-child onset anticipation with the offspring exhibiting symptoms at an earlier age than the parents, and even non-genetic factors (33) (Table 1, Row B). However, the parent-child transmission of a low penetrance mutation may cause an increased number of repeats in offspring. Considering the male germline susceptibility to CAG instability (24) and a non-negligible rate of silent low penetrance alleles in the general population (26), pre-test counseling sessions as well as a further update of the international ethical guidelines should address the potentiality of the magnitude of the risk of expansion for future generations.
However, many other environmental and genetic factors, other than the expanded CAG repeats, may contribute to explain the penetrance of the mutation as well as of IAs, and their effects on HD development (31, 34, 35). These, yet still far to be fully elucidated factors, will certainly offer additional clues and discussion to counseling guidelines in future.
Counseling in Case of Markedly Expanded Alleles (> 80 CAG Repeats)
CAG repeat expansions may sometimes greatly increase during the intergenerational parent-child transmission, sometimes reaching over 100 CAG repeats, resulting in PHD with onset before the age 12 (2, 10, 36, 37). PHD children with large-sized and somatically unstable mutations beyond 80 CAG repeats may be characterized by atypical clinical manifestations at onset and overtime, compared to the adult-onset variant and to other juvenile patients with relatively shorter repeats, mental and motor developmental delay, predominant gait disturbance and dystonia, seizures, reduced life span, and more-rapid motor progression (2). The interpretation of the genetic test in case of children should always take into account the international guidelines for testing minors (38) and include counseling of both parents whenever possible. Because the most frequent transmitting parent is the father in case of PHD with large expanded repeats, psychological counseling should take into account the distress not only to the affected individual and the affected parent, but also the caregiver (in this case the mother more often than the father). The genetic test result may, therefore, have a profound impact on the future life of all family members and need to be delivered carefully and compassionately (22) (Table 1, Row C). The interpretation of the most common CAG repeat expansions (e.g., 40–50 CAG) in young adults or minors with psychiatric, but no motor symptoms, is also challenging as the course of the disease is difficult to predict, although the age at onset and the progression often mirrors those in other, affected, adult members of the family (2). These cases will deserve additional in deep analysis for it concerns clinical and biological implications, psychological and genetic counseling management and therapeutic strategies.
Counseling in Case of Subjects Homozygous for CAG Repeat Expansions
This condition occurs very rarely in a dominant disease and is due to the intermarriage of two subjects heterozygous for expanded CAG repeat mutation. In this case subjects inherit two mutations (i.e., one from each mutation carrier parent). Reports of homozygous cases have described relatively frequent occurrence of intermarriages among consanguineous relatives in certain populations with high density of HD (i.e., the large Venezuelan kindred) (34, 39). Such rare condition does not seem to affect age at onset more severely than a heterozygous condition, due to the fully dominant, gain-of-function, effect of the mutation (40). Whether it affects the clinical presentation (i.e., more frequent atypical presentation than heterozygous patients), the progression of HD course, or both (39), still needs to be explored in larger cohorts. However, independent of the phenotype, subjects who carry two expanded mutations always transmit a CAG repeat expansion to their offspring whose risk of inheriting HD becomes of 100%, instead of the typical 50% (41). There are at least two main issues related to such particularly unfortunate condition. (1). How to communicate to at-risk offsprings that they will certainly inherit a HD mutation? (2). How to protect the privacy of the at-risk offspring, considering that the genetic result of their affected parent would unequivocally disclose their own risk to inherit HD mutation, independently from whether they request a genetic test or not? Caution is therefore needed by counselors in these cases and a further update of the international ethical guidelines should address such a rare occurrence (23).
