 Muhammad Wasim1,2†
Muhammad Wasim1,2† Haq Nawaz Khan1,2†
Haq Nawaz Khan1,2† Hina Ayesha3
Hina Ayesha3 Susanna M. I. Goorden4Frederic M. Vaz4
Susanna M. I. Goorden4Frederic M. Vaz4 Clara D. M. van Karnebeek5*
Clara D. M. van Karnebeek5* Fazli Rabbi Awan1,2*
Fazli Rabbi Awan1,2*- 1Health Biotechnology Division, National Institute for Biotechnology and Genetic Engineering (NIBGE), Faisalabad, Pakistan
- 2Pakistan Institute of Engineering and Applied Sciences, Islamabad, Pakistan
- 3Department of Pediatrics, DHQ and Allied Hospitals, Faisalabad Medical University (FMU/PMC), Faisalabad, Pakistan
- 4Laboratory Genetic Metabolic Diseases, Department of Clinical Chemistry, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, Netherlands
- 5Departments of Pediatrics and Clinical Genetics, Emma Children's Hospital, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, Netherlands
Inborn errors of metabolism (IEMs) are rare group of genetic disorders comprising of more than 1,000 different types. Around 200 of IEMs are potentially treatable through diet, pharmacological and other therapies, if diagnosed earlier in life. IEMs can be diagnosed early through newborn screening (NBS) programs, which are in place in most of the developed countries. However, establishing a NBS in a developing country is a challenging task due to scarcity of disease related data, large population size, poor economy, and burden of other common disorders. Since, not enough data is available for the prevalence of IEMs in Pakistan; therefore, in this study, we set out to find the prevalence of various treatable IEMs in a cohort of intellectually disabled patients suspected for IEMs, which will help us to initiate a NBS program for the most frequent IEMs in Pakistan. Therefore, a total of 429 intellectually disabled (IQ < 70) patient samples were collected from Pakistan. A subset of 113 patient samples was selected based on the clinical information for the detailed biochemical screening. Advance analytical techniques like, Amino Acid Analyzer, GC-MS, UHPLC-MS, and MS/MS were used to screen for different treatable IEMs like aminoacidopathies, fatty acid β-oxidation disorders and mucopolysaccharidoses (MPS) etc. A total of 14 patients were diagnosed with an IEM i.e., 9 with homocystinuria, 2 with MPS, 2 with Guanidinoacetate methyltransferase (GAMT) deficiency and 1 with sitosterolemia. These IEMs are found frequent in the collected patient samples from Pakistan. Thus, present study can help to take an initiative step to start a NBS program in Pakistan, especially for the homocystinuria having highest incidence among aminoacidopathies in the studied patients, and which is amenable to treatment. This endeavor will pave the way for a healthier life of affected patients and will lessen the burden on their families and society.
Introduction
Inborn errors of metabolism (IEMs) are phenotypically and genetically heterogeneous disorders which are caused by the deficiency of a protein (most often an enzyme or a transporter) that results in the accumulation of intermediary metabolites which cannot be processed further. Although IEMs are rare when taken individually, but collectively they form a significant group of disorders, which lead to morbidity and mortality of the affected patients (1–3). Overall, prevalence of IEMs is estimated to be 1 in 500 newborns (4). Up till now more than 1,000 IEMs have been reported and about 200 of such disorders are potentially treatable when diagnosed before the appearance of clinical symptoms (5–7). Affected individuals usually appear normal at the time of birth, but symptoms (e.g., poor feeding, seizures, lethargy etc.) may develop within hours to weeks depending upon the disease severity. However, if diagnosed earlier, it allows treatment at the pre-symptomatic stage, thus preventing the subsequent development of intellectual disability. Diagnosis of these disorders can be performed through newborn screening (NBS) programs, which have been established in many developed countries (7–9).
In Pakistan, the prevalence of IEMs is expected to be high due to the high rate of consanguinity (~70% in Pakistan), which plays a significant role in the inherited disorders due to the autosomal recessive inheritance pattern (10). In Pakistan, the common practice of marriages takes place within the closely related communities, tribes, castes and in the same ethnic groups. Thus, the risk of any inherited disease in the offspring of closely related parents is reasonably high. Moreover, research on IEMs is neglected in the developing countries like Pakistan due to the burden of infectious and other common metabolic disorders.
