 Hamdollah Panahpour1,2†
Hamdollah Panahpour1,2† Nicole A. Terpolilli1,3,4†
Nicole A. Terpolilli1,3,4† David Schaffert5
David Schaffert5 Carsten Culmsee6
Carsten Culmsee6 Nikolaus Plesnila1,4*
Nikolaus Plesnila1,4*- 1Laboratory of Experimental Stroke Research, Institute for Stroke and Dementia Research (ISD), Munich University Hospital, Munich, Germany
- 2Department of Physiology, Medical School, Ardabil University of Medical Sciences, Ardabil, Iran
- 3Department of Neurosurgery, Munich University Hospital, Munich, Germany
- 4Munich Cluster for Systems Neurology (SyNergy), Munich, Germany
- 5Pharmaceutical Biotechnology, Department of Pharmacy, Ludwig-Maximilians University, Munich, Germany
- 6Institute for Pharmacology and Clinical Pharmacy, University of Marburg, Marburg, Germany
Epidemiological studies suggest that pharmacological reduction of systemic hypertension lowers incidence and severity of stroke. However, whether the reduction of blood pressure per se or the compounds used to reduce hypertension are responsible for this effect received little attention. In the current study we therefore aimed to investigate whether Aliskiren, a renin-inhibitor used to treat arterial hypertension, may improve outcome in a mouse model of ischemic stroke when applied centrally and in a dose not affecting blood pressure. Male C57BL/6 mice received 0.6, 2.0, or 6.0 μg Aliskiren or vehicle by intracerebroventricular injection as a pre-treatment and were then subjected to 60 min of middle cerebral artery occlusion (MCAo). Infarct volume, brain edema formation, mortality, antioxidant effects, and functional outcome were assessed up to seven days after MCAo. Central administration of Aliskiren (0.6 or 2.0 μg) had no effect on systemic blood pressure but significantly reduced infarct volume and brain edema formation, blunted mortality, and improved neurological outcome up to 1 week after MCAo. Due to the central and prophylactic administration of the compound, we cannot make any conclusions about the potency of Aliskiren for acute stroke treatment, however, our study clearly demonstrates, that in addition to lowering blood pressure Aliskiren seems to have a direct neuroprotective effect. Hence, renin-inhibitors may be an effective addition to prophylactic treatment regimens in stroke patients.
Introduction
Epidemiological studies suggest that treating arterial hypertension reduces the incidence and the severity of ischemic and hemorrhagic stroke (1, 2). This effect is attributed to the reduction of hypertension, however, an alternative explanation could be that pharmacological compounds used to reduce systemic blood pressure may in addition also have neuroprotective effects.
One pathway that has been implicated in the pathophysiology of hypertension and ischemic stroke, is the renin-angiotensin-system (RAS) (3). The RAS plays a crucial role in the maintenance of blood pressure and blood volume. Drops in blood pressure or blood volume lead to secretion of renin, a protease, which hydrolyses angiotensinogen to angiotensin I (Ang I). Ang I is converted to Angiotensin II (Ang II), a strong vasoconstrictor, by angiotensin converting enzyme (ACE). Ang II activates Angiotensin 1 receptors (AT1R), which induce vasoconstriction, inflammatory changes and oxidative stress, and Angiotensin 2 receptors (AT2R) which mediate vasodilation via Angiotensin 1-7 (Ang1-7) and MAS receptors, the so called “alternative axis” (4).
Accordingly, the RAS reduces blood pressure via inhibition of AT1 receptors and/or activation of AT2 receptors, but may well deteriorate ischemic damage thorough vasoconstriction and the other actions of AT1 receptors (4). In fact, inhibition of ACE or angiotensin type 1 receptors were shown to reduce arterial hypertension and to effectively prevent cerebro-vascular events (5, 6), while also being protective after cerebral ischemia. Further, activation of the AT2 axis inferred neuroprotection after experimental stroke (7–9).
Despite this elegant work on the downstream members of the RAS, relatively little is known about the role of renin after stroke, the first step of the RAS-cascade, which is the rate-limiting enzyme of the whole system (10–12). The activity of renin can be inhibited by the small molecule Aliskiren, a clinically frequently used compound, which was shown to effectively reduce arterial hypertension (13–16), and its sequels, that is nephropathy (17, 18) and myocardial infarction (19, 20). With regard to cerebral ischemia, however, it is not known whether Aliskiren prevents stroke only by its blood pressure lowering effect or possibly also by a direct neuroprotective effect on the brain. To investigate this hypothesis we applied not blood pressure lowering doses of Aliskiren by intracerebroventricular injection to mice, subjected them to experimental stroke, and investigated infarct volume, brain edema formation, and neurological function for up to seven days thereafter.
