 Gabriela D. Colpo
Gabriela D. Colpo Venugopal R. Venna
Venugopal R. Venna Louise D. McCullough
Louise D. McCullough Antonio L. Teixeira
Antonio L. Teixeira- 1Neuropsychiatry Program, Department of Psychiatry and Behavioral Sciences, University of Texas Health Science Center at Houston, Houston, TX, United States
- 2BRAINS Lab, Department of Neurology, University of Texas Health Science Center at Houston, Houston, TX, United States
Background: Stroke is the second leading cause of death after ischemic heart disease and the third leading cause of disability-adjusted life-years lost worldwide. There is a great need for developing more effective strategies to treat stroke and its resulting impairments. Among several neuroprotective strategies tested so far, the kynurenine pathway (KP) seems to be promising, but the evidence is still sparse.
Methods: Here, we performed a systematic review of preclinical and clinical studies evaluating the involvement of KP in stroke. We searched for the keywords: (“kynurenine” or “kynurenic acid” or “quinolinic acid”) AND (“ischemia” or “stroke” or “occlusion) in the electronic databases PubMed, Scopus, and Embase. A total of 1,130 papers was initially retrieved.
Results: After careful screening, forty-five studies were included in this systematic review, being 39 pre-clinical and six clinical studies. Despite different experimental models of cerebral ischemia, the results are concordant in implicating the KP in the pathophysiology of stroke. Preclinical evidence also suggests that treatment with kynurenine and KMO inhibitors decrease infarct size and improve behavioral and cognitive outcomes. Few studies have investigated the KP in human stroke, and results are consistent with the experimental findings that the KP is activated after stroke.
Conclusion: Well-designed preclinical studies addressing the expression of KP enzymes and metabolites in specific cell types and their potential effects at cellular levels alongside more clinical studies are warranted to confirm the translational potential of this pathway as a pharmacological target for stroke and related complications.
Introduction
Stroke is clinically defined by the sudden onset of focal neurological symptoms (motor, sensory, cognitive) due to ischemia or hemorrhage in the brain. It is the second leading cause of death after ischemic heart disease and the third leading cause of disability-adjusted life-years lost worldwide (1). In the last two decades, there has been significant advance in the acute management of stroke, including the establishment of dedicated stroke inpatient units and the use of thrombolysis for eligible patients with ischemic stroke. Despite this progress, stroke-related deaths and morbidity remain a major health problem with personal and societal implications.
To address the great need of advancing stroke management, several mechanisms implicated in the pathophysiology of stroke, such as mitochondria dysfunction, glutamate-induced excitotoxicity, neuroinflammation, oxidative stress, among others, have been investigated as therapeutic targets. Among putative candidates, the kynurenine pathway (KP) received attention in the 1990's with a renewed interest recently on the wave of inflammatory-centric perspective of central nervous system diseases, including stroke (2).
The KP is the major route of tryptophan (TRP) catabolism in mammals. TRP is an essential amino acid used in the biosynthesis of proteins, being also a precursor of several bioactive molecules, such as serotonin and melatonin. Around 90% of TRP is metabolized by tryptophan 2,3-dioxygenase (TDO) into kynurenine (KYN) in the liver, with a much lower contribution of extra-hepatic KP on TRP degradation (5–10%) (3). TDO is liver specific, but two TDO variants been identified in mouse brain structures during development (4).
In extrahepatic tissues, especially cells of the immune and central nervous systems, the KP is initiated by the degradation of TRP by indoleamine 2, 3-dioxygenase 1 (IDO), the rate limiting enzyme of the pathway. This enzyme is potently upregulated by pro-inflammatory stimuli (2). After this step, the KP branches into two major pathways–one implicated in neuroprotection, the other in neurotoxicity–that are segregated across cell types (Figure 1). Under physiological conditions, the neuroprotective branch is more active as most of kynurenine in the brain is metabolized into kynurenic acid, a NMDA and α7-nicotinic acetylcholine receptor antagonist, through the action of kynurenine aminotransferases (KATs) expressed mainly in astrocytes (5). Under inflammatory conditions, the metabolism is shifted through kynurenine-3-monooxygenase (KMO) to produce 3-hydroxykynurenine and other toxic metabolites, including quinolinic acid, a NMDA receptor agonist and an oxidative stressor (3, 6). KMO is primarily expressed in microglia, the resident immune cells in the brain, and is also expressed at high levels in peripheral immune cells such as monocytes/macrophages (7).
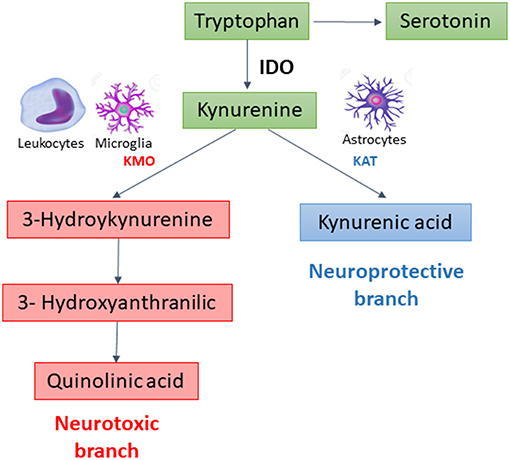
Figure 1. The Kynurenine Pathway (KP) of tryptophan metabolism.
Besides kynurenic acid (KYNA) and quinolinic acid (QUIN), the KP produces several other biologically active metabolites, including the redox cofactors oxidized nicotinamide adenine dinucleotide (NAD+). NAD is a common mediators of various biological processes, including energy metabolism, mitochondrial functions, calcium homeostasis, antioxidation/generation of oxidative stress (8). During cerebral ischemia, NAD is rapidly depleted, and increasing NAD has been proposed as a potential therapeutic strategy against stroke (9). Both NADPH and NAD+ have been reported to display potent neuroprotective effects in ischemia-related neuronal injury (10).
After an ischemic stroke, a series of inflammatory events takes place, leading to activation of resident microglia and mobilization of peripheral leukocytes with their subsequent infiltration into the injured side (11). Infiltrating leukocytes release inflammatory mediators, amplifying the intrinsic brain inflammatory response. Theoretically, these mechanisms could lead to activation of the KP, mainly its “neurotoxic” branch, contributing to neuronal damage and, ultimately, to clinical outcome (3). There is some pre-clinical and clinical evidence indicating activation of KP in stroke. Besides playing a pathophysiological role, KP metabolites have been tested as pharmacological agents to prevent and/or minimize stroke-related brain damage.