Most of the developed countries have NBS programs in place which can screen for different inherited disorders at the time of birth or soon after birth (11–16). However, sadly, there is still no local or national level NBS program for any of the IEM in Pakistan—a country with population of more than 220 million. Indeed, there are many challenges to setup a NBS program for IEMs in the developing countries (17, 18). There are several pros and cons to initiate a NBS program in the developing countries as summarized in Table 1.

Table 1. Pros and Cons to initiate a NBS program in the developing countries.
Recently, in the Hong Kong, it has been advocated that NBS program is essential for the screening of newborns for IEMs, thus setting a reference for policymakers to establish a government funded NBS program to reduce the burden of these disorders (19). Likewise, several studies in China showed substantial occurrence of IEMs with an estimated prevalence of 1 in 3,795 newborns (20). Recently, 364,545 newborns were screened for IEMs in Quanzhou region of China, which showed 130 different IEMs with an incidence of 1 in 2,804 (21). For detailed characterization of various IEMs at the biochemical and genetic levels, tandem mass spectrometry and next generation sequencing was effectively used in China (22, 23). Interestingly, Sri Lanka is the only developing country in Asia which provides free healthcare services through NBS to all its citizens (24). Recently, efforts for NBS for some of the IEMs have also been started in Bangladesh with the help of international collaborators (25). Conversely, India which has the second largest population in the world has the weakest NBS program which is still at an infancy stage (26). There are few reports from India on the screening of IEMs; which showed congenital hypothyroidism as the most frequent disorder (27–29). However, the accurate prevalence of different IEMs is still unknown in India owing to lack of national level policy and NBS program (30). Likewise, due to similar problems, Pakistan also lags behind in setting up an NBS program for any of the IEMs. Although we acknowledge the challenge that costs of treatment pose in a developing country such as Pakistan, many IEMs are treatable with nutritional therapy which is relatively affordable and accessible, and we do believe that this population also has a right to prevention through early diagnosis.
Therefore, in the current study, research was initiated on the identification and characterization of treatable IEMs with the help of international experts, clinicians, geneticists and biochemists, which will pave the way for establishing a NBS program in Pakistan.
Since, special diets, pharmacological, and other therapies are now available for several IEMs; these will be targeted in the identified patients. Thus, for the treatable IEMs, evidence based medicine (EBM) approach would be proposed. Many EBM approaches have been developed and survival rates of patients with such treatable IEMs have improved substantially in recent years. In 2012, 83 drugs were reported, which were used for the treatment of different IEMs (2, 31). Despite the rarity of these disorders, there is growing emphasis on the use of EBM approach in the treatment of such rare metabolic disorders.
Therefore, the aim of this study was to screen a cohort of intellectually disabled patients for the identification of treatable IEMs, and to determine their prevalence for generating data to initiate NBS program in Pakistan.
Materials and Methods
This study was approved by the institutional ethical review committee (National Institute for Biotechnology and Genetic Engineering, NIBGE, Faisalabad, Pakistan). Blood and urine samples were collected from the intellectually disabled patients (IQ < 70), along with important clinical information from January 2016 to December 2018.
In the current study, sample size for the patients was calculated by an online tool “Sample Size Calculator” by creative research systems (www.surveysystem.com/sscalc.htm). By providing relevant information like; confidence interval (95%), confidence level (5%) and population size of infants from two cities (Lahore and Faisalabad, second and third largest cities) of Pakistan (7.6 million). The “Sample Size Calculator” tool showed that 384 patients were needed to conduct this study. Hence, for the effectiveness, we have enrolled the maximum number of patients possible in the given time period comprising of a total of 429 intellectually disabled patient samples (suspected for IEMs), including 5 ID patients from Swat, Pakistan.