Materials and Methods
All protocols used were in accordance with international guidelines, the Basel Declaration, and approved by the Government of Upper Bavaria (protocol number 55.2-1-54-2531-118-05). Results are reported according to the ARRIVE guidelines (21). Mice were randomly assigned to experimental groups by drawing lots. Surgical preparation, physiological monitoring, and data analysis were performed by an investigator (H.P.) blinded to the treatment of the animals.
Transient Cerebral Ischemia
Cerebral ischemia was induced by middle cerebral artery occlusion (MCAo) as previously described (22, 23). Briefly, after induction of isoflurane anesthesia (induction: 30 s at 4%, continuation with 1.0–1.2% in 30% O2 and 70% N2O), a silicone covered 8-0 monofilament (Ethilon®, Ethicon, Germany) was advanced intravascularly via the left common carotid artery and the left internal carotid artery (ICA) until it occluded the middle cerebral artery at the MCA bifurcation. Cerebral blood flow was continuously and non-invasively measured over the left MCA territory via Laser Doppler flowmetry (LDF, Perimed, Sweden) during the procedure. MCAo was confirmed by a drop of CBF to <20% of baseline. The filament was then fixed and animals were allowed to wake up from anesthesia. Later on animals were re-anesthetized and the filament was removed 60 min after the initiation of ischemia. Animals were kept in an incubator heated to 32°C for 6 h after surgery to avoid hypothermia.
Quantification of Infarct Volume
Twenty four hours or seven days after reperfusion mice were sacrificed in deep isoflurane anesthesia and brains were removed and frozen on powdered dry ice. Twelve sequential 10 μm coronal Nissl sections were prepared every 500 μm. Infarct volume was calculated as previously described by histomorphometry using a digital image analysis system (Olympus DP–Soft, Munich, Germany) (23–25). Infarct volumes were corrected for brain swelling and were calculated for the whole brain as well as for subcortical and cortical brain regions.
Measurement of Brain Edema
Brain water content was assessed as previously described (26): after brain removal 24 h after reperfusion olfactory bulb, cerebellum, and medulla oblongata were dissected, cerebral hemispheres divided and weighed (wet weight, WW). After drying the tissue at 110°C for 24 h the dry weight (DW) was obtained. Brain water content was calculated using the following formula: (WW–DW)/WW × 100.
Evaluation of Neurological Deficits
All neurological testing was performed by a researcher blinded to treatment type and group. Neurological deficits were assessed immediately before and 24 h after MCAo using a modified 6 point scale (Neurological deficit score, NDS) assessing spontaneous motor function and tail suspension; in the chronic group the testing was repeated every 24 h. Scoring was performed as described: 1 = Normal motor function (spontaneous, tail suspension), 2 = Normal spontaneous motor function, flexion of contralateral forelimb during tail suspension, 3 = Circling during tail suspension, 4 = Spontaneous circling, 5 = Leaning to contralateral side, loss of righting reflex, 6 = No spontaneous motor activity.
Drug Administration
The specific renin inhibitor Aliskiren was extracted from a commercially available formulation (Novartis, Basel, Switzerland), purified to >99%, and administered at a concentration of 0.6, 2.0, or 6.0 μg in 2 μl saline 45 min before induction of MCAo by intracerebroventricular (i.c.v.) administration (0.9 mm left and 0.1 mm posterior to the bregma; depth: 3.1 mm). Vehicle treated animals received 2 μl of saline (vehicle).
Assessment of Antioxidant Enzyme Activities and Lipid Peroxidation
Twenty four hours after induction of ischemia, animals were sacrificed. The ischemic hemisphere was dissected, weighed and homogenized (1:6) in phosphate buffered saline (pH 7.4) with 0.1 mM EDTA. After centrifugation at 10,000 g for 15 min at 4°C, the resulting supernatant was used for analysis. The concentration of malondialdehyde (MDA)was determined using a colorimetric assay kit (# 10009055, Cayman Chemicals, USA). The enzyme activities of superoxide dismutase (SOD, #706002), catalase (CAT, #707002), and glutathione peroxidase (GPX, #703102) in brain tissue homogenate were measured using respective assay kits (Cayman Chemicals, USA).
