 Franziska Hopfner1,2*
Franziska Hopfner1,2* Markus A. Hobert1
Markus A. Hobert1 Corina Maetzler1
Corina Maetzler1 Clint Hansen1
Clint Hansen1 Minh Hoang Pham1Caroline Moreau3
Minh Hoang Pham1Caroline Moreau3 Daniela Berg1
Daniela Berg1 David Devos4
David Devos4 Walter Maetzler1 and The Fair-Park II study group
Walter Maetzler1 and The Fair-Park II study group- 1Department of Neurology, University Hospital Schleswig-Holstein, Christian-Albrechts-Universität zu Kiel, Kiel, Germany
- 2Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-Universität München, Munich, Germany
- 3Department of Movement Disorders and Neurology, Faculty of Medicine, Lille University Hospital, Lille University, INSERM U1171, Lille, France
- 4Departments of Medical Pharmacology and Movement Disorders, Lille University Hospital, Lille University, INSERM U1171, Lille, France
Background: Recent developments in mobile technology have enabled the investigation of human movements and mobility under natural conditions, i.e., in the home environment. Iron accumulation in the basal ganglia is deleterious in Parkinson's disease (i.e., iron accumulation with lower striatal level of dopamine). The effect of iron chelation (i.e., re-deployment of iron) in Parkinson's disease patients is currently tested in a large investigator-initiated multicenter study. Conversely, restless legs syndrome (RLS) is associated with iron depletion and higher striatal level of dopamine. To determine from animal models which movement and mobility parameters might be associated with iron content modulation and the potential effect of therapeutic chelation inhuman.
Methods: We recapitulated pathophysiological aspects of the association between iron, dopamine, and neuronal dysfunction and deterioration in the basal ganglia, and systematically searched PubMed to identify original articles reporting about quantitatively assessed mobility deficits in animal models of brain iron dyshomeostasis.
Results: We found six original studies using murine and fly models fulfilling the inclusion criteria. Especially postural and trunk stability were altered in animal models with iron overload. Animal models with lowered basal ganglia iron suffered from alterations in physical activity, mobility, and sleep fragmentation.
Conclusion: From preclinical investigations in the animal model, we can deduce that possibly also in humans with iron accumulation in the basal ganglia undergoing therapeutic chelation may primarily show changes in physical activity (such as daily “motor activity”), postural and trunk stability and sleep fragmentation. These changes can readily be monitored with currently available mobile technology.
Background
Mobile technology (mainly reflected by “wearables”) is a dynamically developing field with relevant implications for the health and fitness sector, and also for medical routine and clinical trials (1). Wearables for the assessment of movements and mobility are microelectromechanical systems with multiple degrees of freedom (DOF; e.g., 3D accelerometers, 3D gyroscopes, and 3D magnetometers) (2, 3). They enable continuous assessment during 24 h over many days, and make possible data collection and evaluation of disease progression and response to treatment based on uninstructed, unsupervised, and unobserved movements (1, 3–5). It seems realistic that such data will soon be used as complementary information for treatment decisions in diseases associated with movement deficits, such as Parkinson's disease (PD) (6) and restless legs syndrome (RLS).
This development is, at least partly, driven by accumulating evidence that humans perform similar movements differently in the home environment and in the doctor's practice, hospital and research lab, especially with regard to daily-relevant movements (4, 7). Therefore, clinical assessment may not always reflect daily-relevant information for the affected. Moreover, physical activity and mobility can by definition only be evaluated in the natural environment of the user, and some movement abnormalities, such as those happening during sleep, may occur only in the “unobserved,” but not in the usual professional, environment (8). Specific algorithms for the detection of such movements in the natural environment are currently developed by us and other groups (9–12).
While the usage of mobile technology is widely applied in humans, animal research also benefits from the technological development in movement and mobility assessment, as evidenced by behavioral measures in scientifically used animal models, animal husbandry, or the wild (13, 14). So far translation of quantitative data of movement and mobility from animal models to human has not been performed but it seems likely that quantitatively assessed movement changes in animals correspond to human diseases. Therefore, they may inform about (corresponding) human behavior.
In this review we aim to transfer pathophysiology of disturbed brain iron homeostasis in phenomenology of established PD and RLS animal models (Tables 1, 2). Our approach is based on accumulating evidence that iron accumulation in the basal ganglia leads to a PD-like, hypokinetic movement deficit, and lack of iron may be associated with a certain level of hyperkinetic movements resembling RLS features. We will discuss animal models associated with iron overload (25) and iron deficiency (26). This discussion should help defining a set of promising parameters to monitor changes of movement and mobility in a currently built mobile technology-based dataset obtained from the natural environment of PD patients under investigational treatment with an iron chelator (FAIR PARK II study, www.fairpark2.eu). FAIR PARK II is an investigator-initiated trial testing the effects of a conservative iron chelation therapy -i.e., chelation and redeployment of iron to limit iron sequestration and iron deficit- on the progression of handicap in PD.
