 Kun Huang
Kun Huang Yue-Bei Luo1
Yue-Bei Luo1 Huan Yang
Huan Yang- 1Neurology Department, Xiangya Hospital, Central South University, Changsha, China
- 2Division of Neurogenetics, Center for Neurological Diseases and Cancer, Nagoya University Graduate School of Medicine, Nagoya, Japan
The neuromuscular junction, also called myoneural junction, is a site of chemical communication between a nerve fiber and a muscle cell. There are many types of channels at neuromuscular junction that play indispensable roles in neuromuscular signal transmission, such as voltage-gated calcium channels and voltage-gated potassium channels on presynaptic membrane, and acetylcholine receptors on post-synaptic membrane. Over the last two decades, our understanding of the role that autoantibodies play in neuromuscular junction disorders has been greatly improved. Antibodies against these channels cause a heterogeneous group of diseases, such as Lambert-Eaton syndrome, Isaacs' syndrome and myasthenia gravis. Lambert-Eaton syndrome is characterized by late onset of fatigue, skeletal muscle weakness, and autonomic symptoms. Patients with Isaacs' syndrome demonstrate muscle cramps and fasciculation. Myasthenia gravis is the most common autoimmune neuromuscular junction channelopathy characterized by fluctuation of muscle weakness. All these disorders have a high risk of tumor. Although these channelopathies share some common features, they differ for clinical features, antibodies profile, neurophysiological features, and treatments. The purpose of this review is to give a comprehensive insight on recent advances in autoimmune channelopathies at the neuromuscular junction.
Introduction
Neuromuscular junction (NMJ) is a type of chemical synapse between motor neurons and skeletal muscles, which consists of presynaptic membrane, synaptic cleft, and post-synaptic membrane. The most crucial event at NMJ is neuromuscular transmission that leads to contraction of skeletal muscles. In order to contract skeletal muscles, chemical neurotransmitters, such as acetylcholine (ACh), are released from presynaptic membrane, under the synergy of ion channels, such as voltage-gated calcium channels (VGCCs) and voltage-gated potassium channels (VGKCs), to post-synaptic membrane, binding to acetylcholine receptors (AChRs) of which the clustering and maintenance need muscle-specific kinase (MuSK), lipoprotein-related protein 4 (LRP4), and agrin (1). Neuromuscular junction channelopathies include a variety of disorders of genetic, toxic, and autoimmune origin. Regardless of the causes, these disorders lead to an impaired neuromuscular transmission. Acquired autoimmune channelopathies at neuromuscular junction include Lambert-Eaton syndrome (LEMS), Isaacs' syndrome, and myasthenia gravis (MG).
LEMS is caused by an autoimmune attack against presynaptic VGCCs and is characterized by late onset of fatigue, skeletal muscle weakness, weight loss, autonomic dysfunction, and areflexia. It develops in the context of a malignant neoplasm, usually small cell lung carcinoma (SCLC) (2). Isaacs' syndrome is caused by autoantibodies against VGKCs and patients with Isaacs' syndrome complain of muscle stiffness and cramps, and on physical examination demonstrate fasciculation (3). MG is an autoimmune disease associated with antibodies usually directed against AChRs, MuSK, or LRP4, in the post-synaptic membrane at NMJ, and is characterized by fluctuation of muscle weakness and fatigue (4).
Except for Isaacs' syndrome, although these channelopathies share some symtoms, such as skeletal muscle weakness and fatigue, they differ for clinical features, antibodies profile, neurophysiological features, and treatments. In this paper, we mainly focus on the clinical, laboratory, and pathological features, as well as treatment of these channelopathies, and give a comprehensive insight on recent advances in autoimmune neuromuscular junction channelopathies.
NMJ
Structure and Function of the NMJ
The NMJ, also called myoneural junction, is a specific chemical synapse site between nerve terminal and muscle fiber, causing muscle contraction through transmitting signal from the motor neuron to muscle fiber (5). NMJ, which typically locates near the middle of the muscle fiber, consists of three parts, presynaptic membrane, synaptic cleft, and post-synaptic membrane (Figure 1).
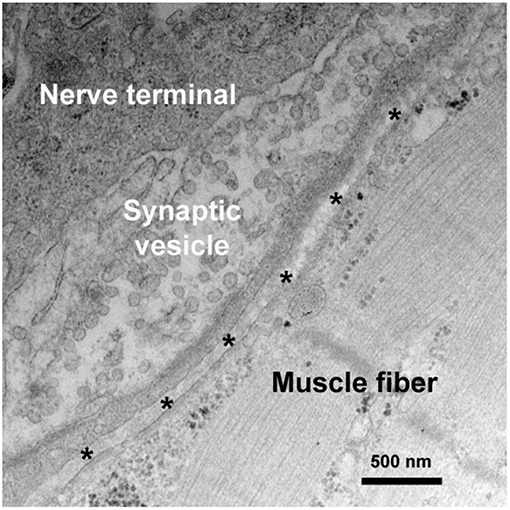
Figure 1. Synaptic ultrastructure at the NMJ. A representative electron micrograph of a human NMJ synapse. Asterisks represent the synaptic cleft.
Presynaptic Membrane Channels
VGCCs
VGCCs is a group of voltage-gated ion channels with a preferential permeability to the calcium ions and are also slightly permeable to sodium ions (Figure 2) (6). One of the essential factors underlying neurotransmitter release and nerve conduction at the presynaptic membrane is the calcium dynamics. VGCC is a complex protein consisting of multiple subunits. The pore-forming α1 subunit is responsible for the biochemical and electrophysiological characteristics of VGCC. At physiological or resting membrane potential, VGCCs are normally closed, the concentration of calcium ions is much lower in inside of the presynaptic membrane than outside (7). During an action potential, VGCCs are activated and open, causing a substantial and temporary influx of the calcium ions and a surge of calcium concentration, then calcium ions flow away from the channel and interact with neurotransmitter release sensors, calcium buffering proteins or kinases (8, 9).
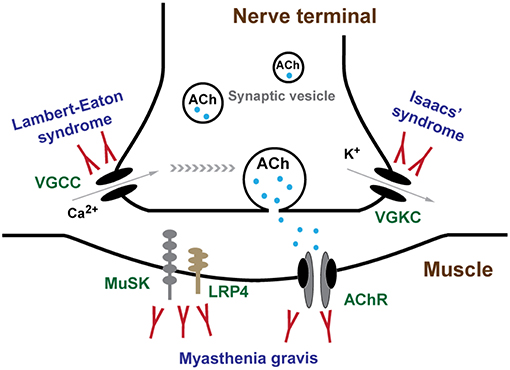
Figure 2. Channels and channelopathies at NMJ. NMJ channels and their associated human diseases are indicated in green and blue letters, respectively. The entry of calcium ions (Ca2+) through VGCCs depolarize the nerve terminal, resulting in the release of ACh from synaptic vesicles into synaptic cleft. In repolarization process, potassium ions (K+) leak out to the extracellular through VGKCs, accompanied by a recovery of resting potential and a halt of substantial ACh release. Diffused ACh are captured by AChR leading to muscle contraction. LEMS and Isaacs' syndrome are caused by antibodies against VGCCs and VGKCs, respectively. MG is mainly caused by antibodies against AChR, MuSK, and LRP4.
VGKCs
VGKCs are transmembrane channels sensitive to voltage changes and specific for potassium in membrane potential (Figure 2). Each subunit of VGKCs has six transmembrane regions named S1 to S6, with N- and C- terminals located at the intracellular side. The channel pore is surrounded by the S5 and S6 regions. Between the region S5 and S6, there is a P region which associates with S6 to form a selectivity filter of the ions (10). VGKCs are involved in determination of the resting potential of cell membranes, controlling thresholds of excitation, modulating wave forms and frequencies of action potentials, and repolarization of depolarized membranes (11). The resting membrane potential of mammalian nerve terminal is generally close to the potassium equilibrium potential owing to the function of background VGKCs. During action potentials, VGKCs play an important role in returning the depolarized cell to a resting state by removing the redundant potassium outside (12). Antibodies of VGKCs in peripheral nervous system cause autoimmune neuromyotonia disorders, such as Isaacs' syndrome, and in central nervous system lead to Morvan syndrome (13) and limbic encephalitis (14).
Post-synaptic Membrane Channels
AChRs
Post-synaptic membrane, namely the sarcolemma, harbors a high density of transmitter receptors, such as AChRs in a density of almost 10,000 AChRs/μm2 (15). Clustering of AChRs at the NMJ is mediated by the agrin-LRP4-MuSK signaling (5, 16). LRP4 is a member of the LDL receptor family. Neural agrin does not directly bind to MuSK, but it activates MuSK through binding with LRP4 (17). MuSK binds to LRP4 to receive neural agrin signaling which is essential for clustering and function maintenance of AChR. AChRs are distributed spatially restricted to the area immediately surrounding the opening of post-junction folds and partially down the sides of the infolded membrane and excluded from the trough of post-junctional folds (18).
There are mainly two types of AChRs, nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs) in human. nAChRs are ion channels which allow the trafficking of sodium, potassium and calcium ions with no selectivity for cations, while the mAChRs are not ion channels (19). The nAChRs are pentamers containing four macromolecules, such as cationic AChRs, cationic serotonergic receptors (5HT3), anionic glycine receptors, and anionic GABAA and GABAC receptors (20). Subunits of nAChRs in neurons and muscles are different. Muscular nAChRs are comprised of, two α subunits, and one each of β, γ and δ in the fetal type, resulting in a stoichiometry of α2βδγ, while in the adult type, the ε subunit replaces the γ subunit with a stoichiometry of α2βδε (21). The relative contents of the two types of AChR channels, depend on innervation of the muscle by spinal motor neurons. Before innervation, the fetal type predominates; at later stages of synapse formation, the fetal type is replaced by the adult type (22, 23). The adult type appears during the first post-natal week and replaces majority of the neonatal form within the second post-natal weeks except for a small part skeletal muscle, such as some extraocular muscles (24). During the time of transition, endplates have both neonatal and adult types of AChR (25, 26). Genes code for these subunits include: CHRNA1 for α, CHRNB for β, CHRND for δ, CHRNG for γ, and CHRNE for ε. Antibodies of AChRs usually lead to MG.
Nerve Impulse Conduction at NMJ
The entry of calcium ions through VGCCs serves as a connection between the depolarization of the nerve terminal and the activation of the neurotransmitter release mechanism (27). A single nerve impulse conducts to nerve terminal, immediately causes the activation of VGCCs which are responsible for calcium action potentials, with an influx of calcium into the intracellular side. Calcium influx causes vesicular exocytosis, leading to ACh release from the vesicles to synaptic cleft (28, 29). The ACh release process triggered by the calcium ion influx is mediated by 100–300 synaptic vesicles, and raises the local concentration of ACh in the synaptic cleft to a concentration of almost 0.3 mM (30). In repolarization process, an important event is potassium ions leak to the extracellular side as a result of activation of VGKCs, accompanied by a recovery of resting potential and a halt of substantial ACh release. AChs, the 146Da small molecules released from the nerve terminal in bursts, diffuse immediately into the synaptic cleft and are captured by AChRs, binding to the α subunits of AChR at their interfaces with surrounding γ and δ subunits (31). Then AChRs are activated and open in microseconds with a flux of cations, mainly sodiums, flowing through by their electrochemical gradients (32). This causes a depolarization potential, which induces an action potential and contraction in the muscle fiber it controls. Normally, a myriad excess of ACh is released from the presynaptic membrane, and several times as many AChRs are activated as would be necessary for an endplate potential (EPP) to reach the muscle-fiber firing threshold. The redundant AChs in the synaptic cleft are hydrolyzed by acetylcholinesterase (AChE) within a millisecond. The opening and closing of AChRs are only too quick to result a prompt initiation and termination of the post-synaptic response (33). Nerve impulse conduction at NMJ are indicated in Figure 2.