Experimental Groups
For dose finding (see Figure 1A for schematic drawing of the experimental setup) Aliskiren was injected i.c.v. in incrementing doses every 20 min (0.6, 1.4, and 4 μg drug in 2 μl physiological saline) to achieve the cumulative doses of 0.6, 2, and 6 μg in CSF; the control group received an equal volume of saline. Mean arterial blood pressure (MAP, measured using a femoral artery catheter), oxygen saturation, heart rate (measured by an oxygen saturation sensor at the hind paw), and body temperature (rectal temperature probe) were continuously measured. At the end of each experiment arterial blood samples were collected for blood gas analysis. Infarct volumes and neurological deficits were investigated 24 h after 60 min MCAo in four groups (n = 7 mice each; see Figure 1B): (1) Vehicle, (2) 0.6 μg Aliskiren, (3) 2 μg Aliskiren, (4) 6 μg Aliskiren. Treatment was administered i.c.v. 45 min before induction of MCAo. Brain edema formation and neurological outcome (Figure 1C) were assessed 24 h after 60 min MCAo in the following groups (n = 7 each): (1) Sham, (2) MCA occlusion only, (3) MCAo and Vehicle, MCAo and (4) 0.6 μg Aliskiren, (5) 2 μg Aliskiren, and (6) 6 μg Aliskiren; again, injections were administered 45 min before induction of MCAo. As both groups were operated under the same conditions, the results for neurological outcome are summarized (n = 7). Lipid peroxidation was assessed 24 h after MCAo induction in animals that received 2 μl Aliskiren or vehicle 45 min before induction of ischemia (n = 7 each, Figure 1D). Lastly, neurological function and bodyweight were assessed before and every 24 h after 60 min of MCA occlusion over 7 days; 45 min before induction of MCAo, Aliskiren (2 μg) in 2 μl or 2 μl saline were injected i.c.v. (n = 8 each group, Figure 1E).
Figure 1. Experimental setup. (A) Dose finding study. (B) Assessment of infarct volume and (C) brain water content 24 h after MCAo. (D) Measurement of antioxidant enzyme activity 24 h after MCAo. (E) Evaluation of outcome as assessed by Neurological Deficit Score for 7 days after MCAo.
Statistical Analysis
Sample size calculations were performed using SigmaStat (Version 13.0, Jandel Scientific, Erkrath, Germany) with the following parameters: alpha error = 0.05, beta error = 0.2, calculated standard deviation ranged from 15 to 20% (depending on the parameter investigated), and a biologically relevant difference of 30%. After performing normality and an equal variance tests the Mann-Whitney rank sum test was used for comparisons between two groups and Analysis of Variance (ANOVA) on ranks was performed for multi group comparisons. Differences between groups were considered significant at p < 0.05. All data are presented as mean ± standard error of the mean (SEM), if not indicated otherwise.
Results
Dose Finding
Repetitive injections of Aliskiren in increasing doses did not affect blood gas parameters (Table 1), heart rate, body temperature, or oxygen saturation (Table 2). Mean arterial blood pressure was slightly lowered after injection of a cumulative dose of 6 μg (Figure 2); higher doses (up to 20 μg, data not shown) induced a significant decrease of blood pressure. Therefore, the first set of experiments on neuroprotection were performed with a maximum dose of 6 μg.

Table 1. Blood gas analysis.
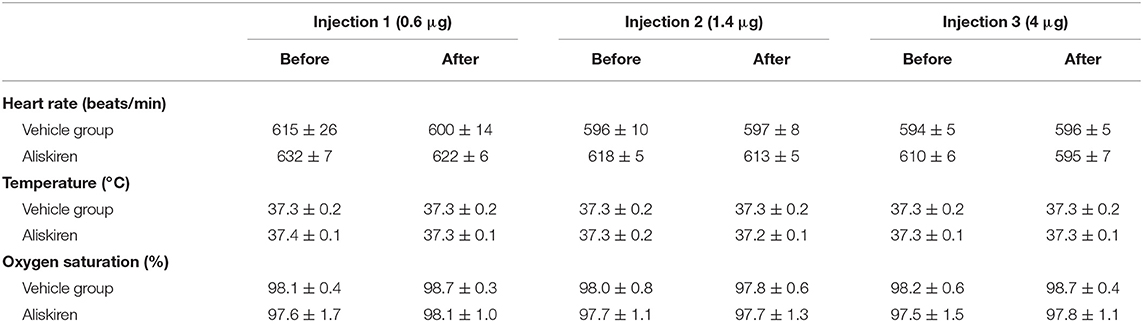
Table 2. Physiological parameters during dose finding.
