 Satoshi Saito
Satoshi Saito Yumi Yamamoto4
Yumi Yamamoto4 Masafumi Ihara
Masafumi Ihara- 1Department of Neurology, National Cerebral and Cardiovascular Center, Suita, Japan
- 2Research Fellow of Japan Society for the Promotion of Science, Tokyo, Japan
- 3Department of Pediatric Dentistry, Osaka University Graduate School of Dentistry, Suita, Japan
- 4Department of Regenerative Medicine and Tissue Engineering, National Cerebral and Cardiovascular Center, Suita, Japan
Recent advances in vascular risk management have successfully reduced the prevalence of Alzheimer's Disease (AD) in several epidemiologic investigations. It is now widely accepted that cerebrovascular disease is both directly and indirectly involved in AD pathogenesis. Herein, we review the non-pharmacological and pharmacological therapeutic approaches for AD treatment. MIND [Mediterranean and DASH (Dietary Approaches to Stop Hypertension) Intervention for Neurodegenerative Delay] diet is an important dietary treatment for prevention of AD. Multi domain intervention including diet, exercise, cognitive training, and intensive risk managements also prevented cognitive decline in the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) study. To confirm these favorable effects of life-style intervention, replica studies are being planned worldwide. Promotion of β-amyloid (Aβ) clearance has emerged as a promising pharmacological approach because insufficient removal of Aβ is more important than excessive Aβ production in the pathogenesis of the majority of AD patients. Most AD brains exhibit accompanying cerebral amyloid angiopathy, and Aβ distribution in cerebral amyloid angiopathy closely corresponds with the intramural periarterial drainage (IPAD) route, emphasizing the importance of Aβ clearance. In view of these facts, promotion of the major vascular-mediated Aβ elimination systems, including capillary transcytosis, the glymphatic system, and IPAD, have emerged as new treatment strategies in AD. In particular, the beneficial effects of cilostazol were shown in several clinical observation studies, and cilostazol facilitated IPAD in a rodent AD model. The COMCID (Cilostazol for prevention of Conversion from MCI to Dementia) trial, evaluating the efficacy of cilostazol for patients with mild cognitive impairment is currently ongoing in Japan. Such therapeutic approaches involving maintenance of cerebrovascular integrity and promotion of vascular-mediated Aβ clearance have the potential to be mainstream treatments for sporadic AD.
Introduction: Alzheimer's Disease and Cerebrovascular Disease
The pathogenesis of cognitive impairment in Alzheimer's disease (AD) cannot be simply explained by the neurodegeneration induced by amyloid β (Aβ) and tau accumulation, although they are the most established characteristics of AD pathology (1, 2). Because these characteristics are detectable by pathological examination, and do not always correlate with cognitive function, antemortem clinical diagnosis of AD is often denied by postmortem diagnosis, while individuals clinically diagnosed as cognitively intact are occasionally found to have AD pathology at postmortem (3).
Accumulating evidence suggests that cognitive impairment in AD patients, especially in elderly subjects, is attributed to both neurodegeneration and cerebrovascular disease (CVD) (4–6). Mixed AD/CVD pathology is commonly seen in patients with clinically diagnosed AD (7, 8). CVD itself impairs cognitive function, increasing the risk of progressing to clinically defined dementia (9–11). More importantly, recent studies indicate that vascular lesions are directly involved in AD pathogenesis (12). One of the mechanisms linking CVD to AD is decreased cerebral blood flow (13), which modulates amyloid precursor protein (APP) cleavage enzymes, such as β- and γ-secretase, leading to increased Aβ production (14). Chronic cerebral hypoperfusion also accelerates tau pathologies (15). Further, in a transgenic AD mouse model, the majority of parenchymal Aβ plaques were deposited adjacent to cerebral blood vessels, implying the involvement of the cerebral vasculature in Aβ accumulation (16). In addition to animal models, a pivotal role of CVD in AD pathogenesis has also been revealed by clinical studies. Cerebrovascular injury, including blood brain barrier (BBB) dysfunction (17, 18) and decreased cerebral blood flow (19), was found to precede neurodegeneration in AD patients. As such, the importance of vascular contributions to AD is now emphasized in a scientific statement from the Alzheimer's Association (20), and the close interrelationship between neurodegeneration and CVD has become a major focus of recent research in dementia (21, 22).
Lifestyle interventions to Prevent Cognitive Impairment
Management of Vascular Risk Factors
AD and CVD share several risk factors (23, 24). For example, hypertension, diabetes mellitus, and dyslipidemia are related to the risk of progression to dementia (25, 26). Conversely, intensive management of vascular risk factors was associated with reduced risk of dementia (25), as well as amelioration of cognitive decline in AD patients (27). Several epidemiologic reports have also revealed a decrease in dementia prevalence over the last few decades, which is considered to be, at least in part, attributable to well-controlled vascular risk factors (28–32). Thus, the importance of risk controls in AD is established.
The FINGER Study
Although pharmacological intervention is a fast and effective approach in treating vascular risk factors, lifestyle intervention remains important for prevention and treatment of AD and CVD. A population based randomized controlled trial, the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) study clearly revealed the importance of life-style modification (33). In that study, a multi domain intervention successfully prevented cognitive decline in elderly subjects. The multi domain intervention consisted of nutritional guidance, exercise, cognitive training, and social activity, while the control group received regular health advice. Metabolic and vascular risk factors were carefully monitored in the intervention group, and when the adjustment of pharmacologic treatment was needed, participants were recommended to contact primary health care centers (34).
Interestingly, the beneficial effects in the FINGER study were not modified by baseline status of sociodemographic, socioeconomic status, cognitive function, cardiovascular factors, or cardiovascular comorbidity, indicating that life-style intervention is universally effective in the elderly (35). Of all the subdomains of cognitive function, substantial positive effects were found in executive function and processing speed (33). However, because no pathological analysis was performed in that study, it remains unclear whether lifestyle intervention has any effect on reducing Aβ and tau pathologies and reducing neurodegeneration. Ongoing replica studies around the world may answer this important question (36).