Safety margin of neuromuscular transmission is generally defined as the ability of neuromuscular transmission to remain effective under various physiological conditions and stresses (34). In the case of NMJ, a large surplus of both ACh and AChR provides a safety margin which allows threshold depolarization across every stimulated NMJ under normal circumstances (35). Once the excess AChRs have been blocked leading to a decreased safety margin, the released ACh cannot produce a sufficient strong signal to generate a signal in the post-synaptic membrane to cause a muscle contraction (36). Autoimmune or genetic defects at the presynaptic region, synaptic basal lamina, or post-synaptic structure of the neuromuscular junction can impair the safety margin of neuromuscular transmission. In MG, antibodies against AChRs lead to a decreased safety margin of neuromuscular transmission, so that slight depletion of ACh results in failure of post-synaptic depolarization for many muscle fibers. Similarly, in LEMS, antibodies against VGCC compromise the safety margin which results in radically decrease ACh release at all times. By comparison, in Isaacs' syndrome, antibodies against VGKC lead to delayed repolarization of the axon after each action potential impairing safety margin which contributes to prolongation of the depolarization of the muscle fiber membrane (34).
Since different channels play varying roles at NMJ, autoimmune antibodies of certain channels cause distinct symptoms. Antibodies against VGCCs and AChRs usually cause similar symptoms, such as skeletal muscle weakness, to list the main feature, due to insufficient AChs released from presynaptic membrane or reduced functional AChR density on post-synaptic membrane, respectively (37). While antibodies against VGKCs often lead to serial symptoms as a result of redundant AChs released from the nerve terminal, such as muscle stiffness and cramps (3).
LEMS
LEMS is an autoimmune neuromuscular junction channelopathy caused by antibodies against VGCCs. Symptoms mainly include late onset muscle fatigue and weakness, weight loss, and autonomic symptoms, such as dry mouth, male impotence, and constipation, usually in association with malignant tumor (38). This rare channelopathy was first reported by Edward Lambert and Lee Eaton in 1957, with a distinctive electrophysiological abnormalities in repetitive nerve stimulation (RNS) which were remarkably different from that of typical MG (39). Approximately 50% of LEMS patients have a primary autoimmune disorder and 60% of patients with LEMS have a tumor, most often SCLC (40). Since almost half of the LEMS associated with tumor, LEMS was usually categorized as non-tumor LEMS (NT-LEMS) and paraneoplastic LEMS (CA-LEMS). Some LEMS clinical symptoms overlap with those of other myasthenic syndromes, most commonly MG, which may lead to misdiagnosis or delayed diagnosis.
Epidemiology
Since LEMS is a rare channelopathy, the epidemiological data varies with different district, usually with a world-wide prevalence of 2–4 per million, which is ~46 times less than that of MG (41–43). The median age of onset is around 50–60, but LEMS can also affect children (44, 45). Particularly, a female predominance has been found in individuals diagnosed under 45 years. On the contrary, a male predominance in those diagnosed after the age of 60 years (46). In CA-LEMS, the median age of onset is 60 years with a male predominance, while in NT-LEMS, the first peak age of onset is around 35 years old and a second, larger peak is age 60 years. The age and sex distribution in NT-LEMS is similar to that reported for MG (47).
Clinical Features
The clinical triad of LEMS typically consists of proximal muscle weakness, autonomic features, and areflexia (40). Patients with LEMS almost invariably suffer from proximal weakness of lower limbs as a first symptom. Gradually, upper limbs, distal lower limbs and sometimes cranial muscles are also involved. As a hallmark of MG, ptosis can also be detected in LEMS, albeit generally in a mild form and later in the disease course (48). Since the main clinical features are similar with those of subacute myopathy, also electromyography (EMG) and biopsy abnormalities mimicking myopathy may often be found in patients with LEMS, therefore it is of obvious importance to diagnose LEMS patients from myopathy (49). The disease progression is much more malignant in CA-LEMS than in NT-LEMS. Usually legs and arms are implicated since the onset of symptoms in a large percentage of CA-LEMS, while most of NT-LEMS may only have proximal lower limbs weakness. Although artificial ventilation was reported as approximately in 11% of LEMS, respiratory failure, a common manifestation of MG, is infrequent in LEMS and it is always due to paralytic agents, such as pancuronium, atracurium, and vecuronium, use or intercurrent pulmonary pathology (50, 51).
Autonomic dysfunction is reported in up to 96% of patients with LEMS (40, 44, 47, 52, 53). Dry mouth, constipation, and erectile dysfunction in men are particularly common, and loss of sweating, orthostatic hypotension, and pupillary abnormalities can also be found. Another typical symptom is the decreased or absent tendon reflexes in LEMS. Deep tendon reflexes are always reduced or absent, especially in the lower limbs. In up to 40% of patients with LEMS, a previously absent or significantly reduced deep tendon reflex will return to normal, also with a recovery of muscle strength to almost normal level, after 10 s of maximal voluntary contraction, which is a characteristic phenomenon in LEMS (38). Thus, tendon reflexes should be tested after a period of rest because of the post-exercise facilitation phenomenon can disguise the abnormal tendon reflexes.
Pathophysiology
LEMS consists of NT-LEMS and CA-LEMS. Tumor association is estimated in about 60% of patients with LEMS (51). The most common malignant carcinoma of CA-LEMS is SCLC, a smoking-related neuroendocrine lung carcinoma. Other tumors also have been found associated with LEMS, such as non-small cell and mixed lung carcinomas, prostate carcinoma, and thymoma (40, 46, 51). Since these cancers have neuroendocrine characteristic, antibodies against VGCC subunits were generated during the disease duration. Besides, SOX1 protein plays a role in airway epithelial differentiation and is shown to be present in SCLC, which show a relative good value for LEMS diagnosis.
Antibodies
Until 1983, the pathogenic antibodies against VGCCs was first found by Fukunaga (54). The discovery of pathogenic autoantibodies of VGCC has greatly facilitated diagnosis of LEMS and improved the understanding of the underlying pathophysiologic mechanisms. Subsequent researches show the most popular antibodies are that against P/Q-type VGCCs, which cause most of the clinical symptoms of LEMS (55). However, the significance of an elevated antibodies against VGCC titer beyond its original clinicopathological correlate, LEMS remains undetermined.
Traditionally, antibodies against P/Q-type VGCCs are detected in 85–90% of patients with LEMS, in some reports even with up to 100% in LEMS patients with SCLC, which suggests a high specificity of LEMS diagnosis (55–57). Interestingly, recent researches reported that antibodies against VGCC were detected not only in other autoimmune diseases, such as MG, but also in healthy people, which questioned the specificity of antibodies against VGCC for LEMS diagnosis. Di Lorenzo found that antibodies against P/Q-type VGCC had a diagnostic sensitivity of 88.89% and specificity of 36.17% (58). Zalewski also reported that antibodies against P/Q-type VGCC have a compromised specificity on LEMS diagnosis (59). Antibodies against another type of VGCCs, N-type or L-type VGCCs, have also been found in 30–40 and 25% of LEMS patients, respectively, but all of these patients were also be detected the P/Q-type VGCCs antibodies (57, 60). Although antibodies against P/Q-type VGCCs are somehow highly sensitive to LEMS, since it has a low specificity, cautious interpretation of results, particularly medium and low titers, is advised.
SCLC itself expresses three types of VGCCs, the N, L, and P type (61). Because SCLC is of neuroendocrine origin, it expresses the same types of VGCCs and secretory machinery as nerve terminals. Immune system produces antibodies targeting the protein which are secreted by SCLC, also attacking VGCCs on motor nerve terminal. The P/Q type of VGCCs, and also N type of VGCCs, are two main targets of IgG-mediated nerve terminal autoimmunity in LEMS (59).
In recent years, a new marker, SOX1, associated with paraneoplastic neurological disease has been described (62, 63). SOX1 is thought to prevent neural differentiation in progenitor cells and mainly expressed in the developing nervous system and downregulated in adults (64). Two studies showed antibody against SOX1 presents in 64–67% of patients with SCLC-LEMS, compared to 0–5% in NT-LEMS patients (65, 66). Using ELISA assay, SOX1 antibody has a sensitivity of 67% and a specificity of 95% to discriminate between SCLC-LEMS and NT-LEMS (66).
A small part of LEMS patients have no detectable VGCCs antibodies, namely the seronegative LEMS. Although antibodies were undetectable in seronegative LEMS, the clinical phenotype is almost identical to seropositive LEMS patients (67). Since passive transfer of seronegative LEMS sera to mice can also generate the typical symptoms and electrophysiological changes as those passively transferred with seropositive sera, seronegative LEMS might therefore due to the same antibodies of VGCCs but at a relatively lower titer, or other antibodies of VGCCs' epitopes not recognized currently (38). Intriguingly, antibodies against AChRs can also be detected in a small part of LEMS, while these specific antibodies have no diagnostic value (68).
EMG
Needle electrode EMG examinations are necessary for patients suspected of having disorders of synaptic transmission, such as LEMS, MG, and Isaacs' syndrome. RNS is essential for the diagnose of LEMS. In LEMS, the first compound muscle action potential (CMAP) is low, even lower at stimulating frequencies, about 2–5 Hz (69). Mostly, reduction of CMAP amplitude of 10% is considered abnormal. In LEMS, almost all the patients show a massive decrease of CAMP (38, 68). One of the key method to differentiate LEMS and MG is the high-frequency stimulation (50 Hz). An increase of the CMAP amplitude more than 100% is considered specific for LEMS. More recently, it is suggested that the threshold for LEMS diagnose can be decreased to 60% to improve sensitivity to 97% while retaining specificity of 99% to exclude MG (70). Single-fiber electromyography (SFEMG) is slightly more sensitive than RNS for diagnosis of LEMS (70). However, SFEMG is less specific than RNS and requires technical experience (69).
Treatment
Most of the LEMS patients have concomitant cancers, so treatment should include two parts, treatment against the known tumor when applicable and symptomatic management.
Oncological Screening
More than half of LEMS patients are associated with SCLC, thus it is pivotal to screen underlying tumors once the diagnosis of LEMS is established. Almost all of SCLC are found within a year since LEMS diagnosis is made (71). Computed tomography (CT) of the thorax or 18F-fluorodeoxyglucose-positron emission tomography (PET), is recommended for oncological screening (72). Otherwise, paraneoplastic biomarkers may be useful for oncological screening as a supplementary to radiological investigations. Once diagnosis of tumor was made, it is of highest priority to treat the cancer at the same time. Surgical removal of cancers usually leads to a prominent alleviation of the symptoms, in which the underlying mechanism may probably because of the reducing VGCC antibodies and a reduction of the autoimmune response. If clinical remission is compromised and symptoms of LEMS remain, additional treatment, such as immunosuppressive treatment might bring symptomatic improvement.
Symptomatic Treatment
Symptomatic treatment of LEMS should aim to enhance the release of neurotransmitters from presynaptic nerve terminal or prolong the activity or availability of AChs in the synaptic cleft. The fundamental and effective symptomatic treatment of LEMS is 3,4-diaminopyridine (3,4-DAP), a drug that blocks VGKCs, prolongs nerve terminal depolarization and increases ACh release from nerve terminal (73, 74). Starting dose of 3,4-DAP is generally from 5 to 10 mg, 3–4 times per day. Most of the patients have a relatively good response from 40 to 60 mg/d. The suitable dose can gradually increase to 80 mg/d, divided into four to six times. Clinic improvement can always be detected within 30 min and reaches a peak at 90 min after each intake (75). 3,4-DAP is well-tolerated. Perioral tingling, digital paresthesias, and gastrointestinal symptoms are the most common side effects (76). Doses of more than 100 mg/d may increase the risk of seizures (77). Since QT interval prolongation was found in patient taking 3,4-DAP, thus before and during intaking 3,4-DAP, electrocardiogram (ECG) should be examined (74).