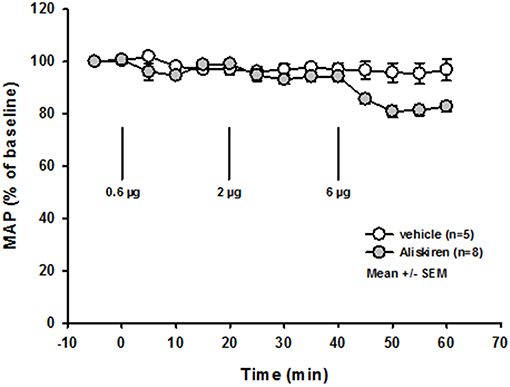
Figure 2. Dose finding. Mean arterial blood pressure is not affected by injection of increasing doses of Aliskiren; after a cumulative dose of 6 μg MAP tends to lower, therefore no higher concentration was used.
Ischemic Brain Damage 24 h After MCAo
Immediately after induction of filament occlusion of the left MCA, cerebral blood flow over the left MCA territory dropped below 20% of baseline cerebral perfusion in all groups (Figure 3A). Twenty four hours after 60 min of transient MCA ischemia, total infarct volume was significantly lower in the 0.6 and 2.0 μg Aliskiren groups as compared to vehicle treated controls (Figure 3B; vehicle: 91.7 ± 7.7 mm3; 0.6 μg Aliskiren: 58.9 ± 8.7 mm3, p < 0.003; 2 μg Aliskiren: 56.0 ± 7.1 mm3, p < 0.003; 6 μg Aliskiren: 63.9 ± 8.5 mm3, p = 0.165). Subcortical infarct volume (Figure 3C) was significantly decreased in both the 0.6 μg as well as the 2 μg Aliskiren group (p < 0.003 vs. vehicle control); cortical infarct volume was only significantly lower in the 2 μg Aliskiren group (Figure 3D, p < 0.008). Figure 3E shows exemplary coronal brain sections for all groups.
Figure 3. Effect of Aliskiren on post-ischemic brain damage 24 h after MCAo. (A) CBF after induction of MCAo. Cerebral blood flow drops to <20% of baseline immediately after filament occlusion of the MCA in all groups. (B–D) Infarct volumes 24 h after MCAo. (B) Total infarct volume is significantly reduced in the 0.6 and 2 μg Aliskiren treatment groups. (C) Subcortical infarct volume was also reduced in these groups while (D) cortical infarct volumes were only significantly smaller in the 2 μg group compared to vehicle treated animals. (E) Exemplary coronal Nissl sections 24 h after MCAo.
Brain water content in the right (non-ischemic) hemisphere (Figure 4A, left side) was not altered in the treatment or vehicle groups as compared to sham operated animals (sham) or animals that underwent MCA occlusion without i.c.v. injection (MCA only). MCA occlusion led to a significant increase of brain water content in the left hemisphere in all MCAo groups compared to sham operated animals indicating the formation of brain edema under these conditions (Figure 4A right side, p < 0.05 vs. sham). Brain edema formation was significantly reduced after 0.6 μg Aliskiren (p < 0.007 vs. vehicle); higher doses had no significant effect.