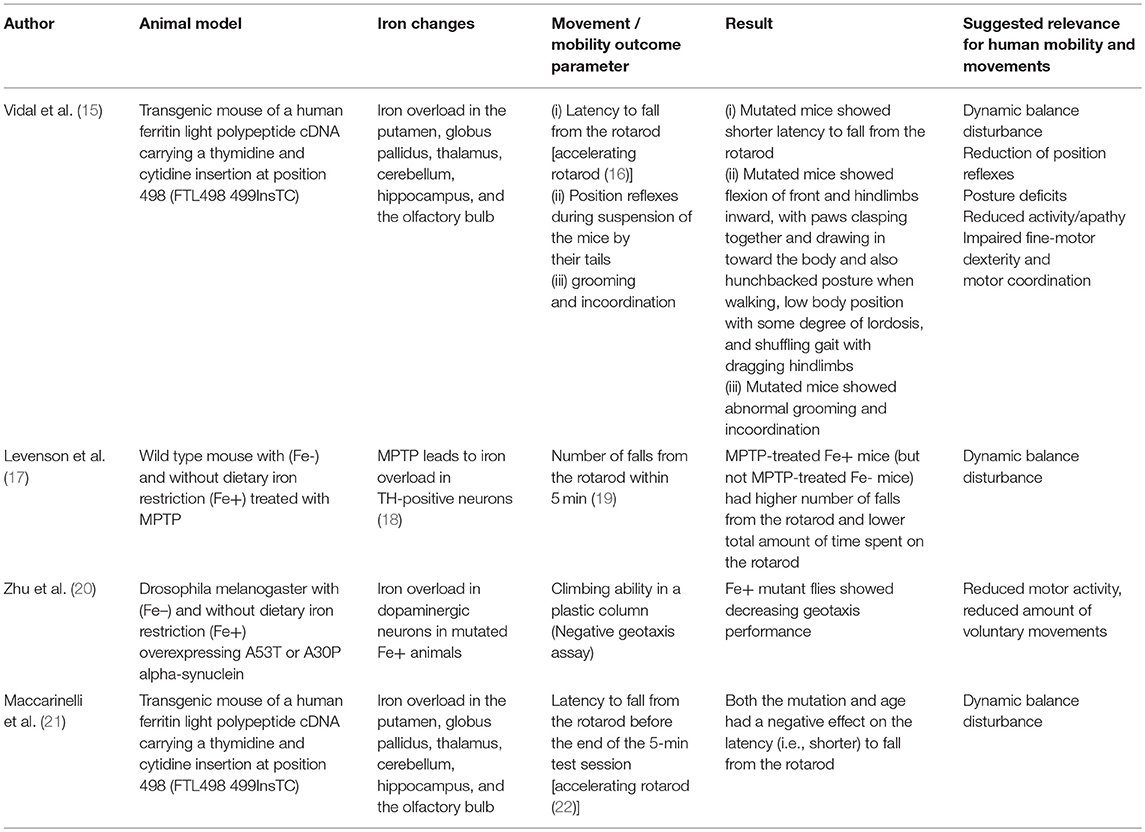
Table 1. Animal models for elevated brain iron.
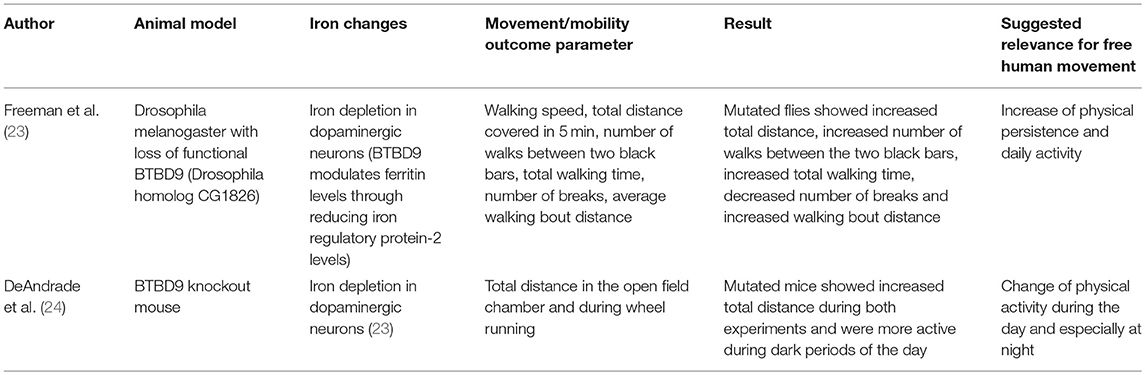
Table 2. Animal models for reduced brain iron.
Main Text
Iron in PD
Several post mortem and imaging (MRI and ultrasound) studies have shown that iron concentration is an important susceptibility factor for nigrostriatal degeneration and that basal ganglia iron concentration is higher in PD patients compared to controls (27, 28). Increased echogenicity of the substantia nigra (SN) on ultrasound is a typical sonographic finding in PD, and sonographic signal intensity of the SN is related to tissue iron content with higher iron levels being associated with increased echogenicity in both animal models and post mortem findings in human (29–31). Recent studies showed that mutant and wild-type alpha-synuclein may have differential interaction with iron and mutant alpha-synuclein toxicity could be preferentially exacerbated by iron. Iron overload could selectively influence mutant alpha-synuclein toxicity and disease phenotypes (32). Overall, the iron content is elevated in the SN of PD patients (33, 34). A conservative repositioning strategy of iron in an animal model using the iron chelator desferrioxamine retarded 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons (35). In PD application of deferiprone improved the motor part of the Unified PD rating scale (25).
Iron and Dopamine in PD
Although brain iron pathways and their relevance to PD are not entirely clarified, it is suggestive that iron is important in PD pathophysiology (25, 36). Iron is a significant co-factor in dopamine biosynthesis and is intricately involved in the regulation of dopamine levels in the brain. Several pathways are involved in dopamine metabolism. Iron concentration in neurons is high so that toxic dopamine metabolites occur (37).