In this systematic review, our main objective was to summarize the evidence on the involvement of KP in both experimental models of brain ischemia and human patients with stroke.
Methods
Systematic Search
We undertook a comprehensive systematic search to identify all published studies evaluating KP in clinical studies and animal models of brain ischemia or stroke using the electronic databases PubMed, Scopus, and Embase. We conducted the search based on the keywords: (“kynurenine” or “kynurenic acid” or “quinolinic acid”) AND (“ischemia” or “stroke” or “occlusion”). Studies published through November 2018 were included.
A total of 1,130 papers was retrieved after removing duplicate manuscripts. Reference management software (EndNote X7 for Windows from Thomson Reuters, 2013) was used for screening purposes. The systematic review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) statement (12) as depicted in Figure 2.
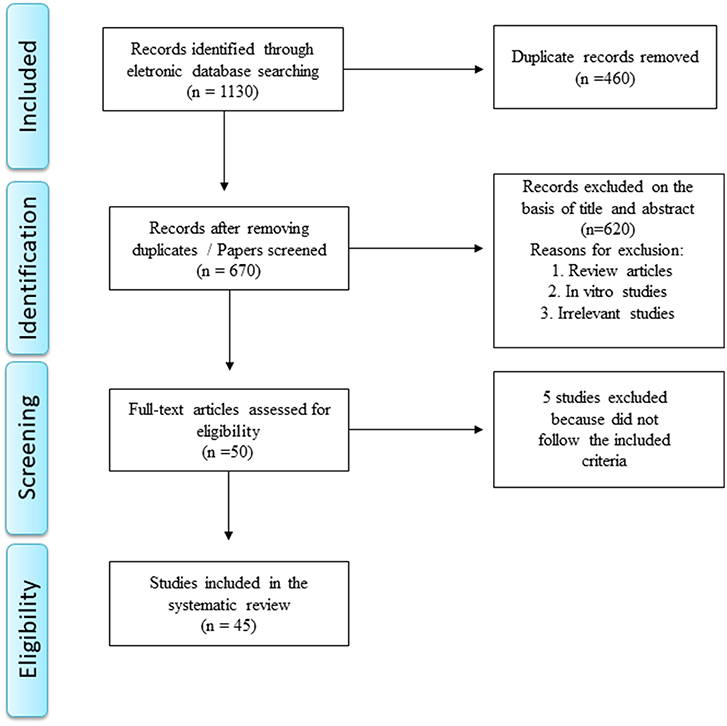
Figure 2. Flow diagram.
Eligibility Criteria, Data Extraction, and Quality Assessment
Studies were considered eligible to be included in the review if they met the following criteria: (1) animal model of stroke or brain ischemia and evaluation of KP metabolites or enzymes; (2) study with stroke patients and evaluation of KP metabolites or enzymes; (3) available in English; and (4) original data (not a review). The exclusion criteria were: (1) review manuscripts, (2) in vitro studies, (3) only abstracts and (4) non-relevant studies. During December 2018 and January 2019, one author (GDC) screened the titles and abstracts of all articles. Full texts were obtained for all articles that met the inclusion criteria (n = 50). Full-texts of the eligible studies were independently screened by two authors (GDC and ALT) who identified aims, methods, results and conclusions, extracting the respective information. Five papers were excluded in this phase.
To assess the risk of bias of the pre-clinical studies included in this systematic review, we used the SYRCLE's RoB tool (13). This tool evaluates specific features that might be source of bias in animal studies such as group allocation, if animals were randomly housed during the experiment, blinded investigators, animals selected at random for outcome assessment.
Results
In the end of the screening process, 45 studies met the inclusion criteria and they were included in this systematic review. Among them, 39 studies involved animal models of stroke/ brain ischemia and six were clinical studies comprising patients with stroke. These studies are summarized in Tables 1, 2, while the main results are summarized in Table 3.
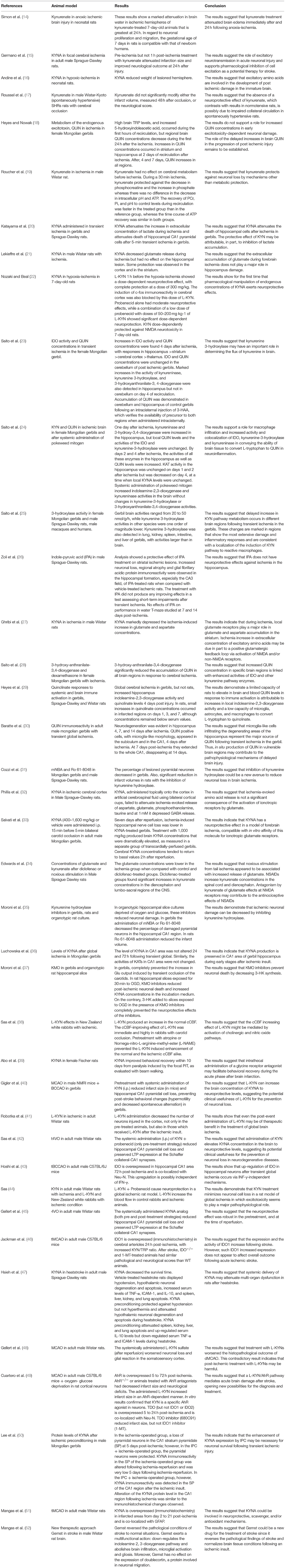
Table 1. Animal model studies with stroke/ischemia and kynurenine pathway.