Theoretically, pyridostigmine, an AChE inhibitor used to prolong the AChs activity in the synapse cleft, is in synergy with 3,4-DAP, but many patients of LEMS seldom benefit from pyridostigmine either on its own or in combination with 3,4-DAP, which largely compromise the clinic use of pyridostigmine in LEMS (78, 79).
Immunosuppressive Agents
Since LEMS is caused by the antibodies against VGCCs, treatment suppressing the immune system is effective. If 3,4-DAP preferably manage the symptoms of LEMS, no further treatment is needed. If symptoms remain, long-term treatment of immunosuppressors, such as prednisone and azathioprine, should be considered, although the direct evidence for their efficacy in treating LEMS is somehow uncertain (80).
Other Treatment
Clinical guidelines of American Academy of Neurology (AAN) review the use of intravenous immunoglobulin (IVIg) in the treatment of neuromuscular disorders, including LEMS and MG (81). According to clinical studies, AAN has endorsed the clinical use of IVIg as supported by evidence of efficacy in the treatment of MG (level B) and LEMS (level C). Some reports and single randomized placebo-controlled crossover studies found clinical improvement in LEMS patients after treatment with IVIg, peaking at 2–4 weeks, and declining by 8 weeks (76, 82–84). Plasma exchange (PE) has been reported in case series and case reports but are lack of clinical trials in LEMS patients. Patients with LEMS respond more slowly to PE than do patients with MG, with a peak effect at almost 2 weeks, and the duration of effect may vary from 1 to 6 weeks (85). PE may result in short-term improvement of LEMS, but is not particularly effective in the management of LEMS without immunosuppressive medications and the other pharmaceutical approaches already mentioned (80, 86).
Isaacs' Syndrome
Isaacs' syndrome is a rare autoimmune channelopathy at NMJ first characterized in 1961 by Hyam Isaacs in two patients with continuous muscle fiber activity (87). Since the patients were not ameliorated by peripheral nerve blockade but could benefit from curare, an inhibitor of AChRs, Isaacs later proposed that the reason of the spontaneous motor activity was due to the distal segments of peripheral nerves. Currently, Isaacs' syndrome is one of the most well-known peripheral nerve hyperexcitability (PNH) which causes persistent muscle fiber contraction characterized by muscle stiffness at rest and impaired muscle relaxation after voluntary contraction, yet different from myotonia (88). Clinically, Isaacs' syndrome is deemed as an autoimmune neuromyotonia disorder.
Clinical Features
Isaacs' syndrome is a channelopathy with heterogeneity which affects patients at any age and varies significantly in severity. Little epidemiological data could be reviewed in previous literatures due to its rare occurrence and potential underestimation. Although Isaacs' syndrome is an autoimmune channelopathy, intriguingly, it was reported that male is more susceptible than female by ~2-folds. The average onset age is in the mid-40s (89–91).
Interestingly, several decades ago, Isaacs' syndrome was defined as “cramp-fasciculation syndrome” because of the chief complaint of cramps and fasciculation (92). About one third of patients have slow muscle relaxation after voluntary contraction, such as handgrip, eye and jaw closure, which is termed as pseudomyotonia (93, 94). In most cases, it manifests with muscle stiffness and muscle cramps worsen by voluntary muscle movement, which commonly without muscle weakness and muscle atrophy at beginning. On physical examination, marked fasciculation and myokymia can be noticed. Fasciculations are spontaneous discharges of a single motor axon which cause focal or multifocal single twitches in a group of muscle fibers, while on the other hand, myokymia are a numerous involuntary, undulating muscle twitches in wavelike style which are visible on the muscle surface. Visible myokymia is one of the most characteristic symptoms in Isaacs' syndrome, almost observed in 90% of patients (94). Even when myokymia is not visible, it is often palpable by clinician. Generally, it can be observed in the limbs, but also can be detected in other muscles, such as truncal and facial muscles (89, 95). Muscle cramps are also one of the frequent signs observed in Isaacs' syndrome in more than 70% of cases and usually can be painful (94). Muscle stiffness can be associated with cramps, which can also be present in rest or sleep and may improve after repeated exercise (96).
Other clinical manifestations include muscle hypertrophy which most often occurs in but not limit to calf muscles (97), sensory disorders which often manifest as distal hypesthesia in a small number of patients (89), and autonomic dysfunction, such as hyperhidrosis, sialorrhea, palpitations, flush, and abdominal pain (87).
Pathophysiology
The fundamental pathophysiology of Isaacs' syndrome is dysfunction of VGKCs in presynaptic terminals due to acquired causes (98). Normally, Isaacs' syndrome is deemed as an autoimmune channelopathy while those neuromyotonia caused by genetic factors were usually classified as genetic diseases.
Antibody
Isaacs' syndrome is an autoimmune channelopathy at the NMJ caused by a group of autoantibodies. Several antibodies have been reported. However, there are almost 40% of patients have no defined targets (89, 99). Some VGKCs antibodies were detected in Isaacs' syndrome, however, positivity of antibodies against VGKCs in the absence of antibodies to leucine-rich glioma inactivated 1 (LGI1) and contactin-associated protein-like 2 (CASPR2) is not a clear disease biomarker for autoimmune inflammation and seems not to contribute in clinical practice (100). Antibodies against LGI1 and CASPR2 are antibodies against VGKCs-associated proteins rather than directly against VGKCs subunits, which were identified in 2010 (101). These antibodies do not directly block VGKCs, but rather decrease channel density either through increased degradation or decreased expression of VGKCs (102).
LGI1 is a secreted neuronal protein mainly expressed in the hippocampus specifically associated with VGKCs subunits in central nervous system presynaptic terminals (103). CASPR2 is a transmembrane protein expressed both in the central and peripheral nervous system with a large extracellular sequence which is vital for localization of subunits of VGKCs at juxtaparanodes (104). Not only detected in Isaacs' syndrome, both LGI1 and CASPR2 antibodies can also be discovered in other diseases, such as Morvan's syndrome, neuropathic pain, epilepsy, limbic encephalitis, and cerebellar dysfunction (105). LGI1 antibody seems to be more strongly associated with limbic encephalitis than Isaacs' syndrome and less seropositive in Isaacs' syndrome compared with CASPR2 antibody (106). It has been reported that these two antibodies highly correlate with clinical measures and have little correlation with cancers in Isaacs' syndrome (107). Since VGKC antibodies have little specificity in Isaacs' syndrome, the titers of antibodies should be considered cautiously during the clinical evaluation especially for those low positive tilters.
Paraneoplastic Association
Since male is more susceptible than female to Isaacs' syndrome by almost 2-folds, suggesting that paraneoplastic syndrome may be a cause of Isaacs' syndrome. More and more researches report that malignancies are found in patients with Isaacs' syndrome, supporting the hypothesis that tumor antigens trigger an autoimmune response and result in antibodies against VGKCs (94). The possible pathogenesis of paraneoplastic Isaacs' syndrome may be the activation of immune response by tumor-related antigens leading to autoantibodies, such as those targeting components of the VGKC complex (108). Thymoma and SCLC are the tumors most commonly associated with Isaacs' syndrome (109, 110).
EMG
EMG shows characteristic myokymic and neuromyotonic discharges (111, 112). Sensory and motor nerve conduction studies are seldomly abnormal, including late responses, such as F waves and H reflexes, except for after discharges on motor nerve conduction studies. Myokymic discharges are spontaneous, continuous, rhythmic, irregularly occurring doublet, triplet or multiplet single motor unit discharges, with a frequency of around 30–40 Hz, followed by a short interval of silence, always up to a few seconds, and then recurrence of the burst at regular intervals (113). On the contrary, neuromyotonic discharges are composed of firing of single myofibers at high frequencies of 150–300 Hz. They can be spontaneous or be provoked by needle movement or muscle contraction. Repetitive supramaximal stimulation of a peripheral nerve at 10 Hz shows a sensitivity of 79% and specificity of 88% for identifying patients with Isaacs' syndrome. No direct evidence shows that SFEMG helps for detection of Isaacs' syndrome, thus SFEMG needs not be performed unless concerning exists for a defect of neuromuscular transmission, such as MG and LEMS (114).
Treatment
Screening of tumor should be positively performed, especially thymoma (115), SCLC (109), and hematological tumor (116). Once tumors were detected, it is better to remove the cancer if it is possible. If no underlying tumors are detected, initial treatment should better include only symptomatic treatment.
Symptomatic Treatment
Currently, there are no FDA-approved drugs for symptomatic treatment of Isaacs' syndrome. Anticonvulsants are often used to moderate the symptoms of Isaacs' syndrome, such as cramps. Carbamazepine and phenytoin, which mainly work through sodium channel blockage, have been shown to be effective for Isaacs' syndrome (117). Gabapentin at a dose of up to 900 mg/day also appears to be beneficial, by predominantly affecting the central pain pathways through binding to calcium channel subunits (118–120). Carbamazepine is recommended as a first-line agent for symptomatic therapy at 400–600 mg/day in divided doses initially, with up to 1,200 mg/day in divided doses as tolerated (3). Efficacy of therapy should be assessed by monitoring the clinical response, rather than electrodiagnosis which can only be used as a secondary outcome measure.
Other Treatment
Beneficial effect of PE have been shown in many studies (121). PE can also be used in combination with prednisolone and azathioprine (122). PE is recommended as the first-line immunomodulating treatment for Isaacs' syndrome (3). IVIg, another common treatment for autoimmune disorders, has been reported to be less effective for Isaacs' syndrome (122).
MG
MG is the most common autoimmune neuromuscular junction channelopathy caused by pathogenic autoantibodies to components, mostly are AChR, MuSK, and LRP4 on the post-synaptic muscle membrane (123). Patients usually complain about muscle weakness with fluctuations in severity in 1 day, which is a remarkable feature of MG. Increased muscle weakness after continued muscle activity represents a strong diagnostic clue for diagnosis of MG. The course of the disease is highly variable, symptoms and signs may change rapidly due to infection or pregnancy. Respiratory muscles may be involved leading to respiratory failure. Diagnosis should be based on confirmatory diagnostic testing, including serum antibodies tests and EMG. Treatment for MG traditionally contains thymectomy, AChE inhibitors, immunosuppressors, PE, and IVIg.
Epidemiology
MG is the most common autoimmune NMJ channelopathy with a worldwide prevalence of 40–180 and an annual incidence of 4–12 per million people (124). AChR seropositive MG has an obvious age pattern of incidence, with a peak age of third decade which is a strong female predominance, and another peak in the elderly with a slight male predominance (125, 126). The incidence peak in young adults is partly due to of the high frequency in female which is typical for many autoimmune disorders, while late-onset MG is slightly more frequent in male (124, 127). The incidence of MuSK-associated MG in Netherland is estimated at 0.1 patients per million per year, with a prevalence of 1.9 per million people (128). In contrast to AChR seropositive MG, where the peak incidence is the third decade, age at symptom onset of MuSK-associated MG is distributed around a peak in the fourth decade, with another smaller peak in the second decade (129). MG rarely coincides in members of the same family (130, 131).
Clinical Feature
Muscle weakness is the most common symptom in MG. Combination of fluctuation in muscle weakness over time and exercise-induced muscle weakness strongly implies the diagnosis of MG.
Muscle weakness in MG can occur in all the skeletal muscles including extraocular, bulbar, limb, and axial muscles. Over half of patients have prominent ptosis or diplopia, and in 20% patients, the muscle weakness is restricted in extraocular muscle without any other muscle weakness (132). Interestingly, weakness of extraocular muscles tends to be asymmetrical, while limb weakness is mostly symmetrical and more severe in proximal than distal (133). Ocular MG (oMG) is a more common form of juvenile MG in Asian populations than in other populations (134, 135). Patients may have eyelid retraction, most prominent upon awakening. If respiratory muscle weakness occurs, patients may develop respiratory failure requiring intubation (136). Premonitory signs usually include difficulty breathing, swallowing, and chocking. Speech can also be affected leading to a change in voice characteristics. Severity of MG can be quantified according to the Myasthenia Gravis Foundation of America's classification system (137).