In neurons a high iron content is detected in the cytoplasm. In this cellular compartment the formation of toxic dopamine metabolites takes place (38, 39). Iron accumulation is found in several brain regions in synucleinopathies whereat only specific regions are vulnerable for neuronal loss (40). Factors triggering neuronal loss associated are still poorly understood. Neuromelanin is produced in the substantia nigra, pars compacta SNc whereas dopamine is converted to o-quinones (41). Neurotoxic intermediates may result from iron mediated oxidation reactions. The interaction of iron and dopamine has been suggested as major player in the development of neuronal cell loss in defined brain regions (i.e., SNc).I. Neuromelanin as intracellular iron storage is of particular importance in PD (42). Accumulation and overload of iron suggested to increase oxidative stress has be demonstrated in the diseased PD brain (43, 44). The direct interaction of neuromelanin with the protein alpha-synuclein has also been accused to trigger neuronal damage. Alpha-synuclein gene dosages are higher in individual melanized neurons, triggering cell death to a greater extent in PD compared the healthy controls (45–47). Finally, microglia might be activated by neuromelanin leading to an outspread of neuronal degeneration in brain tissue (48).
Iron in RLS
RLS is the most frequent movement disorder with potentially severe disease burden due to physical and mental health. The clinical key features include sensory symptoms (restlessness, urge to move), unpleasant sensations (paresthesia, pain), and motor symptoms (periodic limb movements, further motor manifestations) (49). Low brain iron (reflected by, e.g., ferritin) despite normal peripheral iron plays a major role through its effects on the dopaminergic system (26, 50, 51). Cerebrospinal fluid (CSF) and serum ferritin are positively correlated. Low CSF ferritin is accompanied by low serum ferritin, particularly in cases diagnosed with iron deficiency anemia. RLS prevalence, as expected, is about five times more prevalent in patients with iron deficiency anemia than in the general population (52, 53). Iron deficiency anemia may predispose to the development of RLS but is no precondition as about 5% of the general population are affected from RLS (53). Beyond iron deficiency itself, the interaction of low CSF ferritin with other secondary biochemical changes and genetic factors lead to RLS symptoms. Iron replacement therapy shows inconsistent results so that general guidelines for treatment do not unambiguously commit to iron treatment (54, 55). Iron deficiency in brain may be related to a loss of iron regulatory protein activity, the function of blood-brain barrier, where endothelial cells act as iron reservoir for the brain. A dysfunction of iron regulatory proteins in the microvasculature might lead to a decrease of iron storage in endothelial cells.
Most probably, RLS is determined by environmental factors and genetical modifiers of brain iron homeostasis leading to a dysfunction of the cortico-striatal-thalamic-cortical circuit.
Iron and Dopamine in RLS
Using a post-weaning, diet-induced iron deficiency (ID) condition in rodents to reduce SN iron, studies have demonstrated that iron deficiency increases extracellular striatal dopamine, decreases striatal dopamine-2 receptor density, and diminishes dopamine transporter density and function in vitro but not dopamine release (56). Similar to the findings in the iron deficiency rodent, RLS striatal dopaminergic pathology has shown diminished dopamine-2 receptors (56). Iron deficiency in rodents lead to decreased ventral midbrain iron concentrations and to changes in the dopamine system that mimic many of the dopaminergic changes seen in RLS patient where low SN iron is a known pathology of the disease (56). It is well-known that RLS occurs with increased dopamine in the striatum (57, 58). Adding iron to the ventral midbrain leads to increased extra-cellular dopamine in the striatum (56). Adding iron to the striatum had no effect on its dopamine levels. Thus, the increase in iron seen in ventral midbrain with intravenous iron treatment seems to induce changes in striatal dopamine seen in RLS itself. Biochemical studies on the effect of iron in brain indicated that iron regulates hypoxia inducible co-factor pathways, leading to an upregulation of proteins that are involved in angiogenesis, erythropoiesis, cell survival, and proliferation. The interplay between compromised neuronal iron uptake and the functions of the neuromelanin-containing and dopaminergic cells might lead to RLS pathology (59). Decreased density of dopamine-1 and dopamine-2 receptors are detected in conditions of iron deficiency, indicating that iron affects the brain dopaminergic transmission in different ways (60). In RLS it has additionally been reported that A 10 and A11 dopaminergic neurons, localized in the hypothalamus, might be involved in disease mechanism (1). It has been suggested that these neurons are connected to the spinal cord in mice modulating spinal excitability leading to alterations. These changes lead to disturbed sensory processing of leg afferents projecting in brain stem structures (2).
Iron dyshomeostasis in SN is characteristic for both PD and RLS. The misdistribution of iron leads to iron deposits in PD and iron deficiency in RLS, presenting mechanistically a “relatively inverse” picture between PD and RLS. However, the picture may be much more complex because the misdistribution can also lead to both deposits and deficiency at the level of cell compartments (e.g., mitochondria vs. cytoplasma), cells (neurons vs. astrocytes and microglia) and tissue (e.g., SN vs. striatum and other regions). The complex picture can explain why some patients present both PD and RLS (61).
Animal Models for Movement Disorders Associated With Iron Metabolism
Based on the assumption that animal models associated with iron dyshomeostasis in the brain, more specifically in the basal ganglia, may have the potential to report about mobility and movement patterns that may also be altered by humans affected by the “corresponding” disease, we searched PubMed for studies investing such animal models and including quantitative data on mobility and movement patterns, and have been published until September 2018 (Tables 1, 2). We used the following search terms: “animal + iron,” “Parkinson disease iron animal model,” “Parkinson disease iron mouse model,” “Parkinson disease iron rat model,” “Parkinson disease iron drosophila model,” “Parkinson disease iron activity levels mouse,” “Iron dopaminergic system mouse model,” “iron metabolism mouse model,” “Restless legs animal model,” “Restless legs iron mouse model,” “Restless legs iron rat model,” “Restless legs iron drosophila model,” and “Restless legs iron activity levels mouse.” Articles were then checked for duplicates and for fulfilling the inclusion criteria (animal model, direct/indirect evidence of altered iron content in the basal ganglia, quantitative movement, and mobility assessment). We identified nine studies whereof six studies fulfilled these inclusion criteria and were thus included in the final evaluation (Tables 1, 2). Four studies investigated models with iron overload (15, 17, 20, 21) and two studies investigated models with iron deficiency (23, 24). All but two [drosophila melanogaster (20, 23)] were mouse models. Two studies used the identical mouse mutant (15, 21). The six studies are presented in following in more detail.