Prenatal Counseling
Genetic predictive counseling should include the 5Cs: Consent, Counseling, Confidentiality, Cost, and Consequences. Prenatal counseling represents a unique example how advances in genetic and in-utero techniques must be translated into genetic counseling based on ethical and scientific principles before any important decision is considered, including the interruption of pregnancy (42). Reproductive choices for at risk individuals include: (i) the wish to have a pregnancy free of HD and without the risk of a mutation carrier offspring; (ii) the decision to test an ongoing pregnancy by chorionic villous sampling, which may be performed at 8–10 weeks after conception (the sample obtained from chorionic villi can be tested), with the option of termination of the pregnancy if the fetus carries the HD mutation; (iii) the decision to eventually perform the predictive testing before requesting the prenatal diagnosis (by contrast, in case of positive result in the fetus before the at risk parent undergoes the presymptomatic test, the genetic status of the at-risk parent would be indirectly revealed); (iv) the request of pre-implantation genetic diagnosis (PGD) that is performed as part of an in vitro fertilization (IVF) procedure at specialized IVF centers (42). The counselor should discuss each of these options so that the at risk couple has a chance to make the most informed reproductive decision since the pre-pregnancy stage, in agreement with recently reports (43). Clear and unambiguous communication, as well as a proper psychological support, are essential features to prevent any misunderstanding (Table 1, Row D). Unfortunately, miscommunication during counseling may result in unexpected outcome and may risk to lead to wrong decision makings by parents.
There are many ethical issues that must be considered before performing prenatal testing in an attempt to prevent HD. These include variability of HD mutation penetrance and the role of potential modifiers acting to modulate onset and progression severity of the disease. The understanding of natural history of HD is improving as a result of many published worldwide observational studies (2, 25). Accordingly, the genetic and psychological counseling should be performed by professionals who are well-informed about and experienced in HD.
Counseling and HD Family Organizations
One of the most challenging issues for all HD symptomatic and at-risk persons is the profound denial of the presence of HD in the family and discrimination and shunning of the affected individuals from their own family and from the society. In the United States, the Genetic Information Non-discrimination Act (GINA), enacted in 2008, was an important step toward preventing discrimination based on diagnosis of a neurodegenerative disease, such as HD. This act forbids employers from denying employment, promotions, or health insurance coverage to people when genetic tests show they have predisposition to an inherited disease. It also forbids health insurance companies to use genetic tests to determine an applicant's risk profile. Unfortunately, the act does not apply to life, disability, and long-term care insurance.
Lay organizations and support groups play a crucial role not only in creating bridges between scientists and patients, but also in spreading important information among families, including the importance of following the proper “procedure” if and when somebody discover or want to discover if he/she is gene positive. Moreover, they can also play a role—together with researchers and clinicians—in motivating patients and relatives to participate into research programs, i.e., clinical observational studies (i.e., ENROLL-HD) and therapeutic trials. HD Cope is an international coalition connecting three umbrella organizations: the HD Society of America (HDSA), the European Huntington Association (EHA), and the Huntington Society of Canada (HSC). These and other initiatives and organizations can play a critical role in improving quality of life of HD patients and their families.
Conclusion and Recommendations
In this review we attempt to summarize current knowledge about the HD mutation and how information about CAG repeats in the HTT gene can be used to provide sound genetic counseling, which can possibly extend to other neurodegenerative diseases (44). By involving families in observational (i.e., ENROLL-HD platform) and interventional trials, clinicians have an opportunity to include adult patients and their relatives (e.g., partners, symptomatic minors, premanifest adults) in research-based networks that provide current knowledge about developments in genetic testing and novel therapies in HD (45).
With advances in research and improved understanding of mechanisms of dysfunction, degeneration and abnormal development of brain in HD, innovative experimental therapies, such as antisense oligonucleotide (ASO), gene editing by CRSPR/CAS, or suppression of gene modifiers of age at onset may, hopefully, be transferred from basic research into clinics in a not so far future (14, 46, 47). Theoretically, such strategies will consequently require new competencies in the pre- and post-test counseling as well as in pre- and post-conceptional approaches. Moreover, we need to remind that we still need to fully understand how HD develops and why there is such a wide spectrum of heterogeneous clinical manifestations among patients. Accordingly, new potential challenges in genetic diagnosis will require further and constant updates of guidelines and the psychological support should be carefully offered during all counseling procedures.
Author Contributions
SM contributed to manuscript preparation, review, and critique to the project. JJ contributed to design, conception, organization, design, review, and critique to the project. FS contributed to design, conception, organization, execution, design, review and Critique to the project, and wrote the first draft.