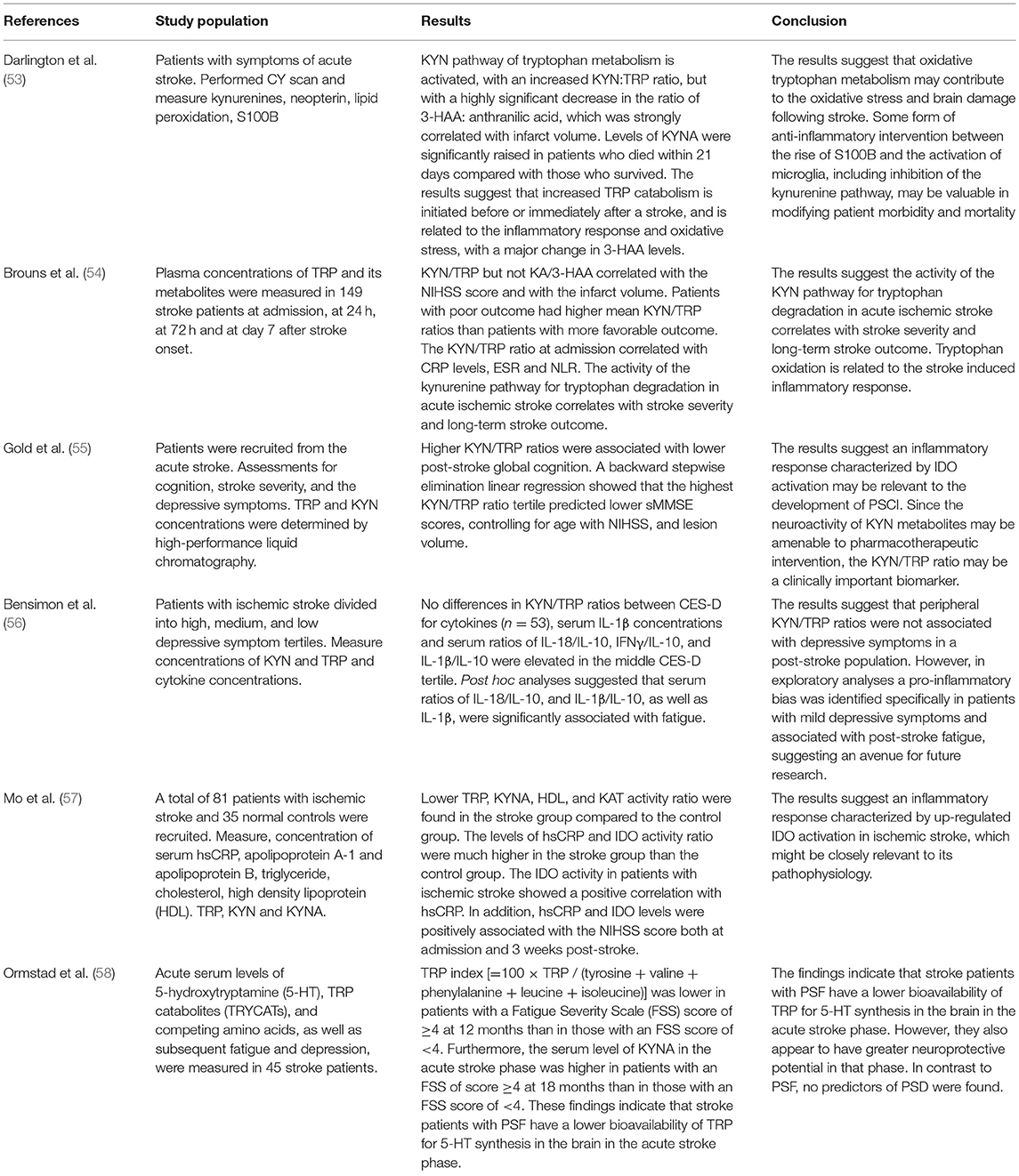
Table 2. Humans studies with stroke/ischemia and kynurenine pathway.
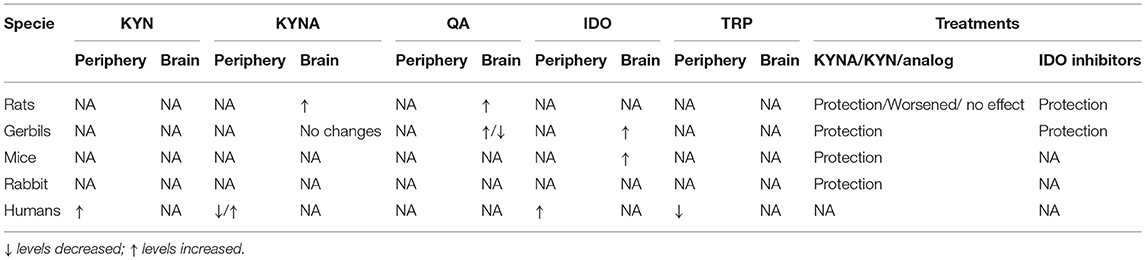
Table 3. Main results of KYN pathway and stroke.
Pre-clinical Studies
Regarding the quality assessment of the pre-clinical studies, only five papers reported that randomization was carried out, but without stating the method of group allocation. All studies reported that age and/or weight were similar among groups. None of the studies described whether animals were randomly selected for outcome assessment.
The type of animal model varied among the studies. More than 25 studies used rats while the second most used animal was the Mongolian gerbil followed by mice. Two studies used rabbits. Only six studies used females: one study with rats and five studies with gerbils, but none specifically examined the effects by sex. No study assessed aged animals.
The main aim of pre-clinical investigation was to assess the neuroprotective effects of KP metabolites (kynurenine or kynurenic acid) and/or KMO inhibitors (mNBA and Ro 61-8048) in different models of ischemia. Overall, studies with kynurenine and KMO inhibitors showed positive results, i.e., decrease in the infarct size and increased survival with treatment.
Gigler et al. (40) showed that intraperitoneal (i.p.) administration of kynurenine before stroke by middle cerebral artery occlusion reduced infarct size and hippocampal CA1 pyramidal cell loss in adult male mice. This same strategy also prevented post-stroke behavioral changes in gerbils in the bilateral carotid occlusion model (40). Pre- and post-treatment with kynurenine decreased cortical neuron damage in adult rats subjected to four-vessel occlusion (41).
Cozzi et al. evaluated the neuroprotective effects of KMO inhibitors. The KMO inhibitors mNBA and Ro 61-8048 were tested as neuroprotective strategies against ischemia induced by bilateral carotid occlusion in male Mongolian gerbils or middle cerebral artery occlusion in male rats. In gerbils, the percentage of lesioned pyramidal neurons in the hippocampal CAl region decreased significantly from around 90% in vehicle-treated animals to 7% after mNBA (400 mg/kg i.p., three times at 1, 30, and 180 min after occlusion) or 10% after Ro61-8048 (40 mg/kg i.p., three times at 1, 30, and 360 min after occlusion). A major reduction in infarct volumes was also found in rats after middle cerebral artery occlusion treated with mNBA or Ro 61-8048 administrated 30 min after the occlusion (31).