Motor Behavior in Animal Models With Elevated Brain Iron Content
Old neuroferritinopathy mutant mice, which have increased accumulation of iron aggregates in the striatum among other brain regions, showed a shorter latency to fall from the rotarod compared to WT mice (21) (Table 1). These mutant mice presented with a progressive behavioral and motor phenotype, consisting of abnormal grooming, tremor, posture, and incoordination (21). Feet clasping was observed after suspension of the mice by their tails. Moreover, they flexed their front and hindlimbs inward, with paws clasped together and drawn in toward the body. In contrast, WT mice demonstrated normal limb posture when suspended by their tails. Toward the end of the life span, mutant mice displayed a hunchbacked posture when walking and low body position with some degree of lordosis. They also showed a shuffling gait with dragging hindlimbs, suggestive of a reduction of position reflexes. In these mice, iron was elevated in multiple brain regions but highly accumulated in the dorsal striatum that is in humans known to mediate motor and executive functions (e.g., inhibitory control). The connection between the location of the damage and observed mobility deficits possess a degree of similarity to motor symptoms occurring in PD.
In another study using the same transgenic mice, the analysis of motor performance on a rotating rod revealed a significant decrease in performance at the earliest age tested (4 months), before an obvious clinical phenotype was observed (Table 1). This effect was somewhat stronger after 13 months of age, suggesting indirect iron-dependent disturbances in dynamic balance (15). Accordingly, rise of iron deposits within a brain region and across regions was positively associated with disease progression in PD patients (62). Taken together, the observed changes in mobility and movement performance of this mutant mouse strain reflect dynamic balance disturbance and motor coordination, reduced position reflexes, postural deficits, and reduced physical activity. Of note, the best surrogate parameter currently known for dynamic balance is gait variability, which deterioration is associated with increased frequency of falls in PD (63). This parameter (63, 64) as well as physical activity (65, 66) can be measured with high accuracy using mobile technology.
In a study aiming to evaluate the role of dietary iron restriction in a mouse model of PD (Table 1), mice were fed diets containing low (Fe-, 4 ppm) or adequate (Fe+, 48 ppm) amounts of iron for 6 weeks before the administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (17). MPTP is a mitochondrial toxin that damages nigrostriatal dopaminergic neurons and induces PD-like symptoms. Administration of MPTP resulted in reduced levels of dopamine and its metabolites in the striatum of Fe+ mice but not Fe- mice. Consistently, MPTP impaired postural control as determined by the mean number of falls from the rotarod and total time spent on the rotarod only in Fe+ mice, but not Fe– mice (17). Iron therefore seems to favor formation of cell death in face of other factors damaging dopaminergic neurons. In conclusion, this study suggests that even in non-mutant animal models reflecting (at least some aspects of) PD, iron homeostasis influences the motor phenotype -more specifically dynamic balance- and that dietary iron restriction has a beneficial effect.
Another study used a drosophila melanogaster model overexpressing A53T or A30P alpha-synuclein with (Fe–) and without dietary iron restriction (Fe+) (Table 1) (20). The authors found an iron overload in dopaminergic neurons only in mutated Fe+ animals but not in all other groups (including the mutated Fe– group). The mutated flies without iron restriction also performed worse in a negative geotaxis assay (a plastic column where the flies had to climb above a certain line), suggestive of reduced motor activity and/or reduced voluntary movements. This study also demonstrates that iron overload, in combination with additional factors (here PD-modulating genetic mutations), can promote degeneration of dopaminergic neurons. These results fit well-with the above observations of reduced physical activity, although based on these results it is difficult to exclude potentially contributing symptoms such as apathy and fatigue.
Motor Behavior in Animal Models With Reduced Brain Iron Content
In BTBD9 knockout mice, a RLS model, physical activity levels, sensory perception, sleep architecture were determined (Table 2) (24). Mutant mice showed a significant increase in physical activity compared with WT mice. Interestingly, the mutant mice did not show sensory deficits during the middle of the night (the active phase) but during the middle of the day (the rest phase), suggesting an alteration of the circadian rhythm. To analyze sleep architecture, the mice were implanted with a wireless telemetry system. Mutated mice showed decreased overall sleep time but no change in the amount of rapid eye movement sleep (24). Although the pathophysiological aspects of the above-mentioned association are, to our best knowledge, not yet understood, these mobility, and movement alterations are interesting in the light of mobile technology applied to men. This novel technology is able to evaluate physical activity and sleep performance such as movements during sleep (67).
In a RLS drosophila melanogaster model with loss of functional BTBD9, increased locomotor activity was found in a camera-based assessment of total walking distance, number of walks, total walking time, number of breaks, and average walking bout distance (Table 2) (23). Moreover, the mutated animal model presented with increased sleep fragmentation, including shorter, and more frequent sleeping bouts. The authors hypothesized that these phenomenon are due to the mediation of different sleep and activity phenotypes by BTBD9 (via the alteration of neurotransmission in some dopaminergic neurons (23). Whatever the reasons for the observed phenotypes are, mobile technology can readily and accurately assess such movement patterns in humans (68).