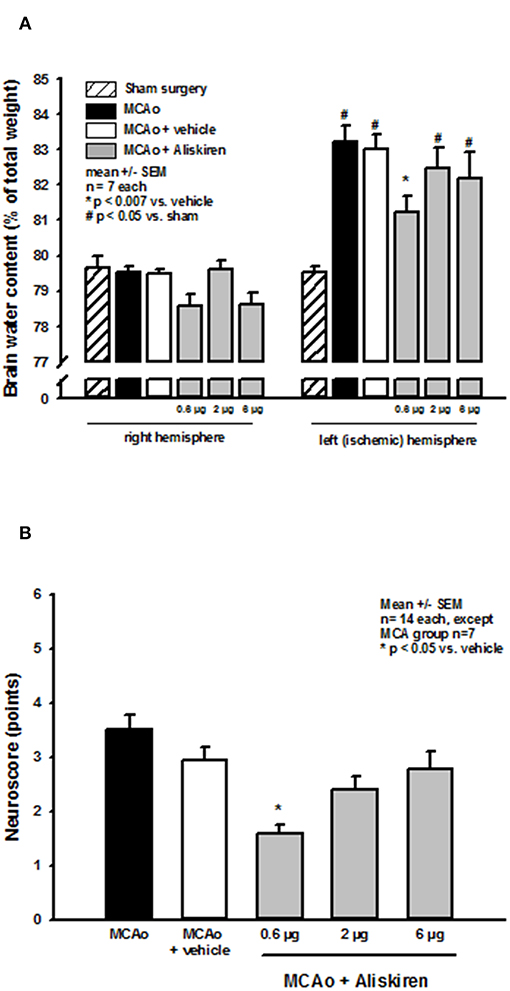
Figure 4. Brain edema formation and neurological outcome after renin inhibition. (A) Brain water content 24 h after MCAo. Brain water content in the non-ischemic right hemisphere of all MCAo groups was not altered compared to sham operated animals; all animals that underwent ischemia showed significant brain edema formation in the left, i.e., ischemic hemisphere. Brain water content was significantly reduced by application of 0.6 μg Aliskiren. (B) Neurological deficit score after renin inhibition. Twenty four hours after MCAo all animals had significant motor deficits; Aliskiren 0.6 μg significantly improved the neurological scores; higher doses tended to reduce the score but did not have a significant effect on the animals performance.
Effect of Renin Inhibition on Post-Ischemic Neurologic Deficits
MCAo lead to significant deterioration of motor function as assessed by the neurological deficit score; mice in the 0.6 μg Aliskiren group achieved significantly better scores than vehicle treated animals (Figure 4B). Higher doses did not improve neurological outcome.
Assessment of Lipid Peroxidation Enzyme Activity
Ischemia and reperfusion resulted in a significantly increased MDA concentration and reduced antioxidant enzyme activities in the ischemic hemispheres as compared to the sham group, consistent with enhanced lipid peroxidation after ischemic stroke. Treatment with Aliskiren significantly reduced the MDA concentration (p < 0.01) and elevated superoxide dismutase and glutathione peroxidase activities (p < 0.05 and p < 0.01 vs. vehicle group, respectively, Figure 5).
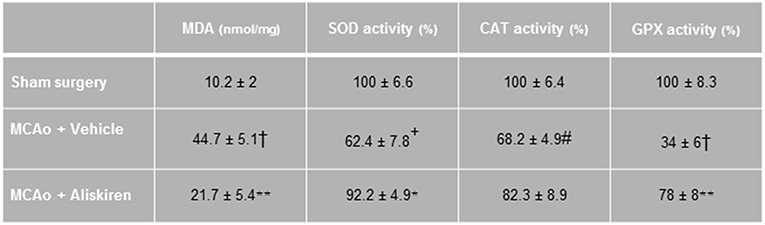
Figure 5. Effects of Aliskiren (2 μg) on lipid peroxidation and brain tissue antioxidant enzyme activities in brain tissue homogenate of studied groups 24 h after MCAO. MDA, malondialdehyde; SOD, superoxide dismutase; CAT, catalase; GPX, glutathione peroxidase (n = 7). Values are mean ± SEM. †p < 0.001, +p < 0.01, and #p < 0.05 compared with sham group; **p < 0.01 and *p < 0.05 compared with vehicle group.
Effects of Renin Inhibition on Outcome Parameters Over 7 Days After MCAo
Based on the observation that Aliskiren improved histopathological and functional outcome 24 h after experimental stroke, we wanted to evaluate if this effect lasted long-term, i.e., for at least 1 week. Since only 2 μg of Aliskiren reduced subcortical and cortical infarct volumes we decided to use this doe for these experiments.
Seven days after 60 min MCAo vehicle treated mice showed a mortality of 50% (Figure 6A). Interestingly, all mice receiving Aliskiren survived. The long-term protective effect of Aliskiren is also impressively exemplified by a less pronounced weight loss in the treatment group 3, 4, and 5 days after MCAo (Figure 6B) and by a significantly better functional outcome staring from day 2 after MCAo until the end of the observation period (Figure 6C).