The results are mixed regarding the effects of kynurenic acid and analogs. Kynurenic acid did not show any protective effects in two studies (17, 48). Roussel et al. for instance, showed that kynurenic acid did not modify neither infarct volume or the neurological score 48 h after middle cerebral artery occlusion in adult male rats (17). Conversely, Gellert et al. (45) showed that administration of a kynurenic acid analog (2-(2-N,N-dimethylaminoethylamine-1-carbonyl)-1H-quinolin-4-one hydrochloride) either before or after ischemia reduced hippocampal CA1 pyramidal cell loss and preserved long-term potentiation expression at the Schaffer collateral-CA1 synapses in adult male rats in a four-vessel occlusion model. Other study showed that pretreatment with systemic administration of KYN (i.p.) reduced infarct size in mice and hippocampal CA1 pyramidal cell loss in gerbils, preventing post-stroke behavioral changes as hypermotility and decreased spontaneous alternation (40).
Pre-clinical studies also addressed the pathophysiological involvement of the KP after stroke, mainly focusing on the expression of the enzyme IDO and the metabolite kynurenic acid. Hoshi et al. showed by mRNA and immunohistochemistry increased expression of IDO in hippocampal CA1 area of adult male mice 72 h after global cerebral ischemia and that IDO is co-localized with Neu-N, a neuronal marker (43). Increased IDO expression was also observed in endothelial cells from cerebral arterioles 24 h post-middle cerebral artery occlusion, with increased plasma kynurenine/tryptophan ratio, an index of IDO activity, in adult male mice (46). Increased IDO activity and quinolinic acid concentrations in hippocampus, striatum, cortex, and thalamus, but not in cerebellum, were observed 4 days after 10 min of ischemia by bilateral common carotid artery occlusion in female Mongolian gerbils. Increased activity of kynureninase, kynurenine 3-hydroxylase, and 3-hydroxyanthranilate-3, 4-dioxygenase was also observed in hippocampus but not in cerebellum on day 4 (23). Most studies found increased expression of kynurenic acid in the stroke area. Kynurenic acid expression was increased in the infarcted areas from day 2 to 21 post-ischemia caused by single transient middle cerebral artery occlusion in adult male rats (52).
Clinical Studies
Only six clinical studies were identified. In these studies, patients were recruited within the first hours of the stroke, and different molecules such as tryptophan, kynurenine and kynurenic acid were measured in parallel with clinical parameters. Only two studies compared stroke patients with healthy controls, while the remaining four investigated whether KP-related molecules could be used as biomarkers of prognosis or clinical outcomes.
Ximing et al. studied 81 patients with ischemic stroke and 35 healthy controls, and reported a decrease in serum levels of tryptophan, kynurenic acid and KAT activity (calculated by kynurenic acid/kynurenine ratio) in the stroke group. Conversely, the levels of highly sensitive C reactive protein (hsCRP), a marker of inflammation, and IDO activity, as assessed by the kynurenine/tryptophan ratio, were significantly higher in the stroke group compared to controls. IDO activity showed a positive correlation with hsCRP, while hsCRP levels and IDO activity were positively correlated with the severity of the stroke as estimated by the NIH Stroke Scale both at admission and 3 weeks post-stroke (57). The other study investigated whether the KP activation correlated with infarct volume in 50 patients with acute stroke and 35 healthy control subjects (53). Tryptophan levels were significantly lower in stroke patients compared to controls at three time points: 1, 7, and 14 days after stroke. Kynurenine levels were increased in patients compared with controls on the first day post-stroke but not at subsequent time points. These results suggest that increased tryptophan catabolism is initiated immediately after the ischemic event, likely triggered by the inflammatory response and oxidative stress, with a major change in 3-hydroxyanthranilic acid levels that strongly correlated with infarct volume (53). Of note, 3-hydroxyanthranilic acid can generate reactive oxygen species, such as hydrogen peroxide and superoxide, in the presence of transition metal ions (59, 60).
Plasma levels of tryptophan and KP metabolites were measured in 149 stroke patients at admission, 24 h, 72 h, and day 7 after stroke. Patients with poor outcome had higher kynurenine/tryptophan ratio, an index of IDO activity and KP activation, compared to patients with favorable outcomes. Moreover, the activity of IDO, as estimated by kynurenine/tryptophan ratio, correlated with stroke severity and clinical outcome (54). In line with this result, Gold et al. reported higher kynurenine/tryptophan ratio in patients with impaired post-stroke cognition as assessed by the Mini-Mental State Examination within 1 month post-stroke (55).
In the study by Bensimon et al. patients with ischemic stroke were categorized based on their depressive symptoms into high, medium, and low severity in the acute phase of the stroke. Kynurenine and tryptophan were also determined. There was no difference in kynurenine/tryptophan ratio among depressive groups (56).
Discussion
There is evidence implicating the KP in the pathophysiology of stroke. Preclinical evidence also suggests that treatment with kynurenine and KMO inhibitors can be neuroprotective strategies to decrease infarct size and the related clinical outcomes.
In the KP, tryptophan is degraded to kynurenine that can be converted to kynurenic acid by KATs expressed in astrocytes (3). Kynurenic acid plays neuroprotective roles in the central nervous system as a NMDA and α7-nicotinic acetylcholine receptor antagonist (61). Corroborating this concept, a study with intrathecal administration of kynurenic acid improved motor outcome in rats subjected to photochemically induced brain thrombosis (39). Nevertheless, peripheral administration of kynurenic acid or its agonists did not lead to similar results because its penetration through the blood-brain barrier is poor (62). Interestingly, peripheral administration of kynurenine—that can cross the blood-brain barrier and then be converted to kynurenic acid in astrocytes—improved neurological outcome after stroke (40, 63, 64). In contrast to these reports, Gellert et al. (48) showed that post-ischemic treatment of adult male rats subjected to distal middle cerebral artery occlusion for 30 min with kynurenine resulted in worsened glial reaction and neuronal death (48). The extension of brain damage in the different models and the timing of treatment could explain this contradicting finding as administered kynurenine can be converted to quinolinic acid by KMO expressed in activated microglia and infiltrating leukocytes in the infarct area (6). Quinolinic acid is a neurotoxic agent, acting through multiple mechanisms including NMDA receptor agonism (65). Supporting these assumptions, there is evidence from human studies showing increased levels of quinolinic acid in microglia from the brain of patients with depression (66). Interestingly enough, depressive disorders are very common post-stroke complications, affecting over 30% of patients, which corroborates the view that KP may play a role in the pathophysiology of stroke (67).