Conclusion
Although we tried to comprehensively analyze/discuss the articles according to our hypothesis, a not negligible limitation of our review is that we could only include six studies to our analysis which fulfilled the inclusion criteria. We tried to set the review in perspective with the state-of-the art methodology in analyzing the influence of iron on mobility in animal models.
Our systematic evaluation of the currently available peer-reviewed studies assessing movement and mobility in animal models with altered brain iron suggests that elevated basal ganglia iron leads to a decrease in motor performance, particularly affecting postural, and trunk stability (Figure 1). Low basal ganglia iron levels were mainly associated with increased physical activity, mobility, and sleep fragmentation as well as with changes of the circadian rhythm (Figure 1). The above-mentioned mobility and movement parameters may be particularly promising to assess effects of conservative iron repositioning on movement and mobility in PD.
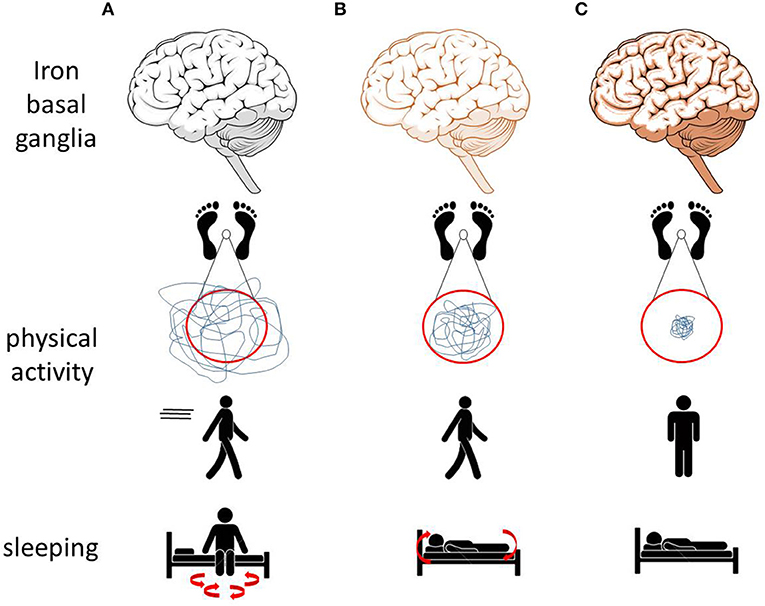
Figure 1. Effects of altered brain (especially basal ganglia) iron homeostasis on movements and mobility observed in animal models may translate to similar alterations in men. (A) Low iron levels in animal models are associated with increased physical activity and with sleep fragmentation. (B) Normal iron levels are associated with normal physical activity, normal sleep behavior, and the ability to move during sleep. (C) Increased iron levels are associated with reduced physical activity, postural and trunk instability, and immobility while sleeping (69, 70).
Author Contributions
FH, MH, WM, MP, and DB drafting and revision of manuscript, Fair-Park II study group. FH, WM, DD, CH, CMa, and CMo study concept and design. FH, MH, WM, DD, CH, and CMo interpretation of data.
Funding
The study was supported by the EU project FAIR-PARK II, funded under the Horizon2020 Program of the European commission (grant N° 633190, PHC13 2014–2015; NCT02655315), intramural funding of the Department of Neurology, Kiel University and the Thiemann foundation. We acknowledge financial support by Land Schleswig-Holstein within the funding program Open Access Publikationsfonds.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
PD, Parkinson's disease; RLS, restless legs syndrome; ID, iron deficiency; CSF, Cerebrospinal fluid; SN, substantia nigra; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
References
2. Artusi CA, Mishra M, Latimer P, Vizcarra JA, Lopiano L, Maetzler W, et al. Integration of technology-based outcome measures in clinical trials of Parkinson and other neurodegenerative diseases. Parkinson Relat Disord. (2018) 46(Suppl 1):S53–6. doi: 10.1016/j.parkreldis.2017.07.022
3. Espay AJAJ, Bonato P, Nahab FBFB, Maetzler W, Dean JMJM, Klucken J, et al. Technology in Parkinson's disease: challenges and opportunities. Mov Disord. (2016) 31:1272–82. doi: 10.1002/mds.26642
4. Haertner LM, Elshehabi M, Zaunbrecher L, Pham HM, Maetzler C, Van Uem JMT, et al. Effect of fear of falling on turning performance in parkinson's disease in the lab and at home. Front Aging Neurosci. (2018) 10:78. doi: 10.3389/fnagi.2018.00078
5. Dorsey ER, Glidden AM, Holloway MR, Birbeck GL, Schwamm LH. Teleneurology and mobile technologies: the future of neurological care. Nat Rev Neurol. (2018) 14: 285–97. doi: 10.1038/nrneurol.2018.31
6. Sánchez-Ferro Á, Elshehabi M, Godinho C, Salkovic D, Hobert MAMA, Domingos J, et al. New methods for the assessment of Parkinson's disease (2005 to 2015): a systematic review. Mov Disord. (2016) 31:1283–92. doi: 10.1002/mds.26723
7. van Lummel RC, Walgaard S, Pijnappels M, Elders PJM, Garcia-Aymerich J, van Dieën JH, et al. Physical performance and physical activity in older adults: associated but separate domains of physical function in old age. PLoS ONE. (2015) 10:e0144048. doi: 10.1371/journal.pone.0144048
8. Maetzler W, Domingos J, Srulijes K, Ferreira JJJJ, Bloem BRBR. Quantitative wearable sensors for objective assessment of Parkinson's disease. Mov Disord. (2013) 28:1628–37. doi: 10.1002/mds.25628
9. Zhang W, Regterschot GRH, Geraedts H, Baldus H, Zijlstra W. Chair rise peak power in daily life measured with a pendant sensor associates with mobility, limitation in activities, and frailty in old people. IEEE J Biomed Heal Inform. (2017) 21:211–7. doi: 10.1109/JBHI.2015.2501828