For patient sample collection, Special Education Centers (SECs) were visited in the two major cities (Lahore and Faisalabad) of Punjab, Pakistan. Permission was taken from the local government and school management as well as informed written consent of parents was obtained prior to sample collection. In each SEC, biofluid (peripheral blood and urine) from each intellectually disabled patient was collected. A total of 18 public SECs have were visited; 11 in Faisalabad and 7 in Lahore. From these SECs and with the support of our clinical collaborator from government (DHQ/Allied) hospitals, Faisalabad, a total of 429 patient blood samples and 340 urine samples were collected. The ratio of boys to girls was 5:2. Plasma and serum were separated from the peripheral blood for the detailed biochemical analyses and saved at −20°C until further analysis. All the experimental analyses were mainly performed during 2018.
To ascertain the presence and estimate prevalence of treatable IEMs, biochemical analyses were started on the collected patient samples. All the patient samples (n = 429) were first analyzed through an analytical HPLC assay for the screening of aminoacidopathies at NIBGE, Faisalabad, Pakistan. From this cohort of all patients, 113 patient samples were selected on the basis of clinically important parameters like, consanguinity, ectopia lentis, hypopigmented hair, hyperactivity, aggressive behavior, developmental delayed milestones, and tall stature etc. For detailed biochemical screening analyses and advance analytical techniques (Amino Acid Analyzer (AAA), GC-MS, UHPLC-MS, and MS/MS) were used at the Amsterdam University Medical Centers (UAMC), University of Amsterdam, The Netherlands. For the diagnosis of various disorders, screening was started for different metabolites like; plasma amino acids, acylcarnitine, chenodeoxycholic acid, cholic acid, ursodeoxycholic acid, di- and tri-hydroxycholestanoic acid, guanidinoacetic acid, creatine, heparan sulfate, dermatan sulfate, keratan sulfate, cholesterol, cholestanol, campesterol, sitosterol, stigmasterol etc., to find the levels of these metabolites in the plasma samples of 113 intellectually disabled patients.
Results
All the important clinical information of the selected 113 intellectually disabled patients is listed in Table 2. After the detailed biochemical analyses through advance techniques, four different IEMs were identified (homocystinuria, mucopolysaccharidoses (MPS), Guanidinoacetate methyltransferase (GAMT) deficiency and sitosterolemia) as listed in Table 3. Interestingly, 9 classical homocystinuria patients from 7 unrelated families were found with the high concentrations of plasma methionine (Mean = 888 μmol/L; Reference range: 11–43 μmol/L) and homocysteine (Mean = 290 μmol/L; Reference range: 6–19 μmol/L).

Table 2. Different clinically important parameters of intellectually disabled patients.
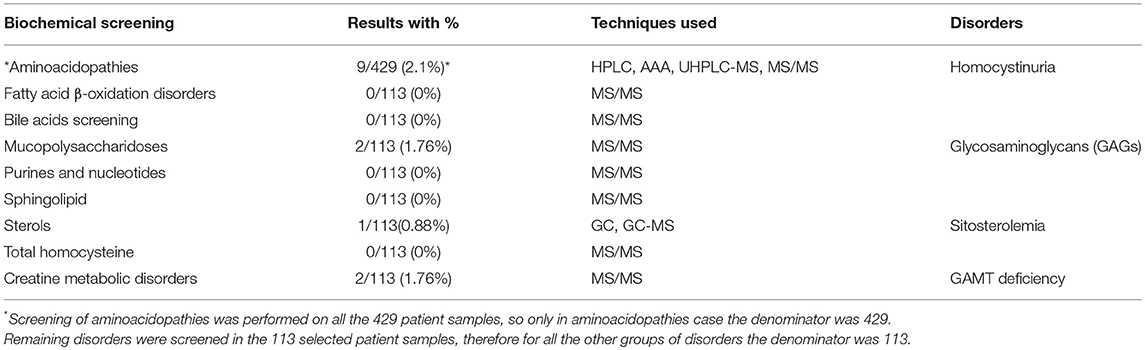
Table 3. Screening of different IEMs with advance analytical techniques and the prevalent disorders in the patient samples.