Evidence from experimental models of stroke, as presented here, neurodegenerative diseases and other neuropsychiatric conditions such as Alzheimer's disease, Huntington's disease and depression, shows that KMO inhibition prevents neuronal loss and the related cognitive and behavioral changes (5, 7). As the available KMO inhibitors do not cross the blood-brain barrier and KMO is highly expressed in peripheral immune cells such as macrophages, it has been proposed that their peripheral action is responsible for these neuroprotective effects (68). In support of this, peripheral inhibition of KMO leads to sustained elevation in the brain levels of kynurenic acid without increasing quinolinic acid levels in the blood or brain (5). However, increased levels of kynurenic acid in the brain may not always be neuroprotective. Kindler et al. showed that patients with schizophrenia had increased kynurenic acid levels that were associated with brain volume loss and attention impairment (69). Accordingly, treatment in stroke should be aimed at “normalizing” and/or rebalancing the levels of kynurenic acid.
IDO is mainly expressed in monocytes, macrophages, dendritic cells and microglia, the tissue-resident phagocytes of the brain (70). IDO is the rate-limiting enzyme of the KP in these cells, and can be upregulated by pro-inflammatory stimuli, especially interferon-gamma (IFN-γ) (71). Accordingly, IDO is highly expressed in infarcted brain areas. Despite that, only one study so far investigated the potential benefits of IDO inhibitors in stroke or ischemia model. The study from Jackman et al. showed that although IDO expression and activity are increased after transient MCAO in mice, genetic and pharmacological methods for IDO inhibition after ischemia (IDO knockout mice and 1-MT treatment, respectively) had no positive effects on infarct volume and neurological outcome, however the authors only examined the effect of IDO1 expression on outcome at 24 h and no later time points (46). IDO inhibitors could be a potential treatment for stroke, however more studies using IDO inhibitors in stroke are warranted to better understand of its mechanism. It is worth mentioning that inhibitors of IDO1 has being used in experiments in oncology (72). As IDO1 has the ability to establish a tumor-promoting inflammatory environment, IDO1 inhibitors decrease tumor-related inflammation and angiogenesis, potentiating the efficacy of cytotoxic or targeted chemotherapies, radiotherapy, immune checkpoint therapies and cancer vaccines (72). This strategy of decreasing the inflammation by IDO inhibitors could be explored in the treatment of stroke and related complications.
Even though several studies were concordant with the concept that KP-based treatments are potentially neuroprotective in stroke, several methodological limitations must be acknowledged. In pre-clinical studies, there were significant differences in the stroke models (focal vs. global ischemia; permanent vs. transient ischemia), in the species used (rats, Gerbils, mice, rabbits) and in the time/dose of pharmacological manipulations. Quality assessment of these studies also suggested a high risk of bias. Moreover, most studies did not take into account sex differences that have been recognized as relevant in the pathophysiology of stroke (73), while no study evaluated aged animals. In addition, there is only one study investigating KP in endothelial cells. Endothelial cells play a critical role in vascular homeostasis and stroke development, with damaged endothelial cells potentially triggering a series of cerebrovascular injuries (74). Finally, most pre-clinical studies only addressed neuroprotection as assessed by infarct size, with limited evaluation of behavioral measures or underlying mechanisms. More carefully designed studies controlling for age and sex differences, and assessing the cells expressing the KP enzymes are definitely warranted.
Human studies corroborate the experimental findings of KP activation after stroke. Interestingly enough, KP-related biomarkers correlated with stroke severity and clinical outcomes, including cognitive impairment. These findings must be seen as preliminary as most studies did not include a control group and did not perform a careful behavioral, cognitive and socio-occupational phenotyping of the patients. In this regard, it would be important to specifically address which clinical parameters (i.e., neurological impairment, cognitive dysfunction, depression) are better predicted by these biomarkers. Another limitation here would be the standard method for KP metabolites assessment, i.e., liquid chromatography–mass spectrometry (LC-MS) that prevents the scaling of their measurement. In addition, only two studies compared patients with health controls.
In conclusion, KP is a promising target for the development of neuroprotective strategies against stroke.
Data Availability
All datasets analyzed for this study are included in the manuscript and/or the supplementary material.
Author Contributions
GC performed the search in the literature, screening of the papers and wrote the manuscript. AT helped screening of the papers, wrote the manuscript and mentored. VV and LM reviewed the manuscript.
Funding
The Neuropsychiatry Program is funded by the Department of Psychiatry and Behavioral Sciences, The University of Texas Health Science Center at Houston.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
3-HK, 3-hydroxykynurenine; AhR, aryl hydrocarbon receptor; BCCAO, Bilateral Common Carotid Artery Occlusion; Ccbf, Corticocerebral blood flow; CNS, Central nervous system; GFAP, Glial fibrillary acidic protein; ICAM-1, Intercellular Adhesion Molecule 1; IDO1, indoleamine 2,3-dioxygenase 1; IDO2, indoleamine 2,3-dioxygenase 2; IFN-γ, Interferon gamma; IL-10, interleukin-10; IPC, Ischemia preconditioning; KATs, Kynurenine aminotransferase; KMO, Kynurenine 3-monooxygenase; KYNA, Kynurenic acid; L-KYN, Kynurenine; LTP, Long-term potentiation; MCAO, Middle Cerebral Artery Occlusion; NMDA, N-methyl-D-aspartate; OGD, Glucose deprivation; PIT, Photochemically induced thrombosis; QUIN, Quinolinic acid; 4VO, Four-vessel occlusion; TDO, Tryptophan-2,3-dioxygenase; TNF-α, Tumor Necrosis Factor Alpha; TRP, Tryptophan; WB, Western blot.