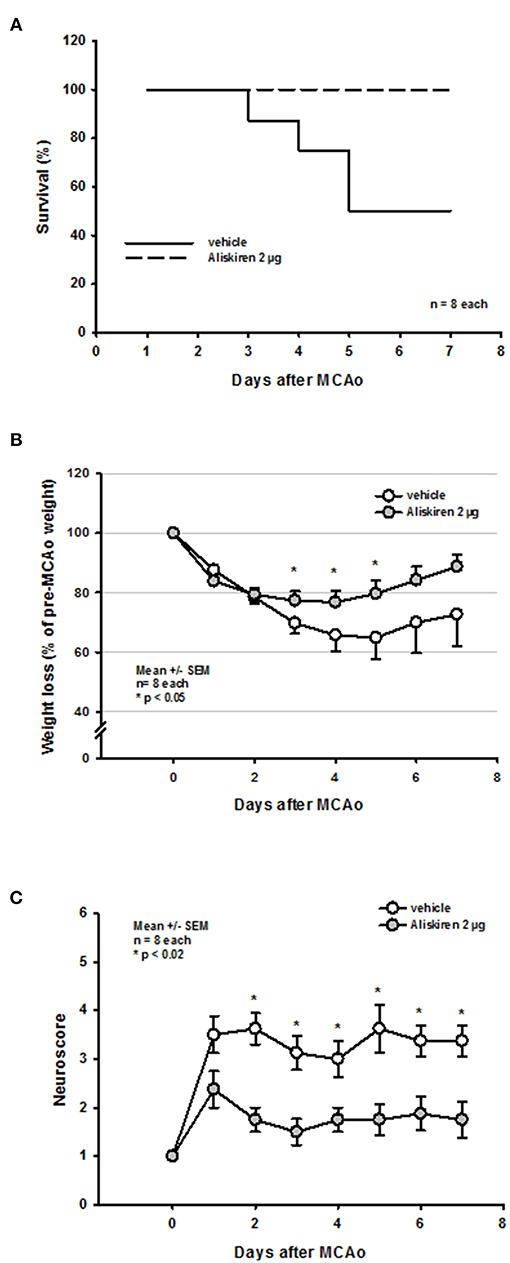
Figure 6. Functional outcome up to 7 days after MCAo. (A) While mortality in the vehicle group was 50% over time all Aliskiren animals survived. (B) Treatment animals recovered significantly quicker from post-ischemic weight loss. (C) Starting 2 days after MCAo, Aliskiren treated mice achieved significantly better scores for motor function.
Discussion
Here, we report that central application of the renin antagonist Aliskiren, a clinically used antihypertensive drug, protects the brain after experimental ischemic stroke independent of its blood pressure lowering activity. Aliskiren significantly attenuated brain edema formation and stroke volume, blunted mortality and improved neurological outcome in a murine stroke model. Hence, the clinically observed effect of Aliskiren on stroke outcome may not only be related to its effect on blood pressure, but also to a direct effect of renin on the pathophysiology of ischemia-induced brain damage.
In order to test the hypothesis of the current study, namely whether the antihypertensive drug Aliskiren may have effects on the ischemic brain beyond its actions on systemic blood pressure, we used the format of a pharmacological study. As a prerequisite for such a study type we tested multiple dosages, took care that the investigated compound reached the target organ (by applying Aliskiren by intracerebroventricular injection), carefully and extensively monitored animal physiology, and performed and analyzed all experiments in a strictly randomized and blinded fashion in order to avoid any investigator-induced bias. To test the short- and long-term effect of Aliskiren in an animal model of stroke we occluded the middle cerebral artery for 60 min using an intraluminal filament, a very well-established and standardized model in our laboratory (24, 26, 27). This was further proven by the fact that in the current study blood flow was reduced to ischemic levels (<20% of baseline) in all investigated animals. The rationale of using a transient ischemia model was that we wanted to be able to investigate the effect of Aliskiren on neurological function over 1 week. In order to induce neurological deficits, a model inducing large infarcts is necessary, which, however, is only survived by mice when the occlusion is transient. Therefore, we could not use a permanent ischemia model, which may have an improved translational profile, for the current study.
Aliskiren has been used in models of cerebral ischemia before, however, it has only been applied systemically (28, 29) and/ or in animals with arterial hypertension (29, 30). Intravenous application of Aliskiren for 5 days before MCA occlusion resulted in improved outcome and decreased mortality in severely hypertensive rats (29) and reduced infarct volume in mice (28). These experiments, however, did not clarify whether the observed neuroprotection was due to a peripheral, i.e., blood pressure lowering, or central effects of Aliskiren. This is particularly true since it is not clear whether Aliskiren crosses the blood-brain barrier and it is well-known that it has a low resorption rate (5%) (31), low bioavailablity (32) and high plasma protein binding (32) after systemic application. Therefore, little is known about the specific central effect of Aliskiren for the development of ischemic brain damage and the current study is the first one to address this issue.