There were 2 MPS patients with high concentrations of heparan sulfate (Mean = 316 μg/ml), keratan sulfate (Mean = 2,144 μg/ml) and dermatan sulfate (Mean = 85 μg/ml). The Whole Exome Sequencing (WES) of these MPS patients is in progress for the confirmation of disease at the genetic level. Moreover, 2 patients were also identified with GAMT deficiency with high concentration of guanidinoacetic acid (Mean = 15 μmol/L; Reference range: 0.4–1.8 μmol/L) and low concentration of creatine (Mean = 2.8 μmol/L; Reference range: 17–109 μmol/L), and 1 sitosterolemia patient was found with the high values of cholesterol (4,969 μmol/L; Reference range: 1,881–4,887 μmol/L), cholestanol (23 μmol/L; Reference range: 3.50–10 μmol/L), stigmasterol (17 μmol/L; Reference range: 0–10 μmol/L), campesterol (173 μmol/L; Reference range: 0–23 μmol/L) and sitosterol (325 μmol/L; Reference range: 0–16 μmol/L). Overall in this cohort of intellectually disabled patients, 14 patients were diagnosed with four distinct types of IEMs.
Discussion
Overall the frequency of IEMs causing in the intellectual disability in different populations is 1 to 3% (32–34). Unfortunately in Pakistan very limited data is available for IEMs in general, let only frequency (35, 36), due to lack of any local, provincial or national level NBS program. For this reason, we initiated to screen intellectually disabled patients who were suspected for IEMs from the second (Lahore) and third (Faisalabad) largest cities of Pakistan. This comprehensive biochemical screening study of a subset of 113 intellectually disabled patients (taken from a bigger cohort of 429 intellectually disabled patients) will be a stepping stone to initiate a NBS program in Pakistan. Hence, screening was started for various treatable IEMs like aminoacidopathies, MPS, fatty acid β-oxidation disorders, creatine metabolic disorders etc. According to these screening analyses, prevalence of IEMs in the studied cohort from Pakistan appeared reasonably high (3.26%), which could be due to high rate of consanguinity.
In the identified IEMs in our cohort, homocystinuria was found as the most frequent aminoacidopathy so far in the intellectually disabled patient cohort from Punjab, Pakistan. Other studies have also reported this condition as the second most prevalent IEM in different populations after phenylketonuria (37–39). In our cohort we did not find any patient with phenylketonuria., but this could be due to the relatively limited sample size for a rare disease study. The reported incidence of homocystinuria varies from 1 in 344,000 worldwide to 1 in 65,000 in the Ireland, with highest in the Qatar as 1 in 1,800 (37, 40, 41). In the present study, 9 patients (2.1%) were found with homocystinuria from the original 429 intellectually disabled patient cohort. Since, early diagnosis of homocystinuria can help the patients to start a timely treatment and avoid disease pathology. Diet and other pharmacological therapies are available like; pyridoxine in combination with folic acid and vitamin B12; a methionine-restricted, cysteine-supplemented diet, and betaine (41–46). Affected patients can live a normal life by using these treatments, which would indeed be a great relief not only for such patients but also their families.
Aside from classical homocystinuria disease, 2 patients were identified with a suggestive phenotype of MPS, a group of rare IEMs in which the accumulation of glycosaminoglycans (GAGs) occur due to enzyme deficiencies (WES data is in progress to find causative mutations).Two more patients were found with GAMT deficiency, which is a disorder of creatine metabolism amenable to high dose creatine and an arginine restricted diet (47). One patient in the current study showed high concentration of cholesterol, cholestanol, stigmasterol, campesterol and sitosterol confirming sitosterolemia disease. Different therapeutic options are available for the treatment of sitosterolemia like, diets low in plant sterols and shellfish, and use of the sterol absorption inhibitor ezetimibe (48, 49). Early diagnosis is important, especially for those IEMs amenable to treatment as irreversible damage can be prevented, but also for ending the diagnostic odyssey, providing accurate genetic counseling on recurrence risk, identification of family members at risk, and providing an answer and optimizing supportive management. With an early diagnosis, preferably by NBS in the future, such patients can live a healthy life and burden of these kinds of IEMs can be minimized from the society.