Since MG is caused by autoantibodies, there is an increased frequency of organ-specific and general autoimmune disorders especially thyroiditis (138). Sixty-five percentage of MG patients have thymic hyperplasia and 10–15%, a thymoma. It is reported that the initial steps triggering humoral immunity in MG take place inside the thymic tissue and thymoma (139).
Pathophysiology
Nowadays, MG is considered as a T-cell-mediated disease. The thymic tissue is able to express epitopes cross-reactive with skeletal muscle proteins, such as AChR, titin, and ryanodine receptor (RyR) (140). Thymic epithelial cells present AChR peptides to T cells in MG patients, resulting in intrathymic immunization (141). The immune response against epitopes expressed on abnormal thymic cells spills over to components at NMJ, mostly like AChR, which causes symptoms of MG (142).
Antibodies
MG is mainly caused by antibodies against AChR or other proteins on the post-synaptic membrane, with a characteristic of impaired signal transduction, muscle weakness, and fatigability. AChR antibodies are found in 85% of all MG patients (143). IgG1 and IgG3 are the prevalent subclass of AChR antibodies which have ability to activate complement and therefore to cause post-synaptic membrane damage and block the signaling pathway (144). Antibodies against AChR α subunit are more pathogenic than those against other subunits, such as β, δ, γ, and ε. Different AChR epitope antibody pattern influences disease severity (145).
MuSK is an AChR related membrane protein which is critical for the formation of NMJ (146). MuSK antibodies occur in <10% of MG patients. In most MuSK-associated MG patients, MuSK antibodies are predominantly against the IgG4 subclass, a minor IgG component without well-defined, but presumably anti-inflammatory roles in immunity. Although IgG4 is deemed to have no activation effect on potent complement, MuSK antibodies bind to the extracellular N-terminal Ig-like domains of the AChR, retaining direct pathogenic capability by reducing post-synaptic AChR density, impairing the alignment between motor nerve terminal and post-synaptic membrane (147).
The prevalence of LRP4 antibodies represents in < 50% of AChR and MuSK antibodies double negative patients (148). In LRP4 immunized mice, LRP4 antibodies induce muscular weakness through disruption of the interaction between LRP4 and agrin, and thereby inhibit AChR-mediated neuromuscular signal transmission (149). Although the presence of anti-LRP4 in MG has been confirmed, their exact prevalence, pathogenic role and associated clinical phenotypes are largely unknown.
Neuronal agrin is an indispensable factor for formation of the NMJ by binding to LRP4 and stimulating MuSK (150). Agrin autoantibodies were detected in some MG patients, either with or without AChR or MuSK antibodies (151, 152). Agrin antibodies can inhibit MuSK phosphorylation and AChR clustering, which is detected in MG patients only (153).
Clinical Classification
According to the age of onset, autoantibodies and thymic pathology, the disease forms are generally divided into several subgroups.
Pure oMG
In this form, muscle weakness is restricted to ocular muscles. Although this type is at risk of progressing to generalized MG (gMG), 90% of those who have had the ocular form for more than 2 years will remain in this subgroup (154). MuSK antibodies very rarely occur in this type (154).
Thymoma-Associated MG
10–15% of all patients associate with thymoma. Thymoma-associated MG is widely deemed as a paraneoplastic disease. Nearly all patients of this type are AChR-associated MG and seldom are ocular MG. Thymoma-associated MG patients usually have a higher prevalence of severe phenotype and also higher anti-AChR antibody titer than non-thymomatous MG patients (155).
AChR-Associated gMG
Nearly 85% of the MG population have detectable AChR antibodies and display this form of the disease. The titer of antibodies has no clear correlation with severity of the disease (156). Thymic abnormalities are more frequently found in this form than other types of MG (157). This form can be further categorized into two types: early onset MG that the onset of the disease before the age of 50, and late onset MG that the onset of the disease after the age of 50.
MuSK-Associated MG
Typically, MuSK antibody positive patients are female predominance, and they have a relatively severe form of the disease with muscular atrophy. The facial, bulbar, and respiratory muscles are frequently affected, while ocular muscle weakness and thymic abnormalities are rare (158, 159).
LRP4-Associated MG
LRP4 antibodies were discovered in ~12–50% of patients who were double seronegative for AChR and MuSK (160). The clinical phenotype of this type is not well-defined.
Antibody-Negative MG
MG patients lack of antibodies of AChR, MuSK, and LRP4 are traditionally called antibody-negative MG or seronegative MG. MG of this type represents a heterogeneous group pathogenically. Patients of this type probably have undefined pathogenic antibodies against proteins in the post-synaptic membrane (161). The diagnosis is more challenging in patients in whom no specific autoantibodies are detected.
Diagnosis
Patients with classical fatigable symptoms need further examination. Ancillary tests include pharmacologic, serologic, and electrophysiologic tests.
Neostigmine Test
Intramuscular injection of 1.0–2.0 mg neostigmine, an AChE inhibitor, has a remarkable ameliorative effect on the deficit signs, such as ptosis, hypernasal voice, and limb weakness from 30 min and persisting for almost 90 min after injection (162). By inhibiting AChE through neostigmine, the amount of AChs is significantly increased in the synaptic cleft, and AChs are capable of binding to the AChRs for a longer period, resulting improved neuromuscular transmission. In MG, 90% patients response positively to AChE inhibitors. A positive reaction to AChE inhibitors can also be observed in congenital myasthenic syndrome, LEMS, amyotrophic lateral sclerosis and Guillain-Barrè syndrome (163). Nevertheless, although neostigmine test is much less used in the past, considering its easy methodology and inexpensive cost, it can still be recommended in developing countries. Although neostigmine test may be one of the first screening of MG, the responsiveness is not necessarily diagnostic for MG.
Serologic Test
The AChR antibodies are highly specific for MG diagnosis (123). If they are negative, it is important to test the anti-MuSK, LRP4 or other clustered AChR antibodies. Their presence is important as it can largely help to make the diagnosis of MG in some uncertain cases.
EMG
Strictly speaking, any AChE inhibitors should be stopped at least 12 h before EMG examination. The examination of EMG is pivotal for MG diagnosis and must be investigated in several proximal and distal nerve and muscle pairs. The classic electrophysiologic demonstration of an NMJ transmission defect is the documentation of a decremental response of the CMAP to slow (2–3 Hz) motor repetitive nerve stimulation (164). In RNS, a gold standard for MG, a decremental response of 10% from the first to the fourth or fifth response while stimulating at 2–5 Hz is valid for the diagnosis of MG.
SFEMG is a highly selective recording technique in which a concentric needle electrode is used to identify and record extracellular action potentials from individual muscle fibers (165). The typical SFEMG finding in MG is that increased jitters with impulse blocking, increased jitter without impulse blocking and also normal jitter can be detected within one muscle. Since SFEMG demonstrates abnormal jitter in virtually all patients with MG, it has been known to be the most sensitive diagnostic procedure for the diagnosis of MG for many years (166–168). Although SFEMG that reveals an elongated jitter is more sensitive than the RNS, it is not specific for MG, for example, in the radiculopathies and neuropathies, the specificity of SFEMG has been questioned (169–171). Besides the diagnostic value for MG, SFEMG is a valuable prognostic factor. In most MG patients, the changes in SFEMG measurements, especially the percentage of abnormal jitter pairs with blocking, correlated with the changes in clinical state as measured by quantitative testing of muscle function (172, 173).
CT Scan
Since a majority of MG patients have thymic diseases, it is essential to take a CT scan especially for gMG patients and those with anti-AChR antibodies (174). It is justifiable to control the thymus every 5 years if the patient was not thymectomized.
Treatment
Therapies for MG include pharmacotherapy, such as symptomatic drugs, immunosuppressors, and other therapies, such as thymectomy, PE, and IVIg.
Symptomatic Treatment
Pyridostigmine, an AChE inhibitor, is the main pharmacologic compound used for MG, both in children and adults. If appropriate usage and dosage of pyridostigmine are prescribed, symptoms and signs of MG still have remission, other immunosuppressive treatment should better to use at the same time (136).
Immunosuppressive Agents
The most common immunosuppressive drug for MG is prednisone which has a good therapeutic effect generally. This medication can easily be administered orally even to children. Additionally, treatment with prednisone can protect the conversion from oMG to gMG (175, 176). Azathioprine can be considered as a second-line treatment for MG patients who respond poorly to prednisone treatment (177). Other immunomodulatory medications can be considered for use in MG, such as rituximab, mycophenolate mofetil, tacrolimus, and eculizumab, which are shown effective therapeutic efficacy and considered as second-line treatment combinated with or without prednisone in some clinic studies (178–182). It is worth mentioning that eculizumab, one of the latest generation treatment, was approved for the treatment of adults with AChR-associated gMG in the USA (183), AChR-associated refractory gMG in the EU (184) or patients with AChR-associated gMG whose symptoms are difficult to control with high-dose IVIg therapy or PE in Japan (185). One of a latest meta-analysis found that eculizumab is the most effective and tolerable therapeutic for refractory MG and tacrolimus is a beneficial therapy for MG extensively (186). Moreover, some new drugs are also under exploration which need further researches, such as efgartigimod (187).
Thymectomy
Many studies have reported a beneficial effect of thymectomy on MG (138). The thymus may trigger autoimmunity against AChRs, thus, its removal may eliminate the main source of antibody production against AChRs which alleviates symptoms of MG. For early-onset MG, thymectomy is recommended for MG, while in late-onset MG, thymectomy is debated (188). The latest researches support that thymectomy improves clinical outcomes even in patients with non-thymomatous gMG (189, 190). Thymectomy is also proved to be safe for juvenile MG, even down to an age of about 5 years (191). All thymus tissue needs to be removed. Since no direct therapeutic effect has been found for patients with MuSK, LRP4, and oMG, thymectomy is not recommended for these patients.
Other Treatment
IVIg and PE are two specific immunosuppressive treatments with a rapid and definite effect occurring often after 2–5 days, and either one often be given to patients with severe MG or MG crisis. IVIg can be administered for MG in an effort to reduce the circulating autoantibodies by decreasing B-cell antibody production and T-cell function. PE works primarily by removing circulating autoantibodies responsible for neuromuscular junction dysfunction, and also removing cytokines responsible for activating lymphocytes, irrespective of antibody status (123). Although IVIg and PE shows a comparable efficacy and duration of effect in MG patients, IVIg is often slightly more convenient, with a lower risk of severe side-effects, and less economic cost, whereas PE might work slightly more rapidly (192, 193). Elderly and those with complex comorbid diseases including acute respiratory failure may be better treated with IVIg (192).
Differential Diagnosis
The autoimmune channelopathies at NMJ, LEMS, Isaacs' syndrome and MG, have overlapped clinical symptoms with each other, which renders the diagnosis more complicated. Auxiliary examinations are necessary for differential diagnosis. Key points of differential diagnosis are concluded in Table 1.
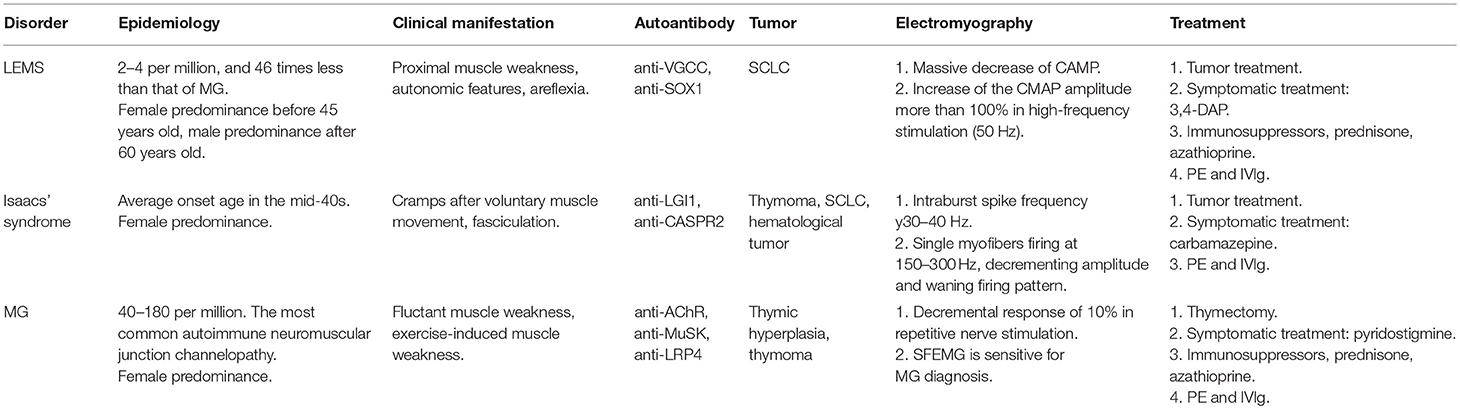
Table 1. Summary of main features of autoimmune neuromuscular junction channelopathies.