10. Del Din S, Godfrey A, Rochester L. Validation of an accelerometer to quantify a comprehensive battery of gait characteristics in healthy older adults and parkinson's disease: toward clinical and at home use. IEEE J Biomed Heal Inform. (2016) 20:838–47. doi: 10.1109/JBHI.2015.2419317
11. Salarian A, Russmann H, Wider C, Burkhard PR, Vingerhoets FJG, Aminian K. Quantification of tremor and bradykinesia in Parkinson's disease using a novel ambulatory monitoring system. IEEE Trans Biomed Eng. (2007) 54:313–22. doi: 10.1109/TBME.2006.886670
12. Pham MH, Elshehabi M, Haertner L, Heger T, Hobert MA, Faber GS, et al. Algorithm for turning detection and analysis validated under home-like conditions in patients with parkinson's disease and older adults using a 6 degree-of-freedom inertial measurement unit at the lower back. Front Neurol. (2017) 8:135. doi: 10.3389/fneur.2017.00135
13. Pasquet MO, Tihy M, Gourgeon A, Pompili MN, Godsil BP, Léna C, et al. Wireless inertial measurement of head kinematics in freely-moving rats. Sci Rep. Nat Publishing Group. (2016) 6:35689. doi: 10.1038/srep35689
14. Sauerbrei BA, Lubenov EV, Siapas AG. Structured variability in purkinje cell activity during locomotion. Neuron. (2015) 87:840–52. doi: 10.1016/j.neuron.2015.08.003
15. Vidal R, Miravalle L, Gao X, Barbeito AG, Baraibar MA, Hekmatyar SK, et al. Expression of a mutant form of the ferritin light chain gene induces neurodegeneration and iron overload in transgenic mice. J Neurosci. (2008) 28:60–7. doi: 10.1523/JNEUROSCI.3962-07.2008
16. Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. (1997) 17:7385–95. doi: 10.1523/JNEUROSCI.17-19-07385.1997
17. Levenson CW, Cutler RG, Ladenheim B, Cadet JL, Hare J, Mattson MP. Role of dietary iron restriction in a mouse model of Parkinson's disease. Exp Neurol. (2004) 190:506–14. doi: 10.1016/j.expneurol.2004.08.014
18. You L-H, Li F, Wang L, Zhao S-E, Wang S-M, Zhang L-L, et al. Brain iron accumulation exacerbates the pathogenesis of MPTP-induced Parkinson's disease. Neuroscience. (2015) 284:234–46. doi: 10.1016/j.neuroscience.2014.09.071
19. Duan W, Zhu X, Ladenheim B, Yu Q, Guo Z, Oyler J, et al. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol. (2002) 52:597–606. doi: 10.1002/ana.10350
20. Zhu Z-J, Wu K-C, Yung W-H, Qian Z-M, Ke Y. Differential interaction between iron and mutant alpha-synuclein causes distinctive Parkinsonian phenotypes in Drosophila. Biochim Biophys Acta. (2016) 1862:518–25. doi: 10.1016/j.bbadis.2016.01.002
21. Maccarinelli F, Pagani A, Cozzi A, Codazzi F, Di Giacomo G, Capoccia S, et al. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol Dis. (2015) 81:119–33. doi: 10.1016/j.nbd.2014.10.023
22. Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. (1999) 835:18–26.
23. Freeman A, Pranski E, Miller RD, Radmard S, Bernhard D, Jinnah HA, et al. Sleep fragmentation and motor restlessness in a drosophila model of restless legs syndrome. Curr Biol. (2012) 22:1142–8. doi: 10.1016/j.cub.2012.04.027
24. DeAndrade MP, Johnson RL, Unger EL, Zhang L, van Groen T, Gamble KL, et al. Motor restlessness, sleep disturbances, thermal sensory alterations and elevated serum iron levels in Btbd9 mutant mice. Hum Mol Genet. (2012) 21:3984–92. doi: 10.1093/hmg/dds221
25. Moreau C, Duce JA, Rascol O, Devedjian J-C, Berg D, Dexter D, et al. Iron as a therapeutic target for Parkinson's disease. Mov Disord. (2018) 33:568–74. doi: 10.1002/mds.27275
26. Trenkwalder C, Paulus W. Restless legs syndrome: pathophysiology, clinical presentation and management. Nat Rev Neurol. (2010) 6:337–46. doi: 10.1038/nrneurol.2010.55
27. Ryvlin P, Broussolle E, Piollet H, Viallet F, Khalfallah Y, Chazot G. Magnetic resonance imaging evidence of decreased putamenal iron content in idiopathic Parkinson's disease. Arch Neurol. (1995) 52:583–8. doi: 10.1001/archneur.1995.00540300057013
28. Ruprecht-Dörfler P, Berg D, Tucha O, Benz P, Meier-Meitinger M, Alders GL, et al. Echogenicity of the substantia nigra in relatives of patients with sporadic Parkinson's disease. Neuroimage. (2003) 18:416–22. doi: 10.1016/S1053-8119(02)00035-6
29. Berg D, Grote C, Rausch WD, Mäurer M, Wesemann W, Riederer P, et al. Iron accumulation in the substantia nigra in rats visualized by ultrasound. Ultrasound Med Biol. (1999) 25: 901–4. doi: 10.1016/S0301-5629(99)00046-0
30. Berg D, Roggendorf W, Schröder U, Klein R, Tatschner T, Benz P, et al. Echogenicity of the substantia nigra: association with increased iron content and marker for susceptibility to nigrostriatal injury. Arch Neurol. (2002) 59:999–1005. doi: 10.1001/archneur.59.6.999