References
2. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. (2012) 13:465–77. doi: 10.1038/nrn3257
3. Badawy AA. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res. (2017) 10:1178646917691938. doi: 10.1177/1178646917691938
4. Kanai M, Nakamura T, Funakoshi H. Identification and characterization of novel variants of the tryptophan 2,3-dioxygenase gene: differential regulation in the mouse nervous system during development. Neurosci Res. (2009) 64:111–7. doi: 10.1016/j.neures.2009.02.004
5. Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. (2011) 145:863–74. doi: 10.1016/j.cell.2011.05.020
6. Cuartero MI, de la Parra J, Garcia-Culebras A, Ballesteros I, Lizasoain I, Moro MA. The kynurenine pathway in the acute and chronic phases of cerebral ischemia. Curr Pharm Des. (2016) 22:1060–73.
7. Parrott JM, O'Connor JC. Kynurenine 3-monooxygenase: an influential mediator of neuropathology. Front Psychiatry. (2015) 6:116. doi: 10.3389/fpsyt.2015.00116
8. Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. (2008) 10:179–206. doi: 10.1089/ars.2007.1672
9. Wang P, Miao CY. NAMPT as a therapeutic target against stroke. Trends Pharmacol Sci. (2015) 36:891–905. doi: 10.1016/j.tips.2015.08.012
10. Huang Q, Sun M, Li M, Zhang D, Han F, Wu JC, et al. Combination of NAD(+) and NADPH offers greater neuroprotection in ischemic stroke models by relieving metabolic stress. Mol Neurobiol. (2018) 55:6063–75. doi: 10.1007/s12035-017-0809-7
11. Ao LY, Yan YY, Zhou L, Li CY, Li WT, Fang WR, et al. Immune cells after ischemic stroke onset: roles, migration, and target intervention. J Mol Neurosci. (2018) 66:342–55. doi: 10.1007/s12031-018-1173-4
12. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
13. Hooijmans CR, Rovers MM, de Vries RB, Leenaars M, Ritskes-Hoitinga M, Langendam MW. SYRCLE's risk of bias tool for animal studies. BMC Med Res Methodol. (2014) 14:43. doi: 10.1186/1471-2288-14-43
14. Simon RP, Young RS, Stout S, Cheng J. Inhibition of excitatory neurotransmission with kynurenate reduces brain edema in neonatal anoxia. Neurosci Lett. (1986) 71:361–4.
15. Germano IM, Pitts LH, Meldrum BS, Bartkowski HM, Simon RP. Kynurenate inhibition of cell excitation decreases stroke size and deficits. Ann Neurol. (1987) 22:730–4. doi: 10.1002/ana.410220609
16. Andine P, Lehmann A, Ellren K, Wennberg E, Kjellmer I, Nielsen T, et al. The excitatory amino acid antagonist kynurenic acid administered after hypoxic-ischemia in neonatal rats offers neuroprotection. Neurosci Lett. (1988) 90:208–12.
17. Roussel S, Pinard E, Seylaz J. Kynurenate does not reduce infarct size after middle cerebral artery occlusion in spontaneously hypertensive rats. Brain Res. (1990) 518:353–5.
18. Heyes MP, Nowak TS Jr. Delayed increases in regional brain quinolinic acid follow transient ischemia in the gerbil. J Cereb Blood Flow Metab. (1990) 10:660–7. doi: 10.1038/jcbfm.1990.119
19. Roucher P, Meric P, Correze JL, Mispelter J, Tiffon B, Lhoste JM, et al. Metabolic effects of kynurenate during reversible forebrain ischemia studied by in vivo 31P-nuclear magnetic resonance spectroscopy. Brain Res. (1991) 550:54–60.
20. Katayama Y, Kawamata T, Kano T, Tsubokawa T. Excitatory amino acid antagonist administered via microdialysis attenuates lactate accumulation during cerebral ischemia and subsequent hippocampal damage. Brain Res. (1992) 584:329–33.
21. Lekieffre D, Ghribi O, Callebert J, Allix M, Plotkine M, Boulu RG. Inhibition of glutamate release in rat hippocampus by kynurenic acid does not protect CA1 cells from forebrain ischemia. Brain Res. (1992) 592:333–7.
22. Nozaki K, Beal MF. Neuroprotective effects of L-kynurenine on hypoxia-ischemia and NMDA lesions in neonatal rats. J Cereb Blood Flow Metab. (1992) 12:400–7. doi: 10.1038/jcbfm.1992.57
23. Saito K, Nowak TS Jr, Markey SP, Heyes MP. Mechanism of delayed increases in kynurenine pathway metabolism in damaged brain regions following transient cerebral ischemia. J Neurochem. (1993) 60:180–92.
24. Saito K, Nowak TS Jr, Suyama K, Quearry BJ, Saito M, Crowley JS, et al. Kynurenine pathway enzymes in brain: responses to ischemic brain injury versus systemic immune activation. J Neurochem. (1993) 61:2061–70.
25. Saito K, Quearry BJ, Saito M, Nowak TS Jr, Markey SP, Heyes MP. Kynurenine 3-hydroxylase in brain: species activity differences and effect of gerbil cerebral ischemia. Arch Biochem Biophys. (1993) 307:104–9. doi: 10.1006/abbi.1993.1567
26. Zoli M, Merlo Pich E, Ferraguti F, Biagini G, Fuxe K, Agnati LF. Indole-pyruvic acid treatment reduces damage in striatum but not in hippocampus after transient forebrain ischemia in the rat. Neurochem Int. (1993) 23:139–48.
27. Ghribi O, Callebert J, Plotkine M, Boulu RG. Effect of kynurenic acid on the ischaemia-induced accumulation of glutamate in rat striatum. NeuroReport. (1994) 5:435–7. doi: 10.1097/00001756-199401120-00016
28. Saito K, Seishima M, Noma A, Suyama K, Markey SP, Heyes MP. 4-chloro-3-hydroxyanthranilate attenuate quinolinic acid accumulation in brain following transient cerebral ischemia in the gerbil. Adv Exp Med Biol. (1996) 398:407–11.