Using the current approach we have proven that centrally applied Aliskiren is neuroprotective after cerebral ischemia independent of its blood pressure lowering effect. Although we did not measure cerebral renin activity, the specificity and the well-known and proven action of Aliskiren as a renin inhibitor (33) allows us to assume with a high level of confidence that the observed neuroprotective effects of Aliskiren were indeed caused by the inhibition of renin. Taking this as given, the next important question is directed toward the mechanisms undelaying the observed functional and histopathological protection of Aliskiren after cerebral ischemia.
Renin is the first and rate limiting component of the RAS. The classical function of the RAS is the regulation of blood pressure and salt/water homeostasis, however, it has also been implicated in pathogenesis and outcome of ischemic injury in various organs such as heart (34, 35) and kidney (36, 37). All components of the RAS are present in the brain and the RAS has also been closely studied in the context of cerebrovascular disease (38) and stroke (4, 39, 40). Specifically, blocking angiotensin-converting enzyme (ACE) (39–43) or AT-1 receptors (41) has been demonstrated to be neuroprotective following ischemic stroke in experimental animals and humans. In recent years, AT-2 (8, 44, 45) or MAS (7) receptor activation have been proposed as another RAS-based therapeutic approach for reducing post-ischemic brain damage after stroke. Therefore, the most straightforward mechanistic explanation of the current results is that the inhibition of renin inhibits the more downstream members of the RAS thereby replicating their well-known neuroprotective effects. This may be a valid, but may not fully explain the quite pronounced neuroprotective effects of Aliskiren. The reason is that inhibition of renin may indeed inhibit the formation of Angiotensin-1 (Ang-1), the subsequent production of Ang-2 by ACE, and activation of damaging AT-1 signaling, however, it would also inhibit neuroprotective AT-2 and MAS signaling. Therefore, reduction of Ang-1 production may not fully explain the neuroprotection currently observed with Aliskiren and suggests that renin may have additional functions. In previous studies using Aliskiren, the protective effects of renin inhibition were hypothesized to be caused by a reduction of apoptotic mechanisms as well as anti-inflammatory effects, however, the link between renin activity and apoptotic signaling remains quite speculative (28, 29). In the present study, we found evidence for a reduction of ischemia-induced lipid peroxidation and less activity impairment of antioxidant enzymes superoxide dismutase and glutathione peroxidase. This is in keeping with previous data obtained by Aliskiren in other organs and models (46–48). As oxidative damage and lipid peroxidation play an important role in the development of postischemic damage (49–51) this finding may partially explain the neuroprotective effect observed. Another mechanism which may have a direct connection to the pathophysiology of cerebral ischemia is the link between renin and the activity of endothelial nitric oxide synthase (eNOS). This link was established since Aliskiren has been described to increase eNOS activity (52) and phosphorylation (53) and its protective effects were eNOS dependent (19) after myocardial ischemia. It is therefore quite possible that the protection observed in the current study is, at least partially, due to restoration of eNOS activity, specifically since the activity of eNOS is significantly reduced after ischemic stroke (54, 55) and increasing the formation of NO has been shown to be neuroprotective after ischemic stroke (26, 54, 56–58). These actions of renin after stroke are quite speculative and will therefore need further experimental validation. Another point that has to be elucidated in further studies is whether the Aliskiren effect observed is different at different doses.
In summary, central application of Aliskiren, a clinically approved renin inhibitor, is neuroprotective and improves functional outcome in a model of ischemic stroke independent of its blood pressure lowering activity. The experimental approach used in the current study (pre-treatment, intracerebroventricular injection) was chosen in order to supply a proof of principle with as little confounding factors as possible and does therefore not allow any conclusion about the effect of Aliskiren for acute stroke treatment. In order to further explore the therapeutic potential of the proposed neuroprotective mechanism of central renin inhibition after ischemic stroke more studies are needed in order to explore a possible therapeutic window; also, pharmacological modification of the drug which may allow for a different route of administration are in planning. Nevertheless, the observed neuroprotection was relevant and reproducible. Therefore, central inhibition of renin may represent a promising approach to protect the brain from ischemic injury when used prophylactically. Hence, developing centrally active renin inhibitors may provide a novel approach toward the prophylactic treatment of ischemic brain injury.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
All protocols used were in accordance with international guidelines, the Basel Declaration, and approved by the Government of Upper Bavaria (protocol number 55.2-1-54-2531-118-05).