Funding
SM in the past year from data of submission, has received financial support, for clinical and research activity unrelated to this research, from Campus Bio-Medico University, Rome and from LIRH Foundation (funds from 5 × 1,000 of taxes). FS in the past year from data of submission, has received research and/or training grants, unrelated to this research, from IRCCS Casa Sollievo della Sofferenza Hospital (funds from Ricerca Corrente 2018 and from Ricerca Finalizzata [RF-2016-02364123] by Italian Ministry of Health to FS) and from LIRH Foundation (funds from 5 × 1,000 of taxes). He has served as a consultant or as an advisory committee member for: Teva Pharmaceutical Industries Ltd., La Roche ltd., Istituto per la Sicurezza Sociale (ISS), San Marino Republic. JJ in the past year from data of submission, has received research and/or training grants unrelated to this research from: Adamas Pharmaceuticals, Inc.; Allergan, Inc.; Biotie Therapies; CHDI Foundation; Civitas/Acorda Therapeutics; Dystonia Coalition; Dystonia Medical Research Foundation; F. Hoffmann-La Roche Ltd.; Huntington Study Group; Kyowa Haako Kirin Pharma, Inc.; Medtronic Neuromodulation; Merz Pharmaceuticals; Michael J. Fox Foundation for Parkinson Research; National Institutes of Health; Neurocrine Biosciences; NeuroDerm Ltd.; Parkinson's Foundation; Nuvelution; Parkinson Study Group; Pfizer Inc.; Prothena Biosciences Inc.; Psyadon Pharmaceuticals, Inc.; Revance Therapeutics, Inc.; Sangamo BioSciences, Inc.; St. Jude Medical; Teva Pharmaceutical Industries Ltd. JJ has served as a consultant or as an advisory committee member for: Adamas Pharmaceuticals, Inc.; Allergan, Inc.; Merz Pharmaceuticals; Pfizer Inc.; Prothena Biosciences; Revance Therapeutics, Inc.; Teva Pharmaceutical Industries Ltd. JJ has received royalties or other payments from: Cambridge; Elsevier; Future Science Group; Hodder Arnold; Medlink: Neurology; Lippincott Williams and Wilkins; Wiley-Blackwell.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Testa CM, Jankovic J. Huntington disease: a quarter century of progress since the gene discovery. J Neurol Sci. (2019) 396:52–68. doi: 10.1016/j.jns.2018.09.022
2. Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, Barbagallo G, et al. Biological and clinical manifestations of juvenile Huntington's disease: a retrospective analysis. Lancet Neurol. (2018) 17:986–93. doi: 10.1016/S1474-4422(18)30294-1
3. Squitieri F, Berardelli A, Nargi E, Castellotti B, Mariotti C, Cannella M, et al. Atypical movement disorders in the early stages of Huntington's disease: clinical and genetic analysis. Clin Genet. (2000) 58:50–6. doi: 10.1034/j.1399-0004.2000.580108.x
4. MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. (1993) 72:971–83. doi: 10.1016/0092-8674(93)90585-E
5. Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, et al. A worldwide study of the Huntington's disease mutation: the sensitivity and specificity of measuring CAG repeats. New Engl J Med. (1994) 330:1401–6. doi: 10.1056/NEJM199405193302001
6. Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet. (1996) 59:16–22.
7. Quarrell OW, Brewer HM, Squitieri F. Juvenile Huntington's Disease: And Other Trinucleotide Repeat Disorders. Oxford, UK: Oxford University Press (2009).
8. Telenius H, Almqvist E, Kremer B, Spence N, Squitieri F, Nichol K, et al. Somatic mosaicism in sperm is associated with intergenerational (CAG) n changes in Huntington disease. Hum Mol Genet. (1995) 2:189–95. doi: 10.1093/hmg/4.2.189
9. Benjamin CM, Adam S, Wiggins S, Theilmann JL, Copley TT, Bloch M, et al. Proceed with care: direct predictive testing for Huntington disease. Am J Hum Genet. (1995) 4:1015.