31. Zecca L, Berg D, Arzberger T, Ruprecht P, Rausch WD, Musicco M, et al. In vivo detection of iron and neuromelanin by transcranial sonography: a new approach for early detection of damage. Mov Disord. (2005) 20:1278–85. doi: 10.1002/mds.20550
32. Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. (2000) 20:6048–54. doi: 10.1523/JNEUROSCI.20-16-06048.2000
33. Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain. (1991) 114(Pt 4):1953–75. doi: 10.1093/brain/114.4.1953
34. Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MB. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem. (1991) 56:978–82. doi: 10.1111/j.1471-4159.1991.tb02017.x
35. Ben-Shachar D, Eshel G, Finberg JP, Youdim MB. The iron chelator desferrioxamine (Desferal) retards 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons. J Neurochem. (1991) 56:1441–4. doi: 10.1111/j.1471-4159.1991.tb11444.x
36. Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P, et al. Brain iron pathways and their relevance to Parkinson's disease. J Neurochem. (2001) 79:225–36. doi: 10.1046/j.1471-4159.2001.00608.x
37. Santiago M, Matarredona ER, Granero L, Cano J, Machado A. Neurotoxic relationship between dopamine and iron in the striatal dopaminergic nerve terminals. Brain Res. (2000) 858:26–32. doi: 10.1016/S0006-8993(99)02485-3
38. Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. (2012) 31:3038–62. doi: 10.1038/emboj.2012.170
39. Hare DJ, Lei P, Ayton S, Roberts BR, Grimm R, George JL, et al. An iron–dopamine index predicts risk of parkinsonian neurodegeneration in the substantia nigra pars compacta. Chem Sci. Royal Society of Chemistry. (2014) 5:2160–9. doi: 10.1039/C3SC53461H
40. McCann H, Cartwright H, Halliday GM. Neuropathology of α-synuclein propagation and braak hypothesis. Mov Disord. (2016) 31:152–60. doi: 10.1002/mds.26421
41. Zucca FA, Segura-Aguilar J, Ferrari E, Muñoz P, Paris I, Sulzer D, et al. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog Neurobiol. (2017) 155:96–119. doi: 10.1016/j.pneurobio.2015.09.012
42. Bohic S, Murphy K, Paulus W, Cloetens P, Salomé M, Susini J, et al. Intracellular chemical imaging of the developmental phases of human neuromelanin using synchrotron X-ray microspectroscopy. Anal Chem. (2008) 80:9557–66. doi: 10.1021/ac801817k
43. Double KL, Gerlach M, Schünemann V, Trautwein AX, Zecca L, Gallorini M, et al. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem Pharmacol. (2003) 66:489–94. doi: 10.1016/S0006-2952(03)00293-4
44. Faucheux BA, Martin M-E, Beaumont C, Hauw JJ, Agid Y, Hirsch EC. Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson's disease. J Neurochem. (2003) 86:1142–8. doi: 10.1046/j.1471-4159.2003.01923.x
45. Halliday GM, Ophof A, Broe M, Jensen PH, Kettle E, Fedorow H, et al. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson's disease. Brain. (2005) 128:2654–64. doi: 10.1093/brain/awh584
46. Li J, Yang J, Zhao P, Li S, Zhang R, Zhang X, et al. Neuromelanin enhances the toxicity of α-synuclein in SK-N-SH cells. J Neural Transm. (2012) 119:685–91. doi: 10.1007/s00702-011-0753-z
47. Xuan Q, Xu S-L, Lu D-H, Yu S, Zhou M, Uéda K, et al. Increased expression of α-synuclein in aged human brain associated with neuromelanin accumulation. J Neural Transm. (2011) 118:1575–83. doi: 10.1007/s00702-011-0636-3
48. Karlsson O, Lindquist NG. Melanin affinity and its possible role in neurodegeneration. J Neural Transm. (2013) 120:1623–30. doi: 10.1007/s00702-013-1062-5
49. Allen R, Picchietti DL, Garcia-Borreguero D, Ondo WG, Walters AS, Winkelman JW. Restless legs syndrome/Willis–Ekbom disease diagnostic criteria: updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria–history. Sleep Med. 15:860–73. doi: 10.1016/j.sleep.2014.03.025
50. Connor JR, Ponnuru P, Wang X-S, Patton SM, Allen RP, Earley CJ. Profile of altered brain iron acquisition in restless legs syndrome. Brain. (2011) 134:959–68. doi: 10.1093/brain/awr012
51. Connor JR, Boyer PJ, Menzies SL, Dellinger B, Allen RP, Ondo WG, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology. (2003) 61:304–9. doi: 10.1212/01.WNL.0000078887.16593.12
52. Akyol A, Kiylioglu N, Kadikoylu G, Bolaman AZ, Ozgel N. Iron deficiency anemia and restless legs syndrome: is there an electrophysiological abnormality? Clin Neurol Neurosurg. (2003) 106:23–7. doi: 10.1016/j.clineuro.2003.07.004
53. Allen RP, Auerbach S, Bahrain H, Auerbach M, Earley CJ. The prevalence and impact of restless legs syndrome on patients with iron deficiency anemia. Am J Hematol. (2013) 88:261–4. doi: 10.1002/ajh.23397