Clinical Features
Among these channelopathies, Isaacs' syndrome is the most easily to differentiate from LEMS and MG. Isaacs' syndrome have prominent symptoms, such as cramp, fasciculation, and myokymia which are rarely detected in LEMS and MG. To differentiate LEMS and MG, the former one typically starts with mild leg weakness, which progresses in a caudocranial direction, while the latter one commonly begins with oculobulbar weakness, and muscle weakness spreads craniocaudally (194). Autonomic dysfunction and diminished tendon reflexes are rarely seen with MG, while are quite normal in LEMS (194). Once autonomic dysfunctions present in one patient, the diagnosis of MG should be doubtful.
Auxiliary Examination
Electromyography
Needle electrode examination of Isaacs' syndrome normally shows an abnormal pattern of motor unit firing which are different with LEMS and MG, consisting of myokymic discharges, doublets and multiplets, neuromyotonic discharges, and fasciculations, which may occur spontaneously or may be activated by voluntary muscle contraction. These abnormalities may occur alone or in combination (195). To differentiate LEMS and MG, RNS is an indispensable test. Traditionally, an increase of the CMAP amplitude more than 100% is considered specific for LEMS. Recently, an increase of the CMAP amplitude more than 60% is considered to have both high sensitivity and specificity for LEMS diagnosis, which is rarely represented in MG (196).
Serological Test
Since all these disorders are autoimmune diseases, serological tests of antibody are necessary for diagnosis and identify the subgroup of disease. More than 85% of LEMS have antibodies against P/Q-type VGCC which are highly specific (55–57). While to MG, antibodies against AChR, MuSK, or LRP4, which also have high specificity for MG, are presented in almost more than 90% patients (123). For Isaacs' syndrome, antibodies have less sensitivity and specificity for diagnosis.
Conclusion
At NMJ, three channels, VGCCs, VGKCs, and AChRs, play fundamental roles in signal transmission. Autoimmune antibodies of certain channels cause distinct symptoms. All the autoimmune neuromuscular junction channelopathies are orphan diseases causing by antibodies against these channels. Different groups of antibodies cause different disorders and symptoms. Although they share some common symptoms, characteristic symptoms or signs can be detected in different channelopathies, such as autonomic dysfunction, myokymia, fasciculation, and fluctuation of muscle weakness. Making a correct diagnosis of these channelopathies may be somehow hard, but when suspicion of neuromuscular junction channelopathies is raised for a fatigable deficit, proper diagnostic tests should be pursued. Besides the typical symptoms and signs, auxiliary examinations including EMG and serologic tests are essential for diagnosis. Since a part of patients with these disorders can associate with tumors, oncological screening tests should not be neglected. After diagnosis, appropriate treatment is pivotal for patients' quality of life and ability to perform daily activities. Due to current limited knowledge of these channelopathies, there is a need for a standard regimen for diagnosis and treatment of autoimmune neuromuscular junction channelopathies to maximize long-term benefits.
Author Contributions
KH and HY conceived and planned the review. KH wrote the manuscript. Y-BL and HY critically revised the manuscript for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The first author, KH, obtained a scholarship from the China Scholarship Council (CSC).
References
1. Plomp JJ, Morsch M, Phillips WD, Verschuuren JJ. Electrophysiological analysis of neuromuscular synaptic function in myasthenia gravis patients and animal models. Exp Neurol. (2015) 270:41–54. doi: 10.1016/j.expneurol.2015.01.007
2. Sabater L, Hoftberger R, Boronat A, Saiz A, Dalmau J, Graus F. Antibody repertoire in paraneoplastic cerebellar degeneration and small cell lung cancer. PLoS ONE. (2013) 8:e60438. doi: 10.1371/journal.pone.0060438
3. Ahmed A, Simmons Z. Isaacs syndrome: a review. Muscle Nerve. (2015) 52:5–12. doi: 10.1002/mus.24632
4. Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B. Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun. (2014) 48–49:143–8. doi: 10.1016/j.jaut.2014.01.003
5. Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. (2010) 137:1017–33. doi: 10.1242/dev.038711
6. Yamakage M, Namiki A. Calcium channels–basic aspects of their structure, function and gene encoding; anesthetic action on the channels–a review. Can J Anaesth. (2002) 49:151–64. doi: 10.1007/BF03020488
7. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. (2005) 57:411–25. doi: 10.1124/pr.57.4.5
8. Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. (1996) 383:431–4. doi: 10.1038/383431a0
9. Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca(2)(+) channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. (2011) 13:7–21. doi: 10.1038/nrn3125
10. Wray D. The roles of intracellular regions in the activation of voltage-dependent potassium channels. Eur Biophys J. (2004) 33:194–200. doi: 10.1007/s00249-003-0363-2
11. Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. (1992) 9:271–84. doi: 10.1016/0896-6273(92)90166-B
12. Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. (1998) 280:69–77. doi: 10.1126/science.280.5360.69
13. Barber PA, Anderson NE, Vincent A. Morvan's syndrome associated with voltage-gated K+ channel antibodies. Neurology. (2000) 54:771–2. doi: 10.1212/WNL.54.3.771
14. Radja GK, Cavanna AE. Treatment of VGKC complex antibody-associated limbic encephalitis: a systematic review. J Neuropsychiatry Clin Neurosci. (2013) 25:264–71. doi: 10.1176/appi.neuropsych.13020022
15. Heuser JE, Salpeter SR. Organization of acetylcholine receptors in quick-frozen, deep-etched, and rotary-replicated Torpedo postsynaptic membrane. J Cell Biol. (1979) 82:150–73. doi: 10.1083/jcb.82.1.150
16. Burden SJ. SnapShot: neuromuscular junction. Cell. (2011) 144:826–826.e1. doi: 10.1016/j.cell.2011.02.037
17. Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, et al. Agrin acts via a MuSK receptor complex. Cell. (1996) 85:513–23. doi: 10.1016/S0092-8674(00)81252-0
18. York AL, Zheng JQ. Super-resolution microscopy reveals a nanoscale organization of acetylcholine receptors for trans-synaptic alignment at neuromuscular synapses. eNeuro. (2017) 4:ENEURO.0232-17.2017. doi: 10.1523/ENEURO.0232-17.2017
19. Bertrand D, Terry AV Jr. The wonderland of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. (2018) 151:214–25. doi: 10.1016/j.bcp.2017.12.008
20. Keramidas A, Moorhouse AJ, Schofield PR, Barry PH. Ligand-gated ion channels: mechanisms underlying ion selectivity. Prog Biophys Mol Biol. (2004) 86:161–204. doi: 10.1016/j.pbiomolbio.2003.09.002
21. Mihailescu S, Drucker-Colin R. Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch Med Res. (2000) 31:131–44. doi: 10.1016/S0188-4409(99)00087-9
22. Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, et al. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature. (1986) 321:406–11. doi: 10.1038/321406a0
23. Missias AC, Chu GC, Klocke BJ, Sanes JR, Merlie JP. Maturation of the acetylcholine receptor in skeletal muscle: regulation of the AChR gamma-to-epsilon switch. Dev Biol. (1996) 179:223–38. doi: 10.1006/dbio.1996.0253
24. Yumoto N, Wakatsuki S, Sehara-Fujisawa A. The acetylcholine receptor gamma-to-epsilon switch occurs in individual endplates. Biochem Biophys Res Commun. (2005) 331:1522–7. doi: 10.1016/j.bbrc.2005.04.081
25. Fischbach GD, Schuetze SM. A post-natal decrease in acetylcholine channel open time at rat end-plates. J Physiol. (1980) 303:125–37. doi: 10.1113/jphysiol.1980.sp013275
26. Gu Y, Hall ZW. Immunological evidence for a change in subunits of the acetylcholine receptor in developing and denervated rat muscle. Neuron. (1988) 1:117–25. doi: 10.1016/0896-6273(88)90195-X
27. Luo F, Dittrich M, Cho S, Stiles JR, Meriney SD. Transmitter release is evoked with low probability predominately by calcium flux through single channel openings at the frog neuromuscular junction. J Neurophysiol. (2015) 113:2480–9. doi: 10.1152/jn.00879.2014
28. Ram JL, Parti R. Regulation of calcium influx into buccal muscles of Aplysia by acetylcholine and serotonin. J Neurobiol. (1985) 16:57–68. doi: 10.1002/neu.480160106
29. Stanley EF. Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron. (1993) 11:1007–11. doi: 10.1016/0896-6273(93)90214-C
30. Kuffler SW, Yoshikami D. The number of transmitter molecules in a quantum: an estimate from iontophoretic application of acetylcholine at the neuromuscular synapse. J Physiol. (1975) 251:465–82. doi: 10.1113/jphysiol.1975.sp011103
31. Unwin N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: insights from Torpedo postsynaptic membranes. Q Rev Biophys. (2013) 46:283–322. doi: 10.1017/S0033583513000061
32. Chakrapani S, Auerbach A. A speed limit for conformational change of an allosteric membrane protein. Proc Natl Acad Sci USA. (2005) 102:87–92. doi: 10.1073/pnas.0406777102
33. Maconochie DJ, Fletcher GH, Steinbach JH. The conductance of the muscle nicotinic receptor channel changes rapidly upon gating. Biophys J. (1995) 68:483–90. doi: 10.1016/S0006-3495(95)80209-2
34. Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol. (2001) 64:393–429. doi: 10.1016/S0301-0082(00)00055-1
35. Trontelj JV, Mihelin M, Khuraibet A. Safety margin at single neuromuscular junctions. Muscle Nerve Suppl. (2002) 11:S21–7. doi: 10.1002/mus.10143
36. Paton WD, Waud DR. The margin of safety of neuromuscular transmission. J Physiol. (1967) 191:59–90. doi: 10.1113/jphysiol.1967.sp008237
37. Sanders DB, Guptill JT. Myasthenia gravis and Lambert-Eaton myasthenic syndrome. Continuum (Minneap Minn). (2014) 20:1413–25. doi: 10.1212/01.CON.0000455873.30438.9b
38. Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. (2011) 10:1098–107. doi: 10.1016/S1474-4422(11)70245-9
39. Eaton LM, Lambert EH. Electromyography and electric stimulation of nerves in diseases of motor unit; observations on myasthenic syndrome associated with malignant tumors. J Am Med Assoc. (1957) 163:1117–24. doi: 10.1001/jama.1957.02970480021005