10. Squitieri F. Neurodegenerative disease:'fifty shades of grey'in the Huntington disease gene. Nat Rev Neurol. (2013) 9:421–2. doi: 10.1038/nrneurol.2013.128
11. Squitieri F, Andrew S, Goldberg Y, Kremer B, Spence N, Zelsler J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet. (1994) 3:2103–14. doi: 10.1093/hmg/3.12.2103
12. Warby SC, Montpetit A, Hayden AR, Carroll JB, Butland SL, Visscher H, et al. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J of Hum Genet. (2009) 84:351–66. doi: 10.1016/j.ajhg.2009.02.003
13. Gargiulo M, du Montcel ST, Jutras MF, Herson A, Cazeneuve C, Durr A. A liminal stage after predictive testing for Huntington disease. J Med Genet. (2017) 8:511–20. doi: 10.1136/jmedgenet-2016-104199
14. Kieburtz K, Reilmann R, Olanow CW. Huntington's disease: current and future therapeutic prospects. Mov Disord. (2018) 7:1033–41. doi: 10.1002/mds.27363
15. Kay C, Collins JA, Wright GE, Baine F, Miedzybrodzka Z, Aminkeng F. The molecular epidemiology of Huntington disease is related to intermediate allele frequency and haplotype in the general population. Am J Med Genet Part B Neuropsychiatric Genet. (2018) 3:346–57. doi: 10.1002/ajmg.b.32618
16. Squitieri F, Jankovic J. Huntington's disease: how intermediate are intermediate repeat lengths? Mov Dis. (2012) 27:1714–7. doi: 10.1002/mds.25172
17. Kenney C, Powell S, Jankovic J. Autopsy-proven Huntington's disease with 29 trinucleotide repeats. Mov Dis. (2007) 22:127–30.
18. Ha AD, Beck CA, Jankovic J. Intermediate CAG repeats in Huntington's disease: analysis of COHORT. Tremor Hyperkinetic Mov. (2012) 2:tre-02-64-287-4. doi: 10.7916/D8FF3R2P
19. Ha AD, Jankovic J. Exploring the correlates of intermediate CAG repeats in Huntington disease. Postgraduate Med. (2011) 123:116–21. doi: 10.3810/pgm.2011.09.2466
20. Killoran A, Biglan KM, Jankovic J, Eberly S, Kayson E, Oakes D, et al. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology. (2013) 80:2022–7. doi: 10.1212/WNL.0b013e318294b304
21. Cubo E, Ramos-Arroyo MA, Martinez-Horta S, Martinez-Descalls A, Gil-Polo C. Intermediate CAG repeats in Huntington' s disease. A longitudinal analysis of the European Huntington' s Disease Network REGISTRY Cohort (S25. 003). Neurology. (2016) 86:571–8.
22. Semaka A, Hayden M. Evidence-based genetic counselling implications for Huntington disease intermediate allele predictive test results. Clin Genet. (2014) 85:303–11. doi: 10.1111/cge.12324
23. MacLeod R, Tibben A, Frontali M, Evers-Kiebooms G, Jones A, Martinez-Descales A, et al. Recommendations for the predictive genetic test in Huntington's disease. Clin Genet. (2013) 83:221–31. doi: 10.1111/j.1399-0004.2012.01900.x
24. Semaka A, Kay C, Doty C, Collins JA, Bijlsma EK, Richards F, et al. CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet. (2013) 10:696–703. doi: 10.1136/jmedgenet-2013-101796
25. Landwehrmeyer GB, Fitzer-Attas CJ, Giuliano JD, Gonçalves N, Anderson KE, Cardoso F, et al. Data Analytics from Enroll-HD, a global clinical research platform for Huntington's disease. Mov Dis Clin Pract. (2017) 4:212–24. doi: 10.1002/mdc3.12388
26. Kay C, Collins JA, Miedzybrodzka Z, Madore SJ, Gordon ES, Gerry N, et al. Huntington disease reduced penetrance alleles occur at high frequency in the general population. Neurology. (2016) 87:282–8. doi: 10.1212/WNL.0000000000002858
27. Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) 1:113. doi: 10.1038/ng0594-113b
28. Gonitel R, Moffitt H, Sathasivam K, Woodman B, Detloff PJ, Faull RL, et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci USA. (2008) 105:3467–72. doi: 10.1073/pnas.0800048105
29. Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M Bates instbility of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat Genet. (1997) 2:197–200. doi: 10.1038/ng0297-197