29. Heyes MP, Saito K, Chen CY, Proescholdt MG, Nowak TS Jr, Li J, et al. Species heterogeneity between gerbils and rats: quinolinate production by microglia and astrocytes and accumulations in response to ischemic brain injury and systemic immune activation. J Neurochem. (1997) 69:1519–29.
30. Baratte S, Molinari A, Veneroni O, Speciale C, Benatti L, Salvati P. Temporal and spatial changes of quinolinic acid immunoreactivity in the gerbil hippocampus following transient cerebral ischemia. Brain Res Mol Brain Res. (1998) 59:50–7.
31. Cozzi A, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61-8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab. (1999) 19:771–7. doi: 10.1097/00004647-199907000-00007
32. Phillis JW, Song D, Guyot LL, O'Regan MH. Failure of kynurenic acid to inhibit amino acid release from the ischemic rat cerebral cortex. Neurosci Lett. (1999) 273:21–4.
33. Salvati P, Ukmar G, Dho L, Rosa B, Cini M, Marconi M, et al. Brain concentrations of kynurenic acid after a systemic neuroprotective dose in the gerbil model of global ischemia. Prog Neuropsychopharmacol Biol Psychiatry. (1999) 23:741–52.
34. Edwards SR, Mather LE, Lin Y, Power I, Cousins MJ. Glutamate and kynurenate in the rat central nervous system following treatments with tail ischaemia or diclofenac. J Pharm Pharmacol. (2000) 52:59–66. doi: 10.1211/0022357001773698
35. Moroni F, Cozzi A, Peruginelli F, Carpenedo R, Pellegrini-Giampietro DE. Neuroprotective effects of kynurenine-3-hydroxylase inhibitors in models of brain ischemia. Adv Exp Med Biol. (2000) 467:199–206. doi: 10.1007/978-1-4615-4709-9_26
36. Luchowska E, Luchowski P, Sarnowska A, Wielosz M, Turski WA, Urbanska EM. Endogenous level of kynurenic acid and activities of kynurenine aminotransferases following transient global ischemia in the gerbil hippocampus. Pol J Pharmacol. (2003) 55:443–7.
37. Moroni F, Carpenedo R, Cozzi A, Meli E, Chiarugi A, Pellegrini-Giampietro DE. Studies on the neuroprotective action of kynurenine mono-oxygenase inhibitors in post-ischemic brain damage. Adv Exp Med Biol. (2003) 527:127–36. doi: 10.1007/978-1-4615-0135-0_15
38. Sas K, Csete K, Vecsei L, Papp JG. Effect of systemic administration of L-kynurenine on corticocerebral blood flow under normal and ischemic conditions of the brain in conscious rabbits. J Cardiovasc Pharmacol. (2003) 42:403–9. doi: 10.1097/00005344-200309000-00012
39. Abo M, Yamauchi H, Suzuki M, Sakuma M, Urashima M. Facilitated beam-walking recovery during acute phase by kynurenic acid treatment in a rat model of photochemically induced thrombosis causing focal cerebral ischemia. Neurosignals. (2006) 15:102–10. doi: 10.1159/000094876
40. Gigler G, Szenasi G, Simo A, Levay G, Harsing LG Jr, Sas K, et al. Neuroprotective effect of L-kynurenine sulfate administered before focal cerebral ischemia in mice and global cerebral ischemia in gerbils. Eur J Pharmacol. (2007) 564:116–22. doi: 10.1016/j.ejphar.2007.02.029
41. Robotka H, Toldi J, Vécsei L. L-kynurenine: metabolism and mechanism of neuroprotection. Future Neurol. (2008) 3:169–88. doi: 10.2217/14796708.3.2.169
42. Sas K, Robotka H, Rózsa É, Ágoston M, Szénási G, Gigler G, et al. Kynurenine diminishes the ischemia-induced histological and electrophysiological deficits in the rat hippocampus. Neurobiol Dis. (2008) 32:302–8. doi: 10.1016/j.nbd.2008.07.013
43. Hoshi M, Saito K, Murakami Y, Taguchi A, Fujigaki H, Tanaka R, et al. Marked increases in hippocampal neuron indoleamine 2, 3–dioxygenase via IFN-gamma-independent pathway following transient global ischemia in mouse. Neurosci Res. (2009) 63:194–8. doi: 10.1016/j.neures.2008.12.003
44. Sas K. Potential role of glutamate neurotransmission in the pathogenesis of ischemic brain damage and of depression. Effects of L-kynurenine on the survival of the hippocampal neurons and on the corticocerebral blood flow in ischemic animal models. Ideggyogy Sz. (2010) 63:61–70.