MIND Diet
In the FINGER study, the intervention group was recommended to consume whole grains, fruits, vegetables, and oils, while low-fat options were recommended over milk and meat products (34). These foods are well-known components of the Mediterranean diet (37). Improvement in cognitive function with the Mediterranean diet was shown in a multicenter randomized clinical trial for community-dwelling people with vascular risk factors (38). The Dietary Approach to Systolic Hypertension (DASH) diet, which aims to reduce blood pressure, was also reported to prevent dysexecutive function in patients with hypertension and obesity (39). Combining these two diets, the Mediterranean and DASH Intervention for Neurodegenerative Delay (MIND) diet was proposed in the Rush Memory and Aging project in the United States. The MIND diet places a strong emphasis on natural, plant-based food, specifically promoting increased consumption of berries and green leafy vegetables, with limited intake of animal-based and highly saturated fat foods (40). Surprisingly, the incidence of AD in participants who strictly followed the MIND diet was reduced by half compared with those who did not follow it (41). The improvement in cognitive function was particularly significant in the domains of episodic memory, semantic memory, and perceptual speed (42). Replica studies are required to verify these favorable effects (43), although some modification may be required when the MIND diet is applied to other countries. For example, larger fish consumption and excessive salt intake are more common in East Asian than in Western countries. Thus, taking the local food culture into consideration is important when designing a feasible diet therapy (Table 1).

Table 1. Original and Japanese-modified version of the MIND diet.
Pharmacological Interventions for the Promotion of Aβ Clearance
Vascular Mediated Aβ Elimination
The amyloid hypothesis suggests that Aβ precedes tau pathology (44). Both Aβ overproduction and elimination failure have been demonstrated as a cause of AD (45), although the latter plays a major role in the pathogenesis of sporadic AD, which is especially more common in the elderly (46, 47). Thus, promotion of Aβ clearance has been proposed as a new therapeutic approach for sporadic AD (48).
In mice, >80% of 125I-labeled Aβ40 was found in the blood or cerebrospinal fluid (CSF) at 5 h after Aβ40 injection into the caudate nucleus, with minimal remaining in the brain parenchyma (49). Several mechanisms of Aβ clearance have been identified (50), with the major routes of Aβ elimination being vascular-mediated clearance systems, including transcytosis and glymphatic/lymphatic drainage. Thus, there is increasing interest in treatments based on promoting vascular-mediated Aβ elimination.
Transcytosis
Transcytosis in the cerebral capillaries is considered a major route of material exchange between the blood and the brain. The capillary lumen and brain parenchyma are separated by the BBB, which is composed of endothelial cells connected by tight junctions (51). The BBB prevents passive exchange between the blood and brain, and only allows controlled carrier-mediated bidirectional transport of nutrients and waste products (52).
Transcytosis is a major Aβ elimination system, and several molecules have been reported to be involved in Aβ transcytosis. Endothelial low-density lipoprotein receptor related protein-1 (LRP-1), a multifunctional scavenger and signaling receptor, is expressed in neural cells and cerebral microvessels (49, 53). Aβ binds to LRP-1 at the abluminal side of the endothelium, either as a free peptide or bound via ApoE2 and ApoE3. Aβ-ApoE2 and Aβ-ApoE3 complexes are rapidly cleared across the BBB into the blood, while Aβ bound to ApoE4 interacts poorly with LRP-1, and is removed from the brain by much slower and less efficient very low-density lipoprotein receptor clearance mechanism. Aβ transcytosis via LRP-2 was facilitated by clusterin in the brain (54–56). The importance of LRP-1 has also been shown by genetic linkage of LRP-1 with AD (57–59), and by the co-localization of LRP-1 with Aβ in senile plaques (60). LRP-1 staining in vessels was reduced in AD patients (49, 60). The transcytosis of Aβ was reported to be regulated by the phosphatidylinositol-binding clathrin assembly (PICALM) protein, a known genetic risk factor for AD (61).
The receptor for advanced glycation end products (RAGE), an immunoglobulin supergene family member, is another key molecule in Aβ transcytosis (62). Strong staining for RAGE was reported in the vessels of AD patients (60, 63), and RAGE was shown to mediate influx of circulating Aβ into the brain across the BBB (64). RAGE expression in microglia is also involved in microglial activation induced by Aβ (63, 65, 66). These findings prompted researchers to target RAGE for the treatment of AD. For example, inhibition of RAGE ameliorated cerebral Aβ burden and normalized cognitive performance in APP transgenic mice (67). A phase II clinical trial of the RAGE inhibitor TTP488 targeting mild-to-moderate AD patients was also associated with improved cognitive function as assessed by Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog) (68). However, a phase III trial was recently terminated as there was no positive effect using TTP488 treatment (69).
Glymphatic and Lymphatic Drainage
The central nervous system was previously thought to be devoid of systemic lymphatic vessels. However, two recent investigations have clearly shown the presence of lymphatic vessels adjacent to meningeal vessels (70, 71). Ablation of meningeal lymphatic vessels also aggravated Aβ accumulation in AD model mice, suggesting that promoting intracranial lymphatic systems may be a potential therapeutic approach for AD (72).
The glymphatic paravascular drainage system is considered one of the Aβ clearance systems (Figure 1), consisting of the para-arterial CSF influx, convective bulk fluid flux, and para-venous interstitial fluid (ISF) efflux routes (73, 74). Tracers injected into the cisterna magna were found to gradually flow into the brain parenchyma through the perivascular space (PVS) around the arteries, and then flow out along the veins (75, 76). The PVS (also known as Virchow-Robin spaces) is a gap between the brain parenchyma derived from the ectoderm and the cerebral perforating arteries derived from the mesoderm. The fluid circulation from the subarachnoid space to the PVS was initially described in the 1980s (77). At first, the paravascular drainage was considered to be dependent on astroglial water transport via the astrocytic aquaporin-4 (AQP-4) water channel, because CSF influx was severely impaired in Aqp-4 knockout mice (75). However, in a replica study, Aqp-4 gene deletion did not affect fluid transport (78). In addition, the PVS is separated from the subarachnoid space by the pia matter (79, 80), and the degree to which these spaces communicate with each other remains unclear. Further studies are required to unravel the molecular mechanism of the glymphatic systems.