Conclusion
IEMs are rare genetic disorders, and more than 200 of these are potentially treatable if diagnosed early. Diet and pharmacological therapies are available for the treatment of several IEMS. Overall prevalence of IEMs in different population is 2 to 3% and according to the current study, prevalence in the Pakistani cohort was 3.26%, due to high rate of consanguinity. It is worth mentioning that the studied patient cohort did not represent all the geographic locations of Pakistan but only the Faisalabad and Lahore regions of Punjab, Pakistan. Thus, more comprehensive large scale studies covering all areas of Pakistan would be required to know the real prevalence of IEMs. Unfortunately, Pakistan has no NBS program up till now, so this detailed biochemical screening study will help to start an initial setup for NBS program.
In this study, after the biochemical screening, different IEMs have been demonstrated in Pakistani patient cohort, thus diagnoses and treatment options were made available for these rare disorders. It is a practical option with benefits for the patients to start screening of treatable IEMs like aminoacidopathies, fatty acid β-oxidation and creatine metabolic disorders etc. Based on this initial screening data, screening of homocystinuria will be initiated due to its highest frequency (2.1%) among IEMs in the collected 429 intellectually disabled patients from Pakistan. As we have also found 5 patients besides classical homocystinuria, two with MPS, two patients with GAMT deficiency and one with sitosterolemia. In future, we would like to setup a NBS program in Pakistan for the screening of most prevalent IEMs because after the early diagnosis and treatment, affected patients can live long with normal life and have a positive impact on their families.
Following sample collection and initial analysis (HPLC) at NIBGE, Faisalabad, Pakistan, all the advanced biochemical screening was performed in the Laboratory Genetic Metabolic Diseases, Department of Clinical Chemistry, Amsterdam UMC, The Netherlands. This collaborative work will be continued to develop capacity for the IEMs research in Pakistan that will help us to setup a NBS program in Pakistan. After receiving a prompt diagnosis, timely initiation of treatment can help to minimize the deleterious symptoms of these rare IEMs.
Data Availability
This manuscript contains previously unpublished data. The name of the repository and accession number are not available.
Ethics Statement
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent of Participants
Informed written consent was obtained for all study participants from their parents/head of special education centers.
Author Contributions
MW and HK collected samples of intellectually disabled patients. MW wrote initial drafts. CvK conceived the idea, revised the manuscript, and approved it. SG and FV have confirmed all the diseases. HA helped in the clinical aspects of this study. FA conducted and supervised this study and revised the manuscript several times and approved it.
Funding
This article originated as a result of research project, Diagnosis of treatable inborn metabolic disorders of intellectual disability (Project No. CRP/PAK14-02; Contract No. CRP/14/012) funded by the International Center for Genetic Engineering and Biotechnology (ICGEB), Italy. Moreover, two PhD students (MW and HK) were funded for their research at the Amsterdam UMC, Amsterdam, the Netherlands by the High Education Commission (HEC), Islamabad, Pakistan specifically through the International Research Support Initiative Program (IRSIP). CvK received a salary award from Stichting Metakids in The Netherlands.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mak CM, Lee HC, Chan AY, Lam CW. Inborn errors of metabolism and expanded newborn screening: review and update. Crit Rev Clin Lab Sci. (2013) 50:142–62. doi: 10.3109/10408363.2013.847896
2. Alfadhel M, Al-Thihli K, Moubayed H, Eyaid W, Al-Jeraisy M. Drug treatment of inborn errors of metabolism: a systematic review. Arch Dis Child. (2013) 98:454–61. doi: 10.1136/archdischild-2012–303131
3. Tarailo-Graovac M, Wasserman WW, Van Karnebeek CD. Impact of next-generation sequencing on diagnosis and management of neurometabolic disorders: current advances and future perspectives. Expert Rev Mol Diagn. (2017) 17:307–9. doi: 10.1080/14737159.2017.1293527
4. Jacob M, Malkawi A, Albast N, Al Bougha S, Lopata A, Dasouki M, et al. A targeted metabolomics approach for clinical diagnosis of inborn errors of metabolism. Anal Chim Acta. (2018) 1025:141–53. doi: 10.1016/j.aca.2018.03.058
5. Ibarra-Gonzalez I, Rodriguez-Valentin R, Lazcano-Ponce E, Vela-Amieva M. Metabolic screening and metabolomics analysis in the intellectual developmental disorders Mexico Study. Salud Publica Mex. (2017) 59:423–8. doi: 10.21149/8668
6. Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med. (2019) 21:102–6. doi: 10.1038/s41436–018-0022–8
7. Graham E, Lee J, Price M, Tarailo-Graovac M, Matthews A, Engelke U, et al. Integration of genomics and metabolomics for prioritization of rare disease variants: a 2018 literature review. J Inherit Metab Dis. (2018) 41:435–45. doi: 10.1007/s10545–018-0139–6
8. Wajner M, Sitta A, Kayser A, Deon M, Groehs AC, Coelho DM, et al. Screening for organic acidurias and aminoacidopathies in high-risk Brazilian patients: eleven-year experience of a reference center. Genet Mol Biol. (2019). doi: 10.1590/1678-4685-GMB-2018-0105. [Epub ahead of print].