45. Gellert L, Fuzik J, Goblos A, Sarkozi K, Marosi M, Kis Z, et al. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur J Pharmacol. (2011) 667:182–7. doi: 10.1016/j.ejphar.2011.05.069
46. Jackman KA, Brait VH, Wang Y, Maghzal GJ, Ball HJ, McKenzie G, et al. Vascular expression, activity and function of indoleamine 2,3-dioxygenase-1 following cerebral ischaemia-reperfusion in mice. Naunyn Schmiedebergs Arch Pharmacol. (2011) 383:471–81. doi: 10.1007/s00210-011-0611-4
47. Hsieh YC, Chen RF, Yeh YS, Lin MT, Hsieh JH, Chen SH. Kynurenic acid attenuates multiorgan dysfunction in rats after heatstroke. Acta Pharmacol Sin. (2011) 32:167–74. doi: 10.1038/aps.2010.191
48. Gellert L, Knapp L, Nemeth K, Heredi J, Varga D, Olah G, et al. Post-ischemic treatment with L-kynurenine sulfate exacerbates neuronal damage after transient middle cerebral artery occlusion. Neuroscience. (2013) 247:95–101. doi: 10.1016/j.neuroscience.2013.04.063
49. Cuartero MI, Ballesteros I, de la Parra J, Harkin AL, Abautret-Daly A, Sherwin E, et al. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation. (2014) 130:2040–51. doi: 10.1161/circulationaha.114.011394
50. Lee JC, Tae HJ, Cho GS, Kim IH, Ahn JH, Park JH, et al. Ischemic preconditioning protects neurons from damage and maintains the immunoreactivity of kynurenic acid in the gerbil hippocampal CA1 region following transient cerebral ischemia. Int J Mol Med. (2015) 35:1537–44. doi: 10.3892/ijmm.2015.2171
51. Mangas A, Yajeya J, Gonzalez N, Ruiz I, Duleu S, Geffard M, et al. Overexpression of kynurenic acid in stroke: an endogenous neuroprotector? Ann Anat. (2017) 211:33–8. doi: 10.1016/j.aanat.2017.01.002
52. Mangas A, Yajeya J, Gonzalez N, Ruiz I, Pernia M, Geffard M, et al. Gemst: a taylor-made combination that reverts neuroanatomical changes in stroke. Eur J Histochem. (2017) 61:2790. doi: 10.4081/ejh.2017.2790
53. Darlington LG, Mackay GM, Forrest CM, Stoy N, George C, Stone TW. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur J Neurosci. (2007) 26:2211–21. doi: 10.1111/j.1460-9568.2007.05838.x
54. Brouns R, Verkerk R, Aerts T, De Surgeloose D, Wauters A, Scharpe S, et al. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem Res. (2010) 35:1315–22. doi: 10.1007/s11064-010-0187-2
55. Gold AB, Herrmann N, Swardfager W, Black SE, Aviv RI, Tennen G, et al. The relationship between indoleamine 2,3-dioxygenase activity and post-stroke cognitive impairment. J Neuroinflamm. (2011) 8:17. doi: 10.1186/1742-2094-8-17
56. Bensimon K, Herrmann N, Swardfager W, Yi H, Black SE, Gao FQ, et al. Kynurenine and depressive symptoms in a poststroke population. Neuropsychiatr Dis Treat. (2014) 10:1827–35. doi: 10.2147/ndt.s65740
57. Mo X, Pi L, Yang J, Xiang Z, Tang A. Serum indoleamine 2,3-dioxygenase and kynurenine aminotransferase enzyme activity in patients with ischemic stroke. J Clin Neurosci. (2014) 21:482–6. doi: 10.1016/j.jocn.2013.08.020
58. Ormstad H, Verkerk R, Amthor KF, Sandvik L. Activation of the kynurenine pathway in the acute phase of stroke and its role in fatigue and depression following stroke. J Mol Neurosci. (2014) 54:181–7. doi: 10.1007/s12031-014-0272-0
59. Goldstein LE, Leopold MC, Huang X, Atwood CS, Saunders AJ, Hartshorn M, et al. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid generate hydrogen peroxide and promote alpha-crystallin cross-linking by metal ion reduction. Biochemistry. (2000) 39:7266–75. doi: 10.1021/bi992997s
60. Leipnitz G, Schumacher C, Dalcin KB, Scussiato K, Solano A, Funchal C, et al. In vitro evidence for an antioxidant role of 3-hydroxykynurenine and 3-hydroxyanthranilic acid in the brain. Neurochem Int. (2007) 50:83–94. doi: 10.1016/j.neuint.2006.04.017
61. Albuquerque EX, Schwarcz R. Kynurenic acid as an antagonist of alpha7 nicotinic acetylcholine receptors in the brain: facts and challenges. Biochem Pharmacol. (2013) 85:1027–32. doi: 10.1016/j.bcp.2012.12.014
62. Varga N, Csapo E, Majlath Z, Ilisz I, Krizbai IA, Wilhelm I, et al. Targeting of the kynurenic acid across the blood-brain barrier by core-shell nanoparticles. Eur J Pharm Sci. (2016) 86:67–74. doi: 10.1016/j.ejps.2016.02.012
63. Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. (1991) 56:2007–17.
64. Robotka H, Sas K, Agoston M, Rozsa E, Szenasi G, Gigler G, et al. Neuroprotection achieved in the ischaemic rat cortex with L-kynurenine sulphate. Life Sci. (2008) 82:915–9. doi: 10.1016/j.lfs.2008.02.014
65. Schwarcz R, Whetsell WO Jr, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. (1983) 219:316–8.
66. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation. (2011) 8:94. doi: 10.1186/1742-2094-8-94
67. Robinson RG, Jorge RE. Post-stroke depression: a review. Am J Psychiatry. (2016) 1:221–31. doi: 10.1176/appi.ajp
68. Phillips RS, Iradukunda EC, Hughes T, Bowen JP. Modulation of enzyme activity in the kynurenine pathway by kynurenine monooxygenase inhibition. Front Mol Biosci. (2019) 6:3. doi: 10.3389/fmolb.2019.00003
69. Kindler J, Lim CK, Weickert CS, Boerrigter D, Galletly C, Liu D, et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol Psychiatry. (2019). doi: 10.1038/s41380-019-0401-9. [Epub ahead of print].
70. Mandi Y, Vecsei L. The kynurenine system and immunoregulation. J Neural Transm. (2012) 119:197–209. doi: 10.1007/s00702-011-0681-y
71. Lestage J, Verrier D, Palin K, Dantzer R. The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain Behav Immun. (2002) 16:596–601. doi: 10.1016/S0889-1591(02)00014-4
72. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. (2017) 77:6795–811. doi: 10.1158/0008-5472.CAN-17-2285
73. Kim T, Chelluboina B, Chokkalla AK, Vemuganti R. Age and sex differences in the pathophysiology of acute CNS injury. Neurochem Int. (2019) 127:22–8. doi: 10.1016/j.neuint.2019.01.012
Keywords: stroke, kynurenine pathway, kynurenic acid, quinolinic acid, KMO, IDO
Citation: Colpo GD, Venna VR, McCullough LD and Teixeira AL (2019) Systematic Review on the Involvement of the Kynurenine Pathway in Stroke: Pre-clinical and Clinical Evidence. Front. Neurol. 10:778. doi: 10.3389/fneur.2019.00778
Received: 26 March 2019; Accepted: 03 July 2019;
Published: 19 July 2019.
Edited by:
Heike Wulff, University of California, Davis, United StatesReviewed by:
Chai K. Lim, Macquarie University, AustraliaGayathri Sundaram, St. Vincent's Hospital, Australia
Copyright © 2019 Colpo, Venna, McCullough and Teixeira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela D. Colpo, Z2FiaWNvbHBvQGdtYWlsLmNvbQ==; Antonio L. Teixeira, YW50b25pby5sLnRlaXhlaXJhQHV0aC50bWMuZWR1