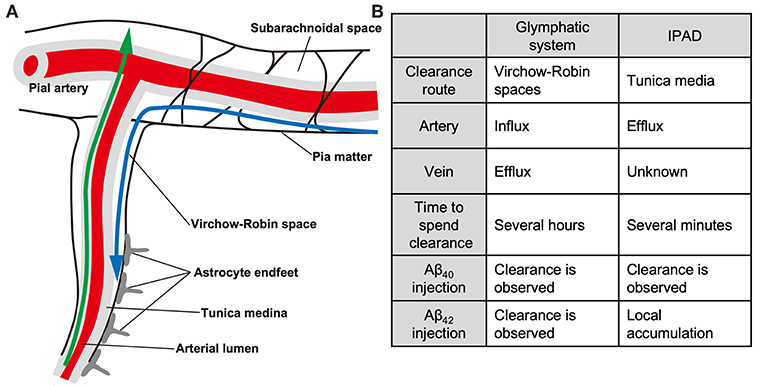
Figure 1. The glymphatic and IPAD systems. Schematic (A) and differences (B) of the glymphatic and IPAD systems. Note that the directions of the two clearance systems are the opposite. Blue, glymphatic system; green, IPAD. The IPAD is a more rapid clearance system than the glymphatic system. Note the differences in the tracer distribution pattern after fluorescent Aβ40 and Aβ42 injection.
Clinical MRI often reveals an enlarged PVS in the basal ganglia in patients with hypertensive arteriopathy (81–84). Morphological changes in the PVS were also reported in patients with hereditary arteriopathy including Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) (85) and Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL) (86). Both sporadic and hereditary arteriopathies are cerebral small vessel diseases (SVD), and expansion of the PVS is considered an imaging marker of SVD (87, 88). The presence of a large PVS was associated with a steeper decline in information processing speed, and an increased risk of developing vascular dementia (89). AD patients were also reported to show an increased ratio of total PVS to white matter volume (90). A mouse model of AD showed suppression of glymphatic paravascular fluid transport caused by accumulation of toxic Aβ (91), suggesting that PVS enlargement in AD patients may be associated with an impaired glymphatic system. An enlarged PVS is commonly observed in the centrum semiovale in both AD and cerebral amyloid angiopathy (CAA) (83, 84). CAA is another subtype of SVD observed in >80% of AD patients, as well as in ~10% of vascular cognitive impairment cases with a general absence of AD pathology (92, 93). After being generated in neurons, Aβ accumulates in the vessel walls (45), resulting in degeneration of smooth muscle cells, vessel wall thickening, luminal narrowing, and concentric wall splitting (double barreling) (94). The degeneration of mural cells and injury to vascular endothelial cells cause various forms of CVD, including ischemic stroke, hemorrhage, and white matter changes. Impaired BBB function likely interferes with Aβ transcytosis, leading to exacerbation of AD and CAA (21, 52).
The driving force behind paravascular clearance is thought to involve cerebral arterial pulsation. Indeed, reduced arterial pulsatility by unilateral carotid artery ligation prevented CSF-ISF exchange, while the opposite effect was observed following administration of the adrenergic agonist dobutamine (95). However, no studies have clearly shown that promotion of the glymphatic system can ameliorate Aβ pathology and cognitive function. In addition, several recent studies argued against the proposed glymphatic mechanism (78, 96–98). As Aβ rarely accumulates in the venous system, and as arterial Aβ accumulation is most prominent within the tunica medina, not the PVS (99), it remains unclear whether promotion of the glymphatic pathway may be a disease-modifying therapy for AD and CAA. Recent technological advances in imaging have enabled visualization and monitoring of PVS structure and change in AD patients (90, 100, 101). Clinical evaluation of the glymphatic system in AD patients may help to determine the relationship between the glymphatic system, PVS, and CAA.
Aβ distribution in CAA closely corresponds to the intramural periarterial drainage (IPAD) route, suggesting that congestion of this drainage pathway plays a major role in the pathogenesis of CAA (102). The IPAD system is also termed perivascular lymphatic drainage (103). ISF and solutes including Aβ are thought to be cleared from the gray matter through the IPAD, which is a space between two basement membranes in the walls of the cerebral capillaries and arteries (104, 105). Interestingly, IPAD was impaired in the aging mouse brain and in the presence of CAA (106). It was also reported that the IPAD and glymphatic systems are not independent, but rather communicate with each other (107). For example, Aβ tracer injected into the cisterna magna flowed into the PVS, while after reaching the deep brain parenchyma, some of the tracer drained out of the brain via the IPAD system. Because use of animal models with tracer injection is far from physiological, the precise relationship between the glymphatic system and IPAD remains elusive, and further studies are required.
The importance of the IPAD was demonstrated in a clinical trial of Aβ immunization. In AN-1792 vaccinated AD patients, parenchymal Aβ plaque was diminished while cerebrovascular Aβ accumulation was increased (108, 109). Considering that decreasing CSF Aβ was correlated with severity of CAA-causing CVD, the ineffectiveness and side-effects of AN-1792 may be partially explained by the excessive antibody-solubilized senile plaque Aβ that is re-deposited in the cerebral vasculature, and impaired Aβ clearance through IPAD (110, 111). Combination therapy of Aβ immunization and promoting Aβ clearance through IPAD may provide a solution for the problem in removal of solubilized Aβ from the brain. The fact that cerebral Aβ clearance was delayed after middle cerebral artery occlusion (112) and after bilateral common carotid artery stenosis (113) emphasizes the importance of cerebrovascular integrity for the promotion of Aβ clearance through IPAD.