9. Gramer G, Haege G, Glahn EM, Hoffmann GF, Lindner M, Burgard P. Living with an inborn error of metabolism detected by newborn screening-parents' perspectives on child development and impact on family life. J Inherit Metab Dis. (2014) 37:189–95. doi: 10.1007/s10545–013-9639–6
10. Ullah MA, Husseni AM, Mahmood SU. Consanguineous marriages and their detrimental outcomes in Pakistan: an urgent need for appropriate measures. Int J Community Med Public Health. (2017) 5:3. doi: 10.18203/2394–6040.ijcmph20175757
11. Kaufmann JO, Krapels IP, Van Brussel BT, Zekveld-Vroon RC, Oosterwijk JC, van Erp F, et al. After the introduction into the national newborn screening program: who is receiving genetic counseling for hemoglobinopathies in the Netherlands? Public Health Genomics. (2014) 17:16–22. doi: 10.1159/000355223
12. Lim JS, Tan ES, John CM, Poh S, Yeo SJ, Ang JS, et al. Inborn Error of Metabolism (IEM) screening in Singapore by electrospray ionization-tandem mass spectrometry (ESI/MS/MS): An 8 year journey from pilot to current program. Mol Genet Metab. (2014) 113:53–61. doi: 10.1016/j.ymgme.2014.07.018
13. Liao HC, Chiang CC, Niu DM, Wang CH, Kao SM, Tsai FJ, et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry–a national newborn screening program in Taiwan. Clin Chim Acta. (2014) 431:80–6. doi: 10.1016/j.cca.2014.01.030
14. De Jesus VR, Mei JV, Cordovado SK, Cuthbert CD. The newborn screening quality assurance program at the centers for disease control and prevention: thirty-five year experience assuring newborn screening laboratory quality. Int J Neonatal Screen. (2015) 1:13–26. doi: 10.3390/ijns1010013
15. Pollak A, Kasper DC. Austrian newborn screening program: a perspective of five decades. J Perinat Med. (2014) 42:151–8. doi: 10.1515/jpm-2013–0113
16. Therrell BL, Padilla CD, Loeber JG, Kneisser I, Saadallah A, Borrajo GJ, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. (2015) 39:171–87. doi: 10.1053/j.semperi.2015.03.002
17. Ozben T. Expanded newborn screening and confirmatory follow-up testing for inborn errors of metabolism detected by tandem mass spectrometry. Clin Chem Lab Med. (2013) 51:157–76. doi: 10.1515/cclm-2012–0472
19. Chong SC, Law LK, Hui J, Lai CY, Leung TY, Yuen YP. Expanded newborn metabolic screening programme in Hong Kong: a three-year journey. Hong Kong Med J. (2017) 23:489–96. doi: 10.12809/hkmj176274
20. Shi XT, Cai J, Wang YY, Tu WJ, Wang WP, Gong LM, et al. Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep. (2012) 6:79–83. doi: 10.1007/8904_2011_119
21. Lin Y, Zheng Q, Zheng T, Zheng Z, Lin W, Fu Q. Expanded newborn screening for inherited metabolic disorders and genetic characteristics in a southern Chinese population. Clin Chim Acta. (2019) 494:106–11. doi: 10.1016/j.cca.2019.03.1622