40. O'neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. (1988) 111 (Pt 3):577–96. doi: 10.1093/brain/111.3.577
41. Wirtz PW, Nijnuis MG, Sotodeh M, Willems LN, Brahim JJ, Putter H, et al. The epidemiology of myasthenia gravis, Lambert-Eaton myasthenic syndrome and their associated tumours in the northern part of the province of South Holland. J Neurol. (2003) 250:698–701. doi: 10.1007/s00415-003-1063-7
42. Wirtz PW, Van Dijk JG, Van Doorn PA, Van Engelen BG, Van Der Kooi AJ, Kuks JB, et al. The epidemiology of the Lambert-Eaton myasthenic syndrome in the Netherlands. Neurology. (2004) 63:397–8. doi: 10.1212/01.WNL.0000130254.27019.14
43. Abenroth DC, Smith AG, Greenlee JE, Austin SD, Clardy SL. Lambert-Eaton myasthenic syndrome: epidemiology and therapeutic response in the national veterans affairs population. Muscle Nerve. (2017) 56:421–6. doi: 10.1002/mus.25520
44. Titulaer MJ, Wirtz PW, Kuks JB, Schelhaas HJ, Van Der Kooi AJ, Faber CG, et al. The Lambert-Eaton myasthenic syndrome 1988–2008: a clinical picture in 97 patients. J Neuroimmunol. (2008) 201–202:153–8. doi: 10.1016/j.jneuroim.2008.05.025
45. Hajjar M, Markowitz J, Darras BT, Kissel JT, Srinivasan J, Jones HR. Lambert-Eaton syndrome, an unrecognized treatable pediatric neuromuscular disorder: three patients and literature review. Pediatr Neurol. (2014) 50:11–7. doi: 10.1016/j.pediatrneurol.2013.08.009
46. Titulaer MJ, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: tumor versus nontumor forms. Ann N Y Acad Sci. (2008) 1132:129–34. doi: 10.1196/annals.1405.030
47. Titulaer MJ, Maddison P, Sont JK, Wirtz PW, Hilton-Jones D, Klooster R, et al. Clinical Dutch-English Lambert-Eaton Myasthenic syndrome (LEMS) tumor association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. (2011) 29:902–8. doi: 10.1200/JCO.2010.32.0440
48. Young JD, Leavitt JA. Lambert-Eaton Myasthenic syndrome: ocular signs and symptoms. J Neuroophthalmol. (2016) 36:20–2. doi: 10.1097/WNO.0000000000000258
49. Crone C, Christiansen I, Vissing J. Myopathic EMG findings and type II muscle fiber atrophy in patients with Lambert-Eaton myasthenic syndrome. Clin Neurophysiol. (2013) 124:1889–92. doi: 10.1016/j.clinph.2013.02.115
50. Smith AG, Wald J. Acute ventilatory failure in Lambert-Eaton myasthenic syndrome and its response to 3,4-diaminopyridine. Neurology. (1996) 46:1143–5. doi: 10.1212/WNL.46.4.1143
51. Wirtz PW, Smallegange TM, Wintzen AR, Verschuuren JJ. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. (2002) 104:359–63. doi: 10.1016/S0303-8467(02)00054-9
52. Pellkofer HL, Armbruster L, Linke R, Schumm F, Voltz R. Managing non-paraneoplastic Lambert-Eaton myasthenic syndrome: clinical characteristics in 25 German patients. J Neuroimmunol. (2009) 217:90–4. doi: 10.1016/j.jneuroim.2009.09.017
53. Lorenzoni PJ, Scola RH, Kay CS, Parolin SF, Werneck LC. Non-paraneoplastic Lambert-Eaton myasthenic syndrome: a brief review of 10 cases. Arq Neuropsiquiatr. (2010) 68:849–54. doi: 10.1590/S0004-282X2010000600004
54. Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci USA. (1983) 80:7636–40. doi: 10.1073/pnas.80.24.7636
55. Lennon VA, Kryzer TJ, Griesmann GE, O'suilleabhain PE, Windebank AJ, Woppmann A, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. (1995) 332:1467–74. doi: 10.1056/NEJM199506013322203
56. Takamori M, Takahashi M, Yasukawa Y, Iwasa K, Nemoto Y, Suenaga A, et al. Antibodies to recombinant synaptotagmin and calcium channel subtypes in Lambert-Eaton myasthenic syndrome. J Neurol Sci. (1995) 133:95–101. doi: 10.1016/0022-510X(95)00162-U
57. Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. (1997) 147:35–42. doi: 10.1016/S0022-510X(96)05303-8
58. Di Lorenzo R, Mente K, Li J, Shayya L, Rae-Grant A, Li Y, et al. Low specificity of voltage-gated calcium channel antibodies in Lambert-Eaton myasthenic syndrome: a call for caution. J Neurol. (2018) 265:2114–9. doi: 10.1007/s00415-018-8959-8
59. Zalewski NL, Lennon VA, Lachance DH, Klein CJ, Pittock SJ, Mckeon A. P/Q- and N-type calcium-channel antibodies: Oncological, neurological, and serological accompaniments. Muscle Nerve. (2016) 54:220–7. doi: 10.1002/mus.25027
60. Johnston I, Lang B, Leys K, Newsom-Davis J. Heterogeneity of calcium channel autoantibodies detected using a small-cell lung cancer line derived from a Lambert-Eaton myasthenic syndrome patient. Neurology. (1994) 44:334–8. doi: 10.1212/WNL.44.2.334
61. Oguro-Okano M, Griesmann GE, Wieben ED, Slaymaker SJ, Snutch TP, Lennon VA. Molecular diversity of neuronal-type calcium channels identified in small cell lung carcinoma. Mayo Clin Proc. (1992) 67:1150–9. doi: 10.1016/S0025-6196(12)61144-6
62. Graus F, Vincent A, Pozo-Rosich P, Sabater L, Saiz A, Lang B, et al. Anti-glial nuclear antibody: marker of lung cancer-related paraneoplastic neurological syndromes. J Neuroimmunol. (2005) 165:166–71. doi: 10.1016/j.jneuroim.2005.03.020
63. Berger B, Dersch R, Ruthardt E, Rasiah C, Rauer S, Stich O. Prevalence of anti-SOX1 reactivity in various neurological disorders. J Neurol Sci. (2016) 369:342–6. doi: 10.1016/j.jns.2016.09.002
64. Lipka AF, Verschuuren JJ, Titulaer MJ. SOX1 antibodies in Lambert-Eaton myasthenic syndrome and screening for small cell lung carcinoma. Ann N Y Acad Sci. (2012) 1275:70–7. doi: 10.1111/j.1749-6632.2012.06772.x
65. Sabater L, Titulaer M, Saiz A, Verschuuren J, Gure AO, Graus F. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. (2008) 70:924–8. doi: 10.1212/01.wnl.0000281663.81079.24
66. Titulaer MJ, Klooster R, Potman M, Sabater L, Graus F, Hegeman IM, et al. SOX antibodies in small-cell lung cancer and Lambert-Eaton myasthenic syndrome: frequency and relation with survival. J Clin Oncol. (2009) 27:4260–7. doi: 10.1200/JCO.2008.20.6169
67. Nakao YK, Motomura M, Fukudome T, Fukuda T, Shiraishi H, Yoshimura T, et al. Seronegative Lambert-Eaton myasthenic syndrome: study of 110 Japanese patients. Neurology. (2002) 59:1773–5. doi: 10.1212/01.WNL.0000037485.56217.5F
68. Evoli A, Liguori R, Romani A, Mantegazza R, Di Muzio A, Giometto B, et al. Italian recommendations for Lambert-Eaton myasthenic syndrome (LEMS) management. Neurol Sci. (2014) 35:515–20. doi: 10.1007/s10072-014-1637-4
69. Medicine AQACAAOE. Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: summary statement. Muscle Nerve. (2001) 24:1236–8. doi: 10.1002/mus.1139
70. Oh SJ, Kurokawa K, Claussen GC, Ryan HF Jr. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve. (2005) 32:515–20. doi: 10.1002/mus.20389
71. Titulaer MJ, Wirtz PW, Willems LN, Van Kralingen KW, Smitt PA, Verschuuren JJ. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. (2008) 26:4276–81. doi: 10.1200/JCO.2008.17.5133
72. Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. (2011) 18:19–e13. doi: 10.1111/j.1468-1331.2010.03220.x
73. Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. (2009) 40:795–800. doi: 10.1002/mus.21422
74. Lindquist S, Stangel M. Update on treatment options for Lambert-Eaton myasthenic syndrome: focus on use of amifampridine. Neuropsychiatr Dis Treat. (2011) 7:341–9. doi: 10.2147/NDT.S10464
75. Nicolle MW. Myasthenia Gravis and Lambert-Eaton Myasthenic syndrome. Continuum (Minneap Minn). (2016) 22:1978–2005. doi: 10.1212/CON.0000000000000415
76. Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. (2011):CD003279. doi: 10.1002/14651858.CD003279.pub3
77. Flet L, Polard E, Guillard O, Leray E, Allain H, Javaudin L, et al. 3,4-diaminopyridine safety in clinical practice: an observational, retrospective cohort study. J Neurol. (2010) 257:937–46. doi: 10.1007/s00415-009-5442-6
78. Verschuuren JJ, Wirtz PW, Titulaer MJ, Willems LN, Van Gerven J. Available treatment options for the management of Lambert-Eaton myasthenic syndrome. Expert Opin Pharmacother. (2006) 7:1323–36. doi: 10.1517/14656566.7.10.1323
79. Wirtz PW, Verschuuren JJ, Van Dijk JG, De Kam ML, Schoemaker RC, Van Hasselt JG, et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther. (2009) 86:44–8. doi: 10.1038/clpt.2009.35
80. Newsom-Davis J, Murray NM. Plasma exchange and immunosuppressive drug treatment in the Lambert-Eaton myasthenic syndrome. Neurology. (1984) 34:480–5. doi: 10.1212/WNL.34.4.480
81. Patwa HS, Chaudhry V, Katzberg H, Rae-Grant AD, So YT. Evidence-based guideline: intravenous immunoglobulin in the treatment of neuromuscular disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. (2012) 78:1009–15. doi: 10.1212/WNL.0b013e31824de293
82. Bird SJ. Clinical and electrophysiologic improvement in Lambert-Eaton syndrome with intravenous immunoglobulin therapy. Neurology. (1992) 42:1422–3. doi: 10.1212/WNL.42.7.1422
83. Bain PG, Motomura M, Newsom-Davis J, Misbah SA, Chapel HM, Lee ML, et al. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. (1996) 47:678–83. doi: 10.1212/WNL.47.3.678
84. Okada A, Koike H, Nakamura T, Motomura M, Sobue G. Efficacy of intravenous immunoglobulin for treatment of Lambert-Eaton myasthenic syndrome without anti-presynaptic P/Q-type voltage-gated calcium channel antibodies: a case report. Neuromuscul Disord. (2015) 25:70–2. doi: 10.1016/j.nmd.2014.08.006
85. Seneviratne U, De Silva R. Lambert-Eaton myasthenic syndrome. Postgrad Med J. (1999) 75:516–20. doi: 10.1136/pgmj.75.887.516
86. Motomura M, Hamasaki S, Nakane S, Fukuda T, Nakao YK. Apheresis treatment in Lambert-Eaton myasthenic syndrome. Ther Apher. (2000) 4:287–90. doi: 10.1046/j.1526-0968.2000.004004287.x
87. Isaacs H. A Syndrome of continuous muscle-fibre activity. J Neurol Neurosurg Psychiatry. (1961) 24:319–25. doi: 10.1136/jnnp.24.4.319
88. Sawlani K, Katirji B. Peripheral nerve hyperexcitability syndromes. Continuum (Minneap Minn). (2017) 23:1437–50. doi: 10.1212/CON.0000000000000520
89. Hart IK, Maddison P, Newsom-Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain. (2002) 125:1887–95. doi: 10.1093/brain/awf178
90. Joubert B, Honnorat J. Autoimmune channelopathies in paraneoplastic neurological syndromes. Biochim Biophys Acta. (2015) 1848:2665–76. doi: 10.1016/j.bbamem.2015.04.003