Promoting IPAD by Cilostazol
Cilostazol is a type 3 phosphodiesterase (PDE) inhibitor that is currently prescribed for secondary prevention of ischemic stroke and peripheral vascular disease, especially in Eastern Asia. PDE-3 can hydrolyze both cAMP and cGMP, while increasing cAMP level is a major pharmacological action of cilostazol (48). PDE-3 is widely expressed in the central nervous system, and is up-regulated in Aβ-positive vessels, especially in vascular smooth muscle cells (114), suggesting that PDE-3 inhibition may be an effective target for treatment of AD and CAA.
We previously demonstrated that cilostazol promoted IPAD, resulting in maintenance of vascular integrity, amelioration of Aβ deposits, and prevention of cognitive decline in AD model mice (114). Further, cilostazol possesses a wide range of pleiotropic effects, including neurogenesis (115, 116), differentiation of oligodendrocyte precursor cells (117), inhibition of lipid peroxidation (118, 119), enhancement of cholesterol efflux from macrophages (120), amelioration of insulin resistance (121), reduction of inflammatory burden (122–124), stabilization of BBB function (125), and improvement of systemic lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells (126). A nationwide cohort study in Taiwan showed that cilostazol treatment was associated with reduced risk of dementia (127). Favorable effects were also described in observational studies, which demonstrated the efficacy of cilostazol in patients with mild cognitive impairment (128) and AD (129–132). A phase-II, randomized clinical trial (the COMCID study) was started in 2015 (133), in which MCI patients received 100 mg daily cilostazol or placebo for 96 weeks. The results will be announced in 2020.
Conclusions
AD, especially in the elderly, is a syndrome concomitant with CVD and neurodegeneration. The commonly used expression amongst clinicians, “a man is as old as his arteries,” is thus as relevant to patients with dementia as for many other age-related disorders (134). The influence of vascular factors should be given more consideration, as it may represent a future therapeutic target for dementia. The existence of multiple Aβ elimination mechanisms suggests the requirement for several different treatment approaches in AD. Further studies are essential for the development of novel treatments for dementia.
Author Contributions
SS and YY wrote the manuscript. MI supervised and made critical revision of manuscript for important intellectual content.
Funding
We gratefully acknowledge grant support from the Ministry of Education, Culture, Sports, Science and Technology [SS, Grant-in-Aid for Young Scientists (B), No 17K14958].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the late Dr. Yoko Okamoto for useful discussions.
References
1. Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer's disease. Acta Neuropathol. (2016) 131:659–85. doi: 10.1007/s00401-016-1571-z
2. Ganz AB, Beker N, Hulsman M, Sikkes S, Netherlands Brain B, Scheltens P, et al. Neuropathology and cognitive performance in self-reported cognitively healthy centenarians. Acta Neuropathol Commun. (2018) 6:64. doi: 10.1186/s40478-018-0558-5
3. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
4. Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. (2009) 72:368–74. doi: 10.1212/01.wnl.0000341271.90478.8e
5. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. (2015a) 163:1064–78. doi: 10.1016/j.cell.2015.10.067
6. Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. (2018) 21:1318–31. doi: 10.1038/s41593-018-0234-x
7. Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain. (2013) 136(Pt 9):2697–706. doi: 10.1093/brain/awt188
8. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. (2018) 83:74–83. doi: 10.1002/ana.25123
9. Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. (1997) 277:813–7.
10. Matthews FE, Brayne C, Lowe J, McKeith I, Wharton SB, Ince P. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. (2009) 6:e1000180. doi: 10.1371/journal.pmed.1000180
11. Provenzano FA, Muraskin J, Tosto G, Narkhede A, Wasserman BT, Griffith EY, et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol. (2013) 70:455–61. doi: 10.1001/jamaneurol.2013.1321
12. Pimentel-Coelho PM, Rivest S. The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer's disease. Eur J Neurosci. (2012) 35:1917–37. doi: 10.1111/j.1460-9568.2012.08126.x
13. Kitaguchi H, Tomimoto H, Ihara M, Shibata M, Uemura K, Kalaria RN, et al. Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. (2009) 1294:202–10. doi: 10.1016/j.brainres.2009.07.078
14. Gupta A, Iadecola C. Impaired Abeta clearance: a potential link between atherosclerosis and Alzheimer's disease. Front Aging Neurosci. (2015) 7:115. doi: 10.3389/fnagi.2015.00115
15. Zhao Y, Gu JH, Dai CL, Liu Q, Iqbal K, Liu F, et al. Chronic cerebral hypoperfusion causes decrease of O-GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front Aging Neurosci. (2014) 6:10. doi: 10.3389/fnagi.2014.00010
16. Hama H, Hioki H, Namiki K, Hoshida T, Kurokawa H, Ishidate F, et al. ScaleS: an optical clearing palette for biological imaging. Nat Neurosci. (2015) 18:1518–29. doi: 10.1038/nn.4107
17. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. (2015) 85:296–302. doi: 10.1016/j.neuron.2014.12.032
18. Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. (2019) 25:270–6. doi: 10.1038/s41591-018-0297-y
19. Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC Alzheimer's Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. (2016) 7:11934. doi: 10.1038/ncomms11934
20. Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement. (2015) 11:710–7. doi: 10.1016/j.jalz.2014.10.008
21. Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. (2016) 1862:887–900. doi: 10.1016/j.bbadis.2015.12.016
22. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. (2019) 99:21–78. doi: 10.1152/physrev.00050.2017
23. Rodrigue KM, Rieck JR, Kennedy KM, Devous MDSr, Diaz-Arrastia R, Park DC. Risk factors for beta-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol. (2013) 70:600–6. doi: 10.1001/jamaneurol.2013.1342