22. Yang Y, Wang L, Wang B, Liu S, Yu B, Wang T. Application of next-generation sequencing following tandem mass spectrometry to expand newborn screening for inborn errors of metabolism: a multicenter study. Front Genet. (2019) 10:86. doi: 10.3389/fgene.2019.00086
23. Guo K, Zhou X, Chen X, Wu Y, Liu C, Kong Q. Expanded newborn screening for inborn errors of metabolism and genetic characteristics in a chinese population. Front Genet. (2018) 9:122. doi: 10.3389/fgene.2018.00122
24. Hettiarachchi M, Amarasena S. Indicators of newborn screening for congenital hypothyroidism in Sri Lanka: program challenges and way forward. BMC Health Serv Res. (2014) 14:385. doi: 10.1186/1472–6963-14–385
25. Murphy MSQ, Chakraborty P, Pervin J, Rahman A, Wilson LA, Lamoureux M, et al. Incidental screen positive findings in a prospective cohort study in Matlab, Bangladesh: insights into expanded newborn screening for low-resource settings. Orphanet J Rare Dis. (2019) 14:25. doi: 10.1186/s13023–018-0993–1
26. Kaur G, Thakur K, Kataria S, Singh TR, Chavan BS, Kaur G, et al. Current and future perspective of newborn screening: an Indian scenario. J Pediatr Endocrinol Metab. (2016) 29:5–13. doi: 10.1515/jpem-2015–0009
27. Kapoor S, Gupta N, Kabra M. National newborn screening program still a hype or a hope now? Indian Pediatr. (2013) 50:639–43.
28. Kapoor S, Kabra M. Newborn screening in India: current perspectives. Indian Pediatr. (2010) 47:219–24. doi: 10.1007/s13312-010-0043-0
29. Mukhopadhyay K, Balachandran B. Universal newborn screening - is it going to be a reality in India? Indian Pediatr. (2014) 51:697–8.
30. Kapoor S, Thelma BK. Status of newborn screening and inborn errors of metabolism in India. Indian J Pediatr. (2018) 85:1110–7. doi: 10.1007/s12098–018-2681–5
31. Yang CJ, Wei N, Li M, Xie K, Li JQ, Huang CG, et al. Diagnosis and therapeutic monitoring of inborn errors of metabolism in 100,077 newborns from Jining city in China. BMC Pediatr. (2018) 18:110. doi: 10.1186/s12887–018-1090–2
32. van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab. (2012) 105:368–81. doi: 10.1016/j.ymgme.2011.11.191
33. van Karnebeek CDM. Evaluation of the child with developmental impairments. Continuum. (2018) 24:228–47. doi: 10.1212/CON.0000000000000564
34. Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, et al. Exome sequencing and the management of neurometabolic disorders. N Engl J Med. (2016) 374:2246–55. doi: 10.1056/NEJMoa1515792
35. Cheema HA, Malik HS, Parkash A, Fayyaz Z. Spectrum of inherited metabolic disorders in Pakistani children presenting at a tertiary care centre. J Coll Physicians Surg Pak. (2016) 26:498–502.
36. Satwani H, Raza J, Hanai J, Nomachi S. Prevalence of selected disorders of inborn errors of metabolism in suspected cases at a tertiary care hospital in Karachi. J Pak Med Assoc. (2009) 59:815–9.