30. Kennedy L, Evans E, Chen CM, Craven L, Detloff PJ, Ennis M, et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. (2003) 12:3359–367. doi: 10.1093/hmg/ddg352
31. Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, et al. Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol. (2017) 16:701–11. doi: 10.1016/S1474-4422(17)30161-8
32. Squitieri F, Griguoli A, Capelli G, Porcellini A, D'alessio B. Epidemiology of Huntington disease: first post-HTT gene analysis of prevalence in Italy. Clin Genet. (2016) 89:367–70. doi: 10.1111/cge.12574
33. Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington's disease. Nat Rev Dis Primers. (2015) 1:15005. doi: 10.1038/nrdp.2015.5
34. Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA. (2004) 10:3498–503. doi: 10.1073/pnas.0308679101
35. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington's disease. Cell. (2015) 3:516–26. doi: 10.1016/j.cell.2015.07.003
36. Quarrell O, O'Donovan KL, Bandmann O, Strong M. The prevalence of juvenile Huntington's disease: a review of the literature and meta-analysis. PLoS Curr. (2012) 4:e4f8606b742ef3. doi: 10.1371/4f8606b742ef3
37. Koutsis G, Karadima G, Kladi A, Panas M. The challenge of juvenile Huntington disease To test or not to test. Neurology. (2013) 80:990–6. doi: 10.1212/WNL.0b013e31828727fa
38. Anderson J, Hayeems R, Shuman C, Szego M, Monfared N, Bowdin S, et al. Predictive genetic testing for adult-onset disorders in minors: a critical analysis of the arguments for and against the 2013 ACMG guidelines. Clin Genet. (2015) 87:301–10. doi: 10.1111/cge.12460
39. Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. (2003) 4:946–55. doi: 10.1093/brain/awg077
40. Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. (2012) 10:690–5. doi: 10.1212/WNL.0b013e318249f683
41. Squitieri F, Almqvist EW, Cannella M, Cislaghi G and Hayden MR. Predictive testing for persons at risk for homozygosity for CAG expansion in the Huntington disease gene. Clin Genet. (2003) 6:524–5. doi: 10.1046/j.1399-0004.2003.00155.x
42. Schulman J, Stern H. Low utilization of prenatal and pre-implantation genetic diagnosis in Huntington disease–risk discounting in preventive genetics. Clin Genet. (2015) 88:220–3. doi: 10.1111/cge.12523
43. Piña-Aguilar RE, Simpson SA, Alsha A, Clarke A, Craufurd D, Dorkins H, et al. 27 years of prenatal diagnosis for Huntington disease in the United Kingdom. Genet Med. (2018). doi: 10.1038/s41436-018-0367-z. [Epub ahead of print].
44. McCusker EA, Loy CT. Huntington disease: the complexities of making and disclosing a clinical diagnosis after premanifest genetic testing. Tremor Other Hyperkinetic Mov. (2017) 7:467. doi: 10.7916/D8PK0TDD
45. Bashir H, Jankovic J. Treatment options for chorea. Expert Rev Neurother. (2018) 18:51–63. doi: 10.1080/14737175.2018.1403899
46. Ghosh R, Tabrizi SJ. Gene suppression approaches to neurodegeneration. Alzheimer's Res Ther. (2017) 9:82. doi: 10.1186/s13195-017-0307-1
Keywords: Huntington disease, pediatric HD, genetic counseling, intermediate alleles, neurodegenerative disease
Citation: Migliore S, Jankovic J and Squitieri F (2019) Genetic Counseling in Huntington's Disease: Potential New Challenges on Horizon? Front. Neurol. 10:453. doi: 10.3389/fneur.2019.00453
Received: 29 November 2018; Accepted: 15 April 2019;
Published: 30 April 2019.
Edited by:
Irene Litvan, University of California, San Diego, United StatesReviewed by:
Amber L. Southwell, University of Central Florida, United StatesFilippo M. Santorelli, Fondazione Stella Maris (IRCCS), Italy
Copyright © 2019 Migliore, Jankovic and Squitieri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ferdinando Squitieri, Zi5zcXVpdGllcmlAY3NzLW1lbmRlbC5pdA==