91. Gonzalez Primomo SN, Blas L, Bertotti AC, Ameri C. Urinary manifestations in Isaacs's syndrome. Our experience in 8 cases. Neurourol Urodyn. (2018) 37:496–500. doi: 10.1002/nau.23336
92. Tahmoush AJ, Alonso RJ, Tahmoush GP, Heiman-Patterson TD. Cramp-fasciculation syndrome: a treatable hyperexcitable peripheral nerve disorder. Neurology. (1991) 41:1021–4. doi: 10.1212/WNL.41.7.1021
93. Hart IK, Waters C, Vincent A, Newland C, Beeson D, Pongs O, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol. (1997) 41:238–46. doi: 10.1002/ana.410410215
94. Maddison P. Neuromyotonia. Clin Neurophysiol. (2006) 117:2118–27. doi: 10.1016/j.clinph.2006.03.008
95. Newsom-Davis J, Mills KR. Immunological associations of acquired neuromyotonia (Isaacs' syndrome). Report of five cases and literature review. Brain. (1993) 116 (Pt 2):453–69. doi: 10.1093/brain/116.2.453
96. Wallis WE, Van Poznak A, Plum F. Generalized muscular stiffness, fasciculations, and myokymia of peripheral nerve origin. Arch Neurol. (1970) 22:430–9. doi: 10.1001/archneur.1970.00480230048005
97. Zisfein J, Sivak M, Aron AM, Bender AN. Isaacs' syndrome with muscle hypertrophy reversed by phenytoin therapy. Arch Neurol. (1983) 40:241–2. doi: 10.1001/archneur.1983.04050040071012
98. Arimura K, Sonoda Y, Watanabe O, Nagado T, Kurono A, Tomimitsu H, et al. Isaacs' syndrome as a potassium channelopathy of the nerve. Muscle Nerve Suppl. (2002) 11:S55–58. doi: 10.1002/mus.10148
99. Vernino S, Lennon VA. Ion channel and striational antibodies define a continuum of autoimmune neuromuscular hyperexcitability. Muscle Nerve. (2002) 26:702–7. doi: 10.1002/mus.10266
100. Van Sonderen A, Schreurs MW, De Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology. (2016) 86:1692–9. doi: 10.1212/WNL.0000000000002637
101. Xiao F. Neuromyotonia as an unusual neurological complication of primary Sjogren's syndrome: case report and literature review. Clin Rheumatol. (2017) 36:481–4. doi: 10.1007/s10067-016-3499-z
102. Arimura K, Arimura Y, Ng A, Uehara A, Nakae M, Osame M, et al. The origin of spontaneous discharges in acquired neuromyotonia. A Macro EMG study Clin Neurophysiol. (2005) 116:1835–9. doi: 10.1016/j.clinph.2005.03.023
103. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. (2010) 9:776–85. doi: 10.1016/S1474-4422(10)70137-X
104. Irani SR, Vincent A. Voltage-gated potassium channel-complex autoimmunity and associated clinical syndromes. Handb Clin Neurol. (2016) 133:185–97. doi: 10.1016/B978-0-444-63432-0.00011-6
105. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain. (2010) 133:2734–48. doi: 10.1093/brain/awq213
106. Song J, Jing S, Quan C, Lu J, Qiao X, Qiao K, et al. Isaacs syndrome with CASPR2 antibody: a series of three cases. J Clin Neurosci. (2017) 41:63–6. doi: 10.1016/j.jocn.2017.02.063
107. Klein CJ, Lennon VA, Aston PA, Mckeon A, O'toole O, Quek A, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol. (2013) 70:229–34. doi: 10.1001/jamaneurol.2013.592
108. Hart IK. Acquired neuromyotonia: a new autoantibody-mediated neuronal potassium channelopathy. Am J Med Sci. (2000) 319:209–16. doi: 10.1016/S0002-9629(15)40732-3
109. Partanen VS, Soininen H, Saksa M, Riekkinen P. Electromyographic and nerve conduction findings in a patient with neuromyotonia, normocalcemic tetany and small-cell lung cancer. Acta Neurol Scand. (1980) 61:216–26. doi: 10.1111/j.1600-0404.1980.tb01486.x
110. Paterson RW, Zandi MS, Armstrong R, Vincent A, Schott JM. Clinical relevance of positive voltage-gated potassium channel (VGKC)-complex antibodies: experience from a tertiary referral centre. J Neurol Neurosurg Psychiatry. (2014) 85:625–30. doi: 10.1136/jnnp-2013-305218
111. Gutmann L, Gutmann L. Myokymia and neuromyotonia 2004. J Neurol. (2004) 251:138–42. doi: 10.1007/s00415-004-0331-5
112. Lukas RV, Rezania K, Malec M, Salgia R. Teaching Video NeuroImages: myokymia and nerve hyperexcitability as components of Morvan syndrome due to malignant thymoma. Neurology. (2013) 80:e55. doi: 10.1212/WNL.0b013e31827f0fa1
113. Liebenthal JA, Rezania K, Nicholas MK, Lukas RV. Paraneoplastic nerve hyperexcitability. Neurol Res. (2015) 37:553–9. doi: 10.1179/1743132815Y.0000000006
114. Maddison P, Mills KR, Newsom-Davis J. Clinical electrophysiological characterization of the acquired neuromyotonia phenotype of autoimmune peripheral nerve hyperexcitability. Muscle Nerve. (2006) 33:801–8. doi: 10.1002/mus.20536
115. Fleisher J, Richie M, Price R, Scherer S, Dalmau J, Lancaster E. Acquired neuromyotonia heralding recurrent thymoma in myasthenia gravis. JAMA Neurol. (2013) 70:1311–4. doi: 10.1001/jamaneurol.2013.2863
116. London F, Bancilhon JB, Tard C, Vermersch P, Hadhoum N. Isaacs' syndrome and Hodgkin lymphoma: a rare association. Acta Neurol Belg. (2017) 117:417–9. doi: 10.1007/s13760-016-0688-0
117. Jamieson PW, Katirji MB. Idiopathic generalized myokymia. Muscle Nerve. (1994) 17:42–51. doi: 10.1002/mus.880170106
118. Pan HL, Eisenach JC, Chen SR. Gabapentin suppresses ectopic nerve discharges and reverses allodynia in neuropathic rats. J Pharmacol Exp Ther. (1999) 288:1026–30.
119. Tremont-Lukats IW, Megeff C, Backonja MM. Anticonvulsants for neuropathic pain syndromes: mechanisms of action and place in therapy. Drugs. (2000) 60:1029–52. doi: 10.2165/00003495-200060050-00005
120. Dhand UK. Isaacs' syndrome: clinical and electrophysiological response to gabapentin. Muscle Nerve. (2006) 34:646–50. doi: 10.1002/mus.20591
121. Hayat GR, Kulkantrakorn K, Campbell WW, Giuliani MJ. Neuromyotonia: autoimmune pathogenesis and response to immune modulating therapy. J Neurol Sci. (2000) 181:38–43. doi: 10.1016/S0022-510X(00)00407-X
122. Jaben EA, Winters JL. Plasma exchange as a therapeutic option in patients with neurologic symptoms due to antibodies to voltage-gated potassium channels: a report of five cases and review of the literature. J Clin Apher. (2012) 27:267–73. doi: 10.1002/jca.21233
123. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. (2015) 14:1023–36. doi: 10.1016/S1474-4422(15)00145-3
124. Heldal AT, Owe JF, Gilhus NE, Romi F. Seropositive myasthenia gravis: a nationwide epidemiologic study. Neurology. (2009) 73:150–1. doi: 10.1212/WNL.0b013e3181ad53c2
125. Montomoli C, Citterio A, Piccolo G, Cioccale R, Ferretti VV, Fratti C, et al. Epidemiology and geographical variation of myasthenia gravis in the province of Pavia, Italy. Neuroepidemiology. (2012) 38:100–5. doi: 10.1159/000336002
126. Lavrnic D, Basta I, Rakocevic-Stojanovic V, Stevic Z, Peric S, Nikolic A, et al. Epidemiological study of adult-onset myasthenia gravis in the area of Belgrade (Serbia) in the period 1979–2008. Neuroepidemiology. (2013) 40:190–4. doi: 10.1159/000342777
127. Andersen JB, Engeland A, Owe JF, Gilhus NE. Myasthenia gravis requiring pyridostigmine treatment in a national population cohort. Eur J Neurol. (2010) 17:1445–50. doi: 10.1111/j.1468-1331.2010.03089x
128. Niks EH, Kuks JB, Verschuuren JJ. Epidemiology of myasthenia gravis with anti-muscle specific kinase antibodies in The Netherlands. J Neurol Neurosurg Psychiatry. (2007) 78:417–8. doi: 10.1136/jnnp.2006.102517
129. Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. (2011) 44:36–40. doi: 10.1002/mus.22006
130. Corda D, Deiana GA, Mulargia M, Pirastru MI, Serra M, Piluzza MG, et al. Familial autoimmune MuSK positive myasthenia gravis. J Neurol. (2011) 258:1559–60. doi: 10.1007/s00415-011-5964-6
131. Feng HY, Liu WB, Luo CM, Yang LX, Fang W, Qiu L, et al. A retrospective review of 15 patients with familial myasthenia gravis over a period of 25 years. Neurol Sci. (2012) 33:771–7. doi: 10.1007/s10072-011-0818-7
132. Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. (2009) 8:475–90. doi: 10.1016/S1474-4422(09)70063-8
133. Rodolico C, Toscano A, Autunno M, Messina S, Nicolosi C, Aguennouz M, et al. Limb-girdle myasthenia: clinical, electrophysiological and morphological features in familial and autoimmune cases. Neuromuscul Disord. (2002) 12:964–9. doi: 10.1016/S0960-8966(02)00137-2
134. Chiu HC, Vincent A, Newsom-Davis J, Hsieh KH, Hung T. Myasthenia gravis: population differences in disease expression and acetylcholine receptor antibody titers between Chinese and Caucasians. Neurology. (1987) 37:1854–7. doi: 10.1212/WNL.37.12.1854
135. Zhang X, Yang M, Xu J, Zhang M, Lang B, Wang W, et al. Clinical and serological study of myasthenia gravis in HuBei Province, China. J Neurol Neurosurg Psychiatry. (2007) 78:386–90. doi: 10.1136/jnnp.2006.100545
136. Andrews PI. Autoimmune myasthenia gravis in childhood. Semin Neurol. (2004) 24:101–10. doi: 10.1055/s-2004-829591
137. Jaretzki A II, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. (2000) 55:16–23. doi: 10.1212/WNL.55.1.16
138. Huang K, Luo YB, Yang H, Yang XS, Li J. Myasthenia gravis accompanied by Graves' disease, thyrotoxic hypokalemic periodic paralysis and thymic hyperplasia. Neurol India. (2016) 64:783–5. doi: 10.4103/0028-3886.185401
139. Marx A, Porubsky S, Belharazem D, Saruhan-Direskeneli G, Schalke B, Strobel P, et al. Thymoma related myasthenia gravis in humans and potential animal models. Exp Neurol. (2015) 270:55–65. doi: 10.1016/j.expneurol.2015.02.010
140. Willcox N, Leite MI, Kadota Y, Jones M, Meager A, Subrahmanyam P, et al. Autoimmunizing mechanisms in thymoma and thymus. Ann N Y Acad Sci. (2008) 1132:163–73. doi: 10.1196/annals.1405.021
141. Agius MA, Richman DP, Fairclough RH, Aarli J, Gilhus NE, Romi F. Three forms of immune myasthenia. Ann N Y Acad Sci. (2003) 998:453–6. doi: 10.1196/annals.1254.059
142. Mygland A, Kuwajima G, Mikoshiba K, Tysnes OB, Aarli JA, Gilhus NE. Thymomas express epitopes shared by the ryanodine receptor. J Neuroimmunol. (1995) 62:79–83. doi: 10.1016/0165-5728(95)00106-C
143. Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis–autoantibody characteristics and their implications for therapy. Nat Rev Neurol. (2016) 12:259–68. doi: 10.1038/nrneurol.2016.44
144. Verschuuren JJ, Huijbers MG, Plomp JJ, Niks EH, Molenaar PC, Martinez-Martinez P, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun Rev. (2013) 12:918–23. doi: 10.1016/j.autrev.2013.03.001