24. Saito S, Yamamoto Y, Ihara M. Mild cognitive impairment: at the crossroad of neurodegeneration and vascular dysfunction. Curr Alzheimer Res. (2015) 12:507–12. doi: 10.2174/1567205012666150530202508
25. Li J, Wang YJ, Zhang M, Xu ZQ, Gao CY, Fang CQ, et al. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology. (2011) 76:1485–91. doi: 10.1212/WNL.0b013e318217e7a4
26. Goldstein FC, Levey AI, Steenland NK. High blood pressure and cognitive decline in mild cognitive impairment. J Am Geriatr Soc. (2013) 61:67–73. doi: 10.1111/jgs.12067
27. Deschaintre Y, Richard F, Leys D, Pasquier F. Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology. (2009) 73:674–80. doi: 10.1212/WNL.0b013e3181b59bf3
28. Schrijvers EM, Verhaaren BF, Koudstaal PJ, Hofman A, Ikram MA, Breteler MM. Is dementia incidence declining?: trends in dementia incidence since 1990 in the Rotterdam Study. Neurology. (2012) 78:1456–63. doi: 10.1212/WNL.0b013e3182553be6
29. Larson EB, Yaffe K, Langa KM. New insights into the dementia epidemic. N Engl J Med. (2013) 369:2275–7. doi: 10.1056/NEJMp1311405
30. Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, et al. A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: results of the Cognitive Function and Ageing Study I and II. Lancet. (2013) 382:1405–12. doi: 10.1016/S0140-6736(13)61570-6
31. Qiu C, von Strauss E, Bäckman L, Winblad B, Fratiglioni L. Twenty-year changes in dementia occurrence suggest decreasing incidence in central Stockholm, Sweden. Neurology. (2013) 80:1888–94. doi: 10.1212/WNL.0b013e318292a2f9
32. Satizabal CL, Beiser AS, Chouraki V, Chene G, Dufouil C, Seshadri S. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med. (2016) 374:523–32. doi: 10.1056/NEJMoa1504327
33. Ngandu T, Lehtisalo J, Solomon A, Levalahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. (2015) 385:2255–63. doi: 10.1016/S0140-6736(15)60461-5
34. Kivipelto M, Solomon A, Ahtiluoto S, Ngandu T, Lehtisalo J, Antikainen R, et al. The Finnish Geriatric Intervention Study to prevent cognitive impairment and disability (FINGER): study design and progress. Alzheimers Dement. (2013) 9:657–65. doi: 10.1016/j.jalz.2012.09.012
35. Rosenberg A, Ngandu T, Rusanen M, Antikainen R, Backman L, Havulinna S, et al. Multidomain lifestyle intervention benefits a large elderly population at risk for cognitive decline and dementia regardless of baseline characteristics: the FINGER trial. Alzheimers Dement. (2018) 14:263–70. doi: 10.1016/j.jalz.2017.09.006
36. Kivipelto M, Mangialasche F, Ngandu T, World Wide Fingers N. World Wide Fingers will advance dementia prevention. Lancet Neurol. (2018) 17:27. doi: 10.1016/S1474-4422(17)30431-3
37. Panagiotakos DB, Pitsavos C, Arvaniti F, Stefanadis C. Adherence to the Mediterranean food pattern predicts the prevalence of hypertension, hypercholesterolemia, diabetes and obesity, among healthy adults; the accuracy of the MedDietScore. Prev Med. (2007) 44:335–40. doi: 10.1016/j.ypmed.2006.12.009
38. Martinez-Lapiscina EH, Clavero P, Toledo E, Estruch R, Salas-Salvado J, San Julian B, et al. Mediterranean diet improves cognition: the PREDIMED-NAVARRA randomised trial. J Neurol Neurosurg Psychiatry. (2013) 84:1318–25. doi: 10.1136/jnnp-2012-304792
39. Smith PJ, Blumenthal JA, Babyak MA, Craighead L, Welsh-Bohmer KA, Browndyke JN, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. (2010) 55:1331–8. doi: 10.1161/HYPERTENSIONAHA.109.146795
40. Marcason W. What are the components to the MIND diet? J Acad Nutr Diet. (2015) 115:1744. doi: 10.1016/j.jand.2015.08.002
41. Morris MC, Tangney CC, Wang Y, Sacks FM, Bennett DA, Aggarwal NT. MIND diet associated with reduced incidence of Alzheimer's disease. Alzheimers Dement. (2015b) 11:1007–14. doi: 10.1016/j.jalz.2014.11.009
42. Morris MC, Tangney CC, Wang Y, Sacks FM, Barnes LL, Bennett DA, et al. MIND diet slows cognitive decline with aging. Alzheimers Dement. (2015a) 11:1015–22. doi: 10.1016/j.jalz.2015.04.011
43. Hosking DE, Eramudugolla R, Cherbuin N, Anstey KJ. MIND not Mediterranean diet related to 12-year incidence of cognitive impairment in an Australian longitudinal cohort study. Alzheimers Dement. (2019)15:581–9. doi: 10.1016/j.jalz.2018.12.011
44. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992) 256:184–5.
45. Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol. (2015) 25:63–78. doi: 10.1111/bpa.12218
46. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science. (2010) 330:1774. doi: 10.1126/science.1197623
47. Iturria-Medina Y, Sotero RC, Toussaint PJ, Evans AC Alzheimer's Disease Neuroimaging Initiative. Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput Biol. (2014) 10:e1003956. doi: 10.1371/journal.pcbi.1003956
48. Saito S, Ihara M. New therapeutic approaches for Alzheimer's disease and cerebral amyloid angiopathy. Front Aging Neurosci. (2014) 6:290. doi: 10.3389/fnagi.2014.00290
49. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. (2000) 106:1489–99. doi: 10.1172/JCI10498
50. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. (2015) 11:457–70. doi: 10.1038/nrneurol.2015.119
51. Iadecola C. Dangerous leaks: blood-brain barrier woes in the aging hippocampus. Neuron. (2015) 85:231–3. doi: 10.1016/j.neuron.2014.12.056
52. Nelson AR, Sagare AP, Zlokovic BV. Role of clusterin in the brain vascular clearance of amyloid-β. Proc Natl Acad Sci USA. (2017) 114:8681–2. doi: 10.1073/pnas.1711357114
53. Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. (2004) 43:333–44. doi: 10.1016/j.neuron.2004.07.017
54. Zlokovic BV, Martel CL, Mackic JB, Matsubara E, Wisniewski T, McComb JG, et al. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloid β. Biochem Biophys Res Commun. (1994) 205:1431–7. doi: 10.1006/bbrc.1994.2825
55. Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, et al. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid β at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA. (1996) 93:4229–34.
56. Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid β-peptide elimination from the brain. J Neurochem. (2010) 115:1077–89. doi: 10.1111/j.1471-4159.2010.07002.x
57. Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, et al. Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer's disease. Neurology. (1997) 49:56–61.
58. Lambert JC, Wavrant-De Vrièze F, Amouyel P, Chartier-Harlin MC. Association at LRP gene locus with sporadic late-onset Alzheimer's disease. Lancet. (1998) 351:1787–8.
59. Wavrant-DeVrièze F, Lambert JC, Stas L, Crook R, Cottel D, Pasquier F, et al. Association between coding variability in the LRP gene and the risk of late-onset Alzheimer's disease. Hum Genet. (1999) 104:432–4.
60. Donahue JE, Flaherty SL, Johanson CE, Duncan JA, Silverberg GD, Miller MC, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. (2006) 112:405–15. doi: 10.1007/s00401-006-0115-3
61. Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. (2015b) 18:978–87. doi: 10.1038/nn.4025
62. Yan SS, Chen D, Yan S, Guo L, Du H, Chen JX. RAGE is a key cellular target for Aβ-induced perturbation in Alzheimer's disease. Front Biosci. (2012) 4:240–50. doi: 10.2741/265
63. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. (1996) 382:685–91. doi: 10.1038/382685a0
64. Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. (2003) 9:907–13. doi: 10.1038/nm890
65. Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. (2008) 28:3521–30. doi: 10.1523/JNEUROSCI.0204-08.2008
66. Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, et al. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci. (2010) 30:11414–25. doi: 10.1523/JNEUROSCI.2127-10.2010
67. Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. (2012) 122:1377–92. doi: 10.1172/JCI58642
68. Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, Mjalli AM. Effect of TTP488 in patients with mild to moderate Alzheimer's disease. BMC Neurol. (2014) 14:12. doi: 10.1186/1471-2377-14-12
69. The U,.S. NIH. 2-Year Extension Study of Azeliragon in Subjects With Alzheimer's Disease (STEADFAST Extension). (2018) Available online at: https://clinicaltrials.gov/ct2/show/NCT02916056 (accessed November 1, 2018).
70. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. (2015) 212:991–9. doi: 10.1084/jem.20142290
71. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
72. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer's disease. Nature. (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
73. Iliff JJ, Nedergaard M. Is there a cerebral lymphatic system? Stroke. (2013) 44(Suppl. 1):S93–95. doi: 10.1161/STROKEAHA.112.678698
74. Nedergaard M. Neuroscience. Garbage truck of the brain. Science. (2013) 340:1529–30. doi: 10.1126/science.1240514
75. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. (2012) 4:147ra111. doi: 10.1126/scitranslmed.3003748
76. Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, et al. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. (2013a) 123:1299–309. doi: 10.1172/JCI67677
77. Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K, Grady PA. Evidence for a 'paravascular' fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. (1985) 326:47–63.
78. Smith AJ, Yao X, Dix JA, Jin BJ, Verkman AS. Test of the 'glymphatic' hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. Elife. (2017) 6:e27679. doi: 10.7554/eLife.27679
79. Wuerfel J, Haertle M, Waiczies H, Tysiak E, Bechmann I, Wernecke KD, et al. Perivascular spaces–MRI marker of inflammatory activity in the brain? Brain. (2008) 131(Pt 9):2332–40. doi: 10.1093/brain/awn171
80. Mestre H, Kostrikov S, Mehta RI, Nedergaard M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin Sci. (2017) 131:2257–74. doi: 10.1042/CS20160381
81. Hurford R, Charidimou A, Fox Z, Cipolotti L, Jager R, Werring DJ. MRI-visible perivascular spaces: relationship to cognition and small vessel disease MRI markers in ischaemic stroke and TIA. J Neurol Neurosurg Psychiatry. (2014) 85:522–5. doi: 10.1136/jnnp-2013-305815
82. Yakushiji Y, Charidimou A, Hara M, Noguchi T, Nishihara M, Eriguchi M, et al. Topography and associations of perivascular spaces in healthy adults: the Kashima scan study. Neurology. (2014) 83:2116–23. doi: 10.1212/WNL.0000000000001054
83. Banerjee G, Kim HJ, Fox Z, Jager HR, Wilson D, Charidimou A, et al. MRI-visible perivascular space location is associated with Alzheimer's disease independently of amyloid burden. Brain. (2017) 140:1107–16. doi: 10.1093/brain/awx003
84. Charidimou A, Boulouis G, Pasi M, Auriel E, van Etten ES, Haley K, et al. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. (2017) 88:1157–64. doi: 10.1212/WNL.0000000000003746
85. Yamamoto Y, Ihara M, Tham C, Low RW, Slade JY, Moss T, et al. Neuropathological correlates of temporal pole white matter hyperintensities in CADASIL. Stroke. (2009) 40:2004–11. doi: 10.1161/STROKEAHA.108.528299
86. Fukutake T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): from discovery to gene identification. J Stroke Cerebrovasc Dis. (2011) 20:85–93. doi: 10.1016/j.jstrokecerebrovasdis.2010.11.008
87. Potter GM, Doubal FN, Jackson CA, Chappell FM, Sudlow CL, Dennis MS, et al. Enlarged perivascular spaces and cerebral small vessel disease. Int J Stroke. (2015) 10:376–81. doi: 10.1111/ijs.12054
88. Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A, et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. (2018) 114:1462–73. doi: 10.1093/cvr/cvy113
89. Ding J, Sigurethsson S, Jonsson PV, Eiriksdottir G, Charidimou A, Lopez OL, et al. Large perivascular spaces visible on magnetic resonance imaging, cerebral small vessel disease progression, and risk of dementia: the age, gene/environment susceptibility-Reykjavik study. JAMA Neurol. (2017) 74:1105–12. doi: 10.1001/jamaneurol.2017.1397
90. Cai K, Tain R, Das S, Damen FC, Sui Y, Valyi-Nagy T, et al. The feasibility of quantitative MRI of perivascular spaces at 7T. J Neurosci Methods. (2015) 256:151–6. doi: 10.1016/j.jneumeth.2015.09.001
91. Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol Dis. (2016) 93:215–25. doi: 10.1016/j.nbd.2016.05.015
92. Kalaria RN. Cerebrovascular disease and mechanisms of cognitive impairment: evidence from clinicopathological studies in humans. Stroke. (2012) 43:2526–34. doi: 10.1161/STROKEAHA.112.655803
93. Esiri M, Chance S, Joachim C, Warden D, Smallwood A, Sloan C, et al. Cerebral amyloid angiopathy, subcortical white matter disease and dementia: literature review and study in OPTIMA. Brain Pathol. (2015) 25:51–62. doi: 10.1111/bpa.12221
94. Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke. (2015) 17:17–30. doi: 10.5853/jos.2015.17.1.17
95. Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. (2013) 33:18190–9. doi: 10.1523/JNEUROSCI.1592-13.2013
96. Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. (2014) 11:26. doi: 10.1186/2045-8118-11-26
97. Spector R, Robert Snodgrass S, Johanson CE. A balanced view of the cerebrospinal fluid composition and functions: focus on adult humans. Exp Neurol. (2015) 273:57–68. doi: 10.1016/j.expneurol.2015.07.027
98. Holter KE, Kehlet B, Devor A, Sejnowski TJ, Dale AM, Omholt SW, et al. Interstitial solute transport in 3D reconstructed neuropil occurs by diffusion rather than bulk flow. Proc Natl Acad Sci USA. (2017) 114:9894–9. doi: 10.1073/pnas.1706942114
99. Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. (2008) 115:599–609. doi: 10.1007/s00401-008-0366-2
100. Naganawa S, Nakane T, Kawai H, Taoka T. Gd-based contrast enhancement of the perivascular spaces in the basal ganglia. Magn Reson Med Sci. (2017) 16:61–5. doi: 10.2463/mrms.mp.2016-0039
101. Taoka T, Masutani Y, Kawai H, Nakane T, Matsuoka K, Yasuno F, et al. Evaluation of glymphatic system activity with the diffusion MR technique: diffusion tensor image analysis along the perivascular space (DTI-ALPS) in Alzheimer's disease cases. Jpn J Radiol. (2017) 35:172–8. doi: 10.1007/s11604-017-0617-z
102. Weller RO, Sharp MM, Christodoulides M, Carare RO, Mollgard K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. (2018) 135:363–85. doi: 10.1007/s00401-018-1809-z
103. Morris AWJ, Carare RO, Schreiber S, Hawkes CA. The Cerebrovascular Basement Membrane: Role in the Clearance of β-amyloid and Cerebral Amyloid Angiopathy. Front Aging Neurosci. (2014) 6:251. doi: 10.3389/fnagi.2014.00251
104. Arbel-Ornath M, Hudry E, Eikermann-Haerter K, Hou S, Gregory JL, Zhao L, et al. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer's disease mouse models. Acta Neuropathol. (2013) 126:353–64. doi: 10.1007/s00401-013-1145-2
105. Morris AWJ, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, et al. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathologica. (2016) 131:725–36. doi: 10.1007/s00401-016-1555-z
106. Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. (2011) 121:431–43. doi: 10.1007/s00401-011-0801-7
107. Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. (2018) 136:139–52. doi: 10.1007/s00401-018-1862-7
108. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. (2003) 9:448–52. doi: 10.1038/nm840
109. Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, et al. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol. (2006) 169:1048–63. doi: 10.2353/ajpath.2006.060269
110. Verbeek MM, Kremer BP, Rikkert MO, Van Domburg PH, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann Neurol. (2009) 66:245–9. doi: 10.1002/ana.21694
111. van Etten ES, Verbeek MM, van der Grond J, Zielman R, van Rooden S, van Zwet EW, et al. beta-Amyloid in CSF: biomarker for preclinical cerebral amyloid angiopathy. Neurology. (2017) 88:169–76. doi: 10.1212/WNL.0000000000003486
112. Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, et al. Cerebrovascular lesions induce transient β-amyloid deposition. Brain. (2011) 134(Pt 12):3697–707. doi: 10.1093/brain/awr300
113. Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. (2012) 123:381–94. doi: 10.1007/s00401-011-0925-9
114. Maki T, Okamoto Y, Carare RO, Hase Y, Hattori Y, Hawkes CA, et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β-amyloid. Ann Clin Transl Neurol. (2014) 1:519–33. doi: 10.1002/acn3.79
115. Lee JH, Shin HK, Park SY, Kim CD, Lee WS, Hong KW. Cilostazol preserves CA1 hippocampus and enhances generation of immature neuroblasts in dentate gyrus after transient forebrain ischemia in rats. Exp Neurol. (2009) 215:87–94. doi: 10.1016/j.expneurol.2008.09.013