37. Yap S. Classical homocystinuria: vascular risk and its prevention. J Inherit Metab Dis. (2003) 26:259–65. doi: 10.1023/A:1024497419821
38. Rezazadeh A, Oveisgharan S, Shahidi G, Naghdi R. A Case report of homocystinuria with dystonia and stroke. Child Neurol Open. (2014) 1:2329048X14545870. doi: 10.1177/2329048X14545870
39. Al Humaidan M, Al Sharkawy I, Al Sanae A, Al Refaee F. Homocystinuria with lower gastrointestinal bleeding: first case report. Med Princ Pract. (2013) 22:500–2. doi: 10.1159/000345639
40. Mazaheri A, Mostofizadeh N, Hashemipour M. Homocystinuria with stroke and positive familial history. Adv Biomed Res. (2017) 6:132. doi: 10.4103/2277–9175.217215
41. El Bashir H, Dekair L, Mahmoud Y, Ben-Omran T. Neurodevelopmental and cognitive outcomes of classical homocystinuria: experience from Qatar. JIMD Rep. (2015) 21:89–95. doi: 10.1007/8904_2014_394
42. Vanzin CS, Mescka CP, Donida B, Hammerschimidt TG, Ribas GS, Kolling J, et al. Lipid, oxidative and inflammatory profile and alterations in the enzymes paraoxonase and butyrylcholinesterase in plasma of patients with homocystinuria due CBS deficiency: The vitamin B12 and folic acid importance. Cell Mol Neurobiol. (2015) 35:899–911. doi: 10.1007/s10571–015-0185–7
43. Maclean KN, Jiang H, Greiner LS, Allen RH, Stabler SP. Long-term betaine therapy in a murine model of cystathionine beta-synthase deficient homocystinuria: decreased efficacy over time reveals a significant threshold effect between elevated homocysteine and thrombotic risk. Mol Genet Metab. (2012) 105:395–403. doi: 10.1016/j.ymgme.2011.11.190
44. Voskoboeva E, Semyachkina A, Yablonskaya M, Nikolaeva E. Homocystinuria due to cystathionine beta-synthase (CBS) deficiency in Russia: Molecular and clinical characterization. Mol Genet Metab Rep. (2018) 14:47–54. doi: 10.1016/j.ymgmr.2017.11.001
45. Lee PJ, Briddon A. A rationale for cystine supplementation in severe homocystinuria. J Inherit Metab Dis. (2007) 30:35–8. doi: 10.1007/s10545–006-0452–3
46. Keller R, Chrastina P, Pavlikova M, Gouveia S, Ribes A, Kolker S, et al. Newborn screening for homocystinurias: recent recommendations versus current practice. J Inherit Metab Dis. (2019) 42:128–39. doi: 10.1002/jimd.12034
47. Stockler-Ipsiroglu S, van Karnebeek C, Longo N, Korenke GC, Mercimek-Mahmutoglu S, Marquart I, et al. Guanidinoacetate methyltransferase (GAMT) deficiency: outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab. (2014) 111:16–25. doi: 10.1016/j.ymgme.2013.10.018
48. Wang Y, Guo YL, Dong QT, Li JJ. Severe aortic valve stenosis in a 14-year-old boy with sitosterolemia. J Clin Lipidol. (2019) 13:49–53. doi: 10.1016/j.jacl.2018.11.002
Keywords: inborn errors of metabolism (IEMs), intellectual disability, newborn screening (NBS), evidence based medicine (EBM), homocystinuria, Pakistan
Citation: Wasim M, Khan HN, Ayesha H, Goorden SMI, Vaz FM, van Karnebeek CDM and Awan FR (2019) Biochemical Screening of Intellectually Disabled Patients: A Stepping Stone to Initiate a Newborn Screening Program in Pakistan. Front. Neurol. 10:762. doi: 10.3389/fneur.2019.00762
Received: 22 February 2019; Accepted: 01 July 2019;
Published: 17 July 2019.
Edited by:
Austen J. Milnerwood, Montreal Neurological Institute and Hospital, McGill University, CanadaReviewed by:
Liena Elbaghir Omer Elsayed, University of Khartoum, SudanLuca Marsili, University of Cincinnati, United States
Copyright © 2019 Wasim, Khan, Ayesha, Goorden, Vaz, van Karnebeek and Awan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clara D. M. van Karnebeek, Yy5kLnZhbmthcm5lYmVla0BhbXN0ZXJkYW11bWMubmw=; Fazli Rabbi Awan, YXdhbi5mckBnbWFpbC5jb20=
†These authors have contributed equally to this work