145. Kordas G, Lagoumintzis G, Sideris S, Poulas K, Tzartos SJ. Direct proof of the in vivo pathogenic role of the AChR autoantibodies from myasthenia gravis patients. PLoS ONE. (2014) 9:e108327. doi: 10.1371/journal.pone.0108327
146. Messeant J, Dobbertin A, Girard E, Delers P, Manuel M, Mangione F, et al. MuSK frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance. J Neurosci. (2015) 35:4926–41. doi: 10.1523/JNEUROSCI.3381-14.2015
147. Plomp JJ, Huijbers MG, Van Der Maarel SM, Verschuuren JJ. Pathogenic IgG4 subclass autoantibodies in MuSK myasthenia gravis. Ann N Y Acad Sci. (2012) 1275:114–22. doi: 10.1111/j.1749-6632.2012.06808.x
148. Zhang B, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, Lewis RA, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. (2012) 69:445–51. doi: 10.1001/archneurol.2011.2393
149. Romi F, Hong Y, Gilhus NE. Pathophysiology and immunological profile of myasthenia gravis and its subgroups. Curr Opin Immunol. (2017) 49:9–13. doi: 10.1016/j.coi.2017.07.006
150. Barik A, Lu Y, Sathyamurthy A, Bowman A, Shen C, Li L, et al. LRP4 is critical for neuromuscular junction maintenance. J Neurosci. (2014) 34:13892–905. doi: 10.1523/JNEUROSCI.1733-14.2014
151. Gasperi C, Melms A, Schoser B, Zhang Y, Meltoranta J, Risson V, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology. (2014) 82:1976–83. doi: 10.1212/WNL.0000000000000478
152. Zhang B, Shen C, Bealmear B, Ragheb S, Xiong WC, Lewis RA, et al. Autoantibodies to agrin in myasthenia gravis patients. PLoS ONE. (2014) 9:e91816. doi: 10.1371/journal.pone.0091816
153. Witzemann V, Chevessier F, Pacifici PG, Yampolsky P. The neuromuscular junction: selective remodeling of synaptic regulators at the nerve/muscle interface. Mech Dev. (2013) 130:402–11. doi: 10.1016/j.mod.2012.09.004
154. Kerty E, Elsais A, Argov Z, Evoli A, Gilhus NE. EFNS/ENS Guidelines for the treatment of ocular myasthenia. Eur J Neurol. (2014) 21:687–93. doi: 10.1111/ene.12359
155. Massa R, Greco G, Testi M, Rastelli E, Terracciano C, Frezza E, et al. Thymomatous myasthenia gravis: novel association with HLA DQB1*05:01 and strengthened evidence of high clinical and serological severity. J Neurol. (2019) 266:982–9. doi: 10.1007/s00415-019-09225-z
156. Vincent A, Newsom-Davis J. Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry. (1985) 48:1246–52. doi: 10.1136/jnnp.48.12.1246
157. Berrih S, Morel E, Gaud C, Raimond F, Le Brigand H, Bach JF. Anti-AChR antibodies, thymic histology, and T cell subsets in myasthenia gravis. Neurology. (1984) 34:66–71. doi: 10.1212/WNL.34.1.66
158. Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, Batocchi AP, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. (2003) 126:2304–11. doi: 10.1093/brain/awg223
159. Leite MI, Strobel P, Jones M, Micklem K, Moritz R, Gold R, et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol. (2005) 57:444–8. doi: 10.1002/ana.20386
160. Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. (2012) 259:427–35. doi: 10.1007/s00415-011-6194-7
161. Cortes-Vicente E, Gallardo E, Martinez MA, Diaz-Manera J, Querol L, Rojas-Garcia R, et al. Clinical characteristics of patients with double-seronegative myasthenia gravis and antibodies to cortactin. JAMA Neurol. (2016) 73:1099–104. doi: 10.1001/jamaneurol.2016.2032
162. Evoli A, Antonini G, Antozzi C, Dimuzio A, Habetswallner F, Iani C, et al. Italian recommendations for the diagnosis and treatment of myasthenia gravis. Neurol Sci. (2019). doi: 10.1007/s10072-019-03746-1
163. Oh SJ, Cho HK. Edrophonium responsiveness not necessarily diagnostic of myasthenia gravis. Muscle Nerve. (1990) 13:187–91. doi: 10.1002/mus.880130302
164. Pasnoor M, Dimachkie MM, Farmakidis C, Barohn RJ. Diagnosis of myasthenia gravis. Neurol Clin. (2018) 36:261–74. doi: 10.1016/j.ncl.2018.01.010
165. Trontelj JV. Muscle fiber conduction velocity changes with length. Muscle Nerve. (1993) 16:506–12. doi: 10.1002/mus.880160512
166. Rouseev R, Ashby P, Basinski A, Sharpe JA. Single fiber EMG in the frontalis muscle in ocular myasthenia: specificity and sensitivity. Muscle Nerve. (1992) 15:399–403. doi: 10.1002/mus.880150322
167. Sarrigiannis PG, Kennett RP, Read S, Farrugia ME. Single-fiber EMG with a concentric needle electrode: validation in myasthenia gravis. Muscle Nerve. (2006) 33:61–5. doi: 10.1002/mus.20435
168. Khuraibet AJ, Rousseff RT, Behbehani R, Al-Shubaili AF, Khan RA. Single-fiber electromyography of masseter muscle in myasthenia gravis. Muscle Nerve. (2008) 37:522–5. doi: 10.1002/mus.20921
169. Padua L, Stalberg E, Lomonaco M, Evoli A, Batocchi A, Tonali P. SFEMG in ocular myasthenia gravis diagnosis. Clin Neurophysiol. (2000) 111:1203–7. doi: 10.1016/S1388-2457(00)00307-2
170. Sanders DB. Clinical impact of single-fiber electromyography. Muscle Nerve Suppl. (2002) 11:S15–20. doi: 10.1002/mus.10141
171. Shi L, Liu HF, Zhang M, Guo YP, Song B, Song CD, et al. Determination of the normative values of the masseter muscle by single-fiber electromyography in myasthenia gravis patients. Int J Clin Exp Med. (2015) 8:19424–9.
172. Konishi T, Nishitani H, Matsubara F, Ohta M. Myasthenia gravis: relation between jitter in single-fiber EMG and antibody to acetylcholine receptor. Neurology. (1981) 31:386–92. doi: 10.1212/WNL.31.4_Part_2.386
173. Baruca M, Leonardis L, Podnar S, Hojs-Fabjan T, Grad A, Jerin A, et al. Single fiber EMG as a prognostic tool in myasthenia gravis. Muscle Nerve. (2016) 54:1034–40. doi: 10.1002/mus.25174
174. Priola AM, Priola SM. Imaging of thymus in myasthenia gravis: from thymic hyperplasia to thymic tumor. Clin Radiol. (2014) 69:e230–45. doi: 10.1016/j.crad.2014.01.005
175. Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol. (2003) 60:243–8. doi: 10.1001/archneur.60.2.243
176. Kupersmith MJ. Ocular myasthenia gravis: treatment successes and failures in patients with long-term follow-up. J Neurol. (2009) 256:1314–20. doi: 10.1007/s00415-009-5120-8
177. Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology. (1998) 50:1778–83. doi: 10.1212/WNL.50.6.1778
178. Sanders DB, Hart IK, Mantegazza R, Shukla SS, Siddiqi ZA, De Baets MH, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. (2008) 71:400–6. doi: 10.1212/01.wnl.0000312374.95186.cc
179. Stieglbauer K, Topakian R, Schaffer V, Aichner FT. Rituximab for myasthenia gravis: three case reports and review of the literature. J Neurol Sci. (2009) 280:120–2. doi: 10.1016/j.jns.2009.02.357
180. Yoshikawa H, Kiuchi T, Saida T, Takamori M. Randomised, double-blind, placebo-controlled study of tacrolimus in myasthenia gravis. J Neurol Neurosurg Psychiatry. (2011) 82:970–7. doi: 10.1136/jnnp-2011-300148
181. Muppidi S, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. (2019). doi: 10.1002/mus.26447
182. Wang L, Xi J, Zhang S, Wu H, Zhou L, Lu J, et al. Effectiveness and safety of tacrolimus therapy for myasthenia gravis: a single arm meta-analysis. J Clin Neurosci. (2019) 63:160–7. doi: 10.1016/j.jocn.2019.02.004
183. Inc AP. Soliris (eculizumab): US Prescribing Information. (2015). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125166s422lbl.pdf (accessed November 10, 2017).
184. Sas AE. Soliris (eculizumab): Summary of Product Characteristics. (2017). Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000791/WC500054208.pdf (accessed November 11, 2017).
185. Welfare JMOHLA. Soliris (eculizumab): Japanese Prescribing Information. (2017). Available online at: http://wwwhourei.mhlw.go.jp/hourei/doc/tsuchi/T171226I0050.pdf (accessed January 10, 2018).
186. Wang L, Huan X, Xi JY, Wu H, Zhou L, Lu JH, et al. Immunosuppressive and monoclonal antibody treatment for myasthenia gravis: a network meta-analysis. CNS Neurosci Ther. (2019) 25:647–58. doi: 10.1111/cns.13110
187. Huijbers MG, Plomp JJ, Van Es IE, Fillie-Grijpma YE, Majidi SK, Ulrichts P, et al. Efgartigimod improves muscle weakness in a mouse model for muscle-specific kinase myasthenia gravis. Exp Neurol. (2019) 317:133–43. doi: 10.1016/j.expneurol.2019.03.001
188. Otsuka R, Ueda K, Tanaka T, Murakami J, Hayashi M, Hamano K. Who will benefit from thymectomy for myasthenia gravis? Is there any role for this procedure in elderly patients? Ann Transl Med. (2019) 7:4. doi: 10.21037/atm.2018.11.66
189. Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, Marx A, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. (2016) 375:511–22. doi: 10.1056/NEJMoa1602489
190. Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, Marx A, et al. Long-term effect of thymectomy plus prednisone versus prednisone alone in patients with non-thymomatous myasthenia gravis: 2-year extension of the MGTX randomised trial. Lancet Neurol. (2019) 18:259–68. doi: 10.1016/S1474-4422(18)30392-2
191. Liew WK, Kang PB. Update on juvenile myasthenia gravis. Curr Opin Pediatr. (2013) 25:694–700. doi: 10.1097/MOP.0b013e328365ad16
192. Mandawat A, Kaminski HJ, Cutter G, Katirji B, Alshekhlee A. Comparative analysis of therapeutic options used for myasthenia gravis. Ann Neurol. (2010) 68:797–805. doi: 10.1002/ana.22139
193. Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. (2011) 76:2017–23. doi: 10.1212/WNL.0b013e31821e5505
194. Hulsbrink R, Hashemolhosseini S. Lambert-Eaton myasthenic syndrome–diagnosis, pathogenesis and therapy. Clin Neurophysiol. (2014) 125:2328–36. doi: 10.1016/j.clinph.2014.06.031
195. Auger RG. AAEM minimonograph #44: diseases associated with excess motor unit activity. Muscle Nerve. (1994) 17:1250–63. doi: 10.1002/mus.880171103
Keywords: neuromuscular junction (NMJ), channelopathies, Lambert-Eaton syndrome (LEMS), Isaacs' syndrome, myasthenia gravis (MG)
Citation: Huang K, Luo Y-B and Yang H (2019) Autoimmune Channelopathies at Neuromuscular Junction. Front. Neurol. 10:516. doi: 10.3389/fneur.2019.00516
Received: 31 January 2019; Accepted: 30 April 2019;
Published: 17 May 2019.
Edited by:
Rosanna Cardani, Policlinico San Donato (IRCCS), ItalyReviewed by:
Carmelo Rodolico, University of Messina, ItalyLorenzo Maggi, Neurological Institute Foundation Carlo Besta, Italy
Copyright © 2019 Huang, Luo and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huan Yang, eWFuZ2g2OUAxMjYuY29t