116. Tanaka Y, Tanaka R, Liu M, Hattori N, Urabe T. Cilostazol attenuates ischemic brain injury and enhances neurogenesis in the subventricular zone of adult mice after transient focal cerebral ischemia. Neuroscience. (2010) 171:1367–76. doi: 10.1016/j.neuroscience.2010.10.008
117. Miyamoto N, Pham LD, Hayakawa K, Matsuzaki T, Seo JH, Magnain C, et al. Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke. (2013) 44:2573–8. doi: 10.1161/STROKEAHA.113.001530
118. Hiramatsu M, Takiguchi O, Nishiyama A, Mori H. Cilostazol prevents amyloid β peptide(25-35)-induced memory impairment and oxidative stress in mice. Br J Pharmacol. (2010) 161:1899–912. doi: 10.1111/j.1476-5381.2010.01014.x
119. Kurtoglu T, Basoglu H, Ozkisacik EA, Cetin NK, Tataroglu C, Yenisey C, et al. Effects of cilostazol on oxidative stress, systemic cytokine release, and spinal cord injury in a rat model of transient aortic occlusion. Ann Vasc Surg. (2014) 28:479–88. doi: 10.1016/j.avsg.2013.08.005
120. Nakaya K, Ayaori M, Uto-Kondo H, Hisada T, Ogura M, Yakushiji E, et al. Cilostazol enhances macrophage reverse cholesterol transport in vitro and in vivo. Atherosclerosis. (2010) 213:135–41. doi: 10.1016/j.atherosclerosis.2010.07.024
121. Wada T, Onogi Y, Kimura Y, Nakano T, Fusanobori H, Ishii Y, et al. Cilostazol ameliorates systemic insulin resistance in diabetic db/db mice by suppressing chronic inflammation in adipose tissue via modulation of both adipocyte and macrophage functions. Eur J Pharmacol. (2013) 707:120–9. doi: 10.1016/j.ejphar.2013.03.016
122. Otsuki M, Saito H, Xu X, Sumitani S, Kouhara H, Kurabayashi M, et al. Cilostazol represses vascular cell adhesion molecule-1 gene transcription via inhibiting NF-κB binding to its recognition sequence. Atherosclerosis. (2001) 158:121–8. doi: 10.1016/S0021-9150(01)00431-2
123. Tsai CS, Lin FY, Chen YH, Yang TL, Wang HJ, Huang GS, et al. Cilostazol attenuates MCP-1 and MMP-9 expression in vivo in LPS-administrated balloon-injured rabbit aorta and in vitro in LPS-treated monocytic THP-1 cells. J Cell Biochem. (2008) 103:54–66. doi: 10.1002/jcb.21388
124. Hattori Y, Suzuki K, Tomizawa A, Hirama N, Okayasu T, Hattori S, et al. Cilostazol inhibits cytokine-induced nuclear factor-κB activation via AMP-activated protein kinase activation in vascular endothelial cells. Cardiovasc Res. (2009) 81:133–9. doi: 10.1093/cvr/cvn226
125. Horai S, Nakagawa S, Tanaka K, Morofuji Y, Couraud PO, Deli MA, et al. Cilostazol strengthens barrier integrity in brain endothelial cells. Cell Mol Neurobiol. (2013) 33:291–307. doi: 10.1007/s10571-012-9896-1
126. Kimura T, Hamazaki TS, Sugaya M, Fukuda S, Chan T, Tamura-Nakano M, et al. Cilostazol improves lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells. J Dermatol Sci. (2014) 74:150–8. doi: 10.1016/j.jdermsci.2014.01.001
127. Tai SY, Chien CY, Chang YH, Yang YH. Cilostazol use is associated with reduced risk of dementia: a Nationwide Cohort Study. Neurotherapeutics. (2017b) 14:784–91. doi: 10.1007/s13311-017-0512-4
128. Taguchi A, Takata Y, Ihara M, Kasahara Y, Tsuji M, Nishino M, et al. Cilostazol improves cognitive function in patients with mild cognitive impairment: a retrospective analysis. Psychogeriatrics. (2013) 13:164–9. doi: 10.1111/psyg.12021
129. Arai H, Takahashi T. A combination therapy of donepezil and cilostazol for patients with moderate Alzheimer disease: pilot follow-up study. Am J Geriatr Psychiatry. (2009) 17:353–4. doi: 10.1097/JGP.0b013e31819431ea
130. Sakurai H, Hanyu H, Sato T, Kume K, Hirao K, Kanetaka H, et al. Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer's disease and cerebrovascular disease: a pilot study. Geriatr Gerontol Int. (2013) 13:90–7. doi: 10.1111/j.1447-0594.2012.00866.x
131. Ihara M, Nishino M, Taguchi A, Yamamoto Y, Hattori Y, Saito S, et al. Cilostazol add-on therapy in patients with mild dementia receiving donepezil: a retrospective study. PLoS ONE. (2014) 9:e89516. doi: 10.1371/journal.pone.0089516
132. Tai SY, Chen CH, Chien CY, Yang YH. Cilostazol as an add-on therapy for patients with Alzheimer's disease in Taiwan: a case control study. BMC Neurol. (2017a) 17:40. doi: 10.1186/s12883-017-0800-y
133. Saito S, Kojima S, Oishi N, Kakuta R, Maki T, Yasuno F, et al. A multicenter, randomized, placebo-controlled trial for cilostazol in patients with mild cognitive impairment: the COMCID study protocol. Alzheimers Dement. (2016) 2:250–7. doi: 10.1016/j.trci.2016.10.001
Keywords: Alzheimer's disease, cerebrovascular disease, cerebral amyloid angiopathy, MIND diet, glymphatic system, IPAD, cilostazol
Citation: Saito S, Yamamoto Y and Ihara M (2019) Development of a Multicomponent Intervention to Prevent Alzheimer's Disease. Front. Neurol. 10:490. doi: 10.3389/fneur.2019.00490
Received: 16 December 2018; Accepted: 23 April 2019;
Published: 08 May 2019.
Edited by:
Stefano L. Sensi, Università degli Studi G. d'Annunzio Chieti e Pescara, ItalyReviewed by:
Berislav Zlokovic, University of Southern California, United StatesElizabeta Blagoja Mukaetova-Ladinska, University of Leicester, United Kingdom
Copyright © 2019 Saito, Yamamoto and Ihara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Saito, c2FpdG9zYUBuY3ZjLmdvLmpw