54. Grote L, Leissner L, Hedner J, Ulfberg J. A randomized, double-blind, placebo controlled, multi-center study of intravenous iron sucrose and placebo in the treatment of restless legs syndrome. Mov Disord. (2009) 24:1445–52. doi: 10.1002/mds.22562
55. Earley CJ, Horská A, Mohamed MA, Barker PB, Beard JL, Allen RP. A randomized, double-blind, placebo-controlled trial of intravenous iron sucrose in restless legs syndrome. Sleep Med. (2009) 10:206–11. doi: 10.1016/j.sleep.2007.12.006
56. Unger EL, Bianco LE, Jones BC, Allen RP, Earley CJ. Low brain iron effects and reversibility on striatal dopamine dynamics. Exp Neurol. (2014) 261:462–8. doi: 10.1016/j.expneurol.2014.06.023
57. Connor JR, Wang X-S, Allen RP, Beard JL, Wiesinger JA, Felt BT, et al. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain. (2009) 132:2403–12. doi: 10.1093/brain/awp125
58. Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas RE, Brašić JR, et al. Increased synaptic dopamine in the putamen in restless legs syndrome. Sleep. (2013) 36:51–7. doi: 10.5665/sleep.2300
59. Michaud M, Dumont M, Selmaoui B, Paquet J, Fantini ML, Montplaisir J. Circadian rhythm of restless legs syndrome: relationship with biological markers. Ann Neurol. (2004) 55:372–80. doi: 10.1002/ana.10843
60. Dauvilliers Y, Winkelmann J. Restless legs syndrome: update on pathogenesis. Curr Opin Pulm Med. (2013) 19:594–600. doi: 10.1097/MCP.0b013e328365ab07
61. Lee SH, Lyoo CH, Ahn SJ, Rinne JO, Lee MS. Brain regional iron contents in progressive supranuclear palsy. Parkinson Relat Disord. (2017) 45:28–32. doi: 10.1016/j.parkreldis.2017.09.020
62. Langkammer C, Pirpamer L, Seiler S, Deistung A, Schweser F, Franthal S, et al. Quantitative Susceptibility Mapping in Parkinson's Disease. Kassubek J, editor. PLoS ONE. (2016) 11:e0162460. doi: 10.1371/journal.pone.0162460
63. Henderson EJ, Lord SR, Brodie MA, Gaunt DM, Lawrence AD, Close JCT, et al. Rivastigmine for gait stability in patients with Parkinson's disease (ReSPonD): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. (2016) 15:249–58. doi: 10.1016/S1474-4422(15)00389-0
64. Pham MH, Elshehabi M, Haertner L, Din SD, Srulijes K, Heger T, et al. Validation of a step detection algorithm during straight walking and turning in Patients with Parkinson's disease and older adults using an inertial measurement unit at the lower back. Front Neurol. (2017) 8:457. doi: 10.3389/fneur.2017.00457
65. van Uem JMT, Cerff B, Kampmeyer M, Prinzen J, Zuidema M, Hobert MA, et al. The association between objectively measured physical activity, depression, cognition, and health-related quality of life in Parkinson's disease. Parkinson Relat Disord. (2018) 48:74–81. doi: 10.1016/j.parkreldis.2017.12.023
66. Lord S, Godfrey A, Galna B, Mhiripiri D, Burn D, Rochester L. Ambulatory activity in incident Parkinson's: more than meets the eye? J Neurol. (2013) 260:2964–72. doi: 10.1007/s00415-013-7037-5
67. Louter M, Maetzler W, Prinzen J, van Lummel RCRC, Hobert M, Arends JBAMJBAM, et al. Accelerometer-based quantitative analysis of axial nocturnal movements differentiates patients with Parkinson's disease, but not high-risk individuals, from controls. J Neurol Neurosurg Psychiatry. (2014) 86:32–7. doi: 10.1136/jnnp-2013-306851
68. Schlachetzki JCM, Barth J, Marxreiter F, Gossler J, Kohl Z, Reinfelder S, et al. Wearable sensors objectively measure gait parameters in Parkinson's disease. Toft M, editor. PLoS ONE. (2017) 12:e0183989. doi: 10.1371/journal.pone.0183989
69. Qu S, Ondo WG, Zhang X, Xie WJ, Pan TH, Le WD. Projections of diencephalic dopamine neurons into the spinal cord in mice. Exp Brain Res. (2006) 168:152–6. doi: 10.1007/s00221-005-0075-1
Keywords: Parkinson's disease, restless legs syndrome, mobility assessment, iron, animal models
Citation: Hopfner F, Hobert MA, Maetzler C, Hansen C, Pham MH, Moreau C, Berg D, Devos D, Maetzler W and The Fair-Park II study group (2019) Mobility Deficits Assessed With Mobile Technology: What Can We Learn From Brain Iron-Altered Animal Models? Front. Neurol. 10:833. doi: 10.3389/fneur.2019.00833
Received: 10 April 2019; Accepted: 18 July 2019;
Published: 08 August 2019.
Edited by:
Antonio Pisani, University of Rome Tor Vergata, ItalyReviewed by:
Thomas Klopstock, Ludwig Maximilian University of Munich, GermanyJong-Min Kim, Seoul National University Bundang Hospital, South Korea
Copyright © 2019 Hopfner, Hobert, Maetzler, Hansen, Pham, Moreau, Berg, Devos, Maetzler and The Fair-Park II study group. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franziska Hopfner, Zi5ob3BmbmVyQG5ldXJvbG9naWUudW5pLWtpZWwuZGU=