Author Contributions
HP performed all the experiments. HP, NT, and NP analyzed and interpreted the data. DS and CC extracted and purified the drug. HP, NT, and NP wrote the manuscript.
Funding
This study was supported by the Solorz-Zak Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Uta Mamrak for technical assistance. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy–ID 390857198).
References
1. Ettehad D, Emdin CA, Kiran A, Anderson SG, Callender T, Emberson J, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet. (2016) 387:957–67. doi: 10.1016/S0140-6736(15)01225-8
2. O'Donnell MJ, Chin SL, Rangarajan S, Xavier D, Liu L, Zhang H, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. (2016) 388:761–75. doi: 10.1016/S0140-6736(16)30506-2
3. Wright JW, Harding JW. The brain renin-angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch. (2013) 465:133–51. doi: 10.1007/s00424-012-1102-2
4. Arroja MM, Reid E, McCabe C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp Transl Stroke Med. (2016) 8:8. doi: 10.1186/s13231-016-0022-1
5. Reboldi G, Angeli F, Cavallini C, Gentile G, Mancia G, Verdecchia P. Comparison between angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on the risk of myocardial infarction, stroke and death: a meta-analysis. J Hypertens. (2008) 26:1282–9. doi: 10.1097/HJH.0b013e328306ebe2
6. Verdecchia P, Reboldi G, Angeli F, Gattobigio R, Bentivoglio M, Thijs L, et al. Angiotensin-converting enzyme inhibitors and calcium channel blockers for coronary heart disease and stroke prevention. Hypertension. (2005) 46:386–92. doi: 10.1161/01.HYP.0000174591.42889.a2
7. Lee S, Evans MA, Chu HX, Kim HA, Widdop RE, Drummond GR, et al. Effect of a selective mas receptor agonist in cerebral ischemia in vitro and in vivo. PLoS ONE. (2015) 10:e0142087. doi: 10.1371/journal.pone.0142087
8. Ma CY, Yin L. Neuroprotective effect of angiotensin II type 2 receptor during cerebral ischemia/reperfusion. Neural Regen Res. (2016) 11:1102–7. doi: 10.4103/1673-5374.187044
9. McCarthy CA, Vinh A, Broughton BR, Sobey CG, Callaway JK, Widdop RE. Angiotensin II type 2 receptor stimulation initiated after stroke causes neuroprotection in conscious rats. Hypertension. (2012) 60:1531–7. doi: 10.1161/HYPERTENSIONAHA.112.199646
10. Gonzalez AA, Prieto MC. Renin and the (pro)renin receptor in the renal collecting duct: role in the pathogenesis of hypertension. Clin Exp Pharmacol Physiol. (2015) 42:14–21. doi: 10.1111/1440-1681.12319
11. Morris BJ. Renin, genes, microRNAs, and renal mechanisms involved in hypertension. Hypertension. (2015) 65:956–62. doi: 10.1161/HYPERTENSIONAHA.114.04366
12. Nguyen G, Burckle CA, Sraer JD. Renin/prorenin-receptor biochemistry and functional significance. Curr Hypertens Rep. (2004) 6:129–32. doi: 10.1007/s11906-004-0088-3
13. Andersen K, Weinberger MH, Egan B, Constance CM, Ali MA, Jin J, et al. Comparative efficacy and safety of aliskiren, an oral direct renin inhibitor, and ramipril in hypertension: a 6-month, randomized, double-blind trial. J.Hypertens. (2008) 26:589–99. doi: 10.1097/HJH.0b013e3282f3ad9a
14. Gradman AH, Schmieder RE, Lins RL, Nussberger J, Chiang Y, Bedigian MP. Aliskiren, a novel orally effective renin inhibitor, provides dose-dependent antihypertensive efficacy and placebo-like tolerability in hypertensive patients. Circulation. (2005) 111:1012–8. doi: 10.1161/01.CIR.0000156466.02908.ED
15. Oh BH, Mitchell J, Herron JR, Chung J, Khan M, Keefe DL. Aliskiren, an oral renin inhibitor, provides dose-dependent efficacy and sustained 24-hour blood pressure control in patients with hypertension. J Am Coll Cardiol. (2007) 49:1157–63. doi: 10.1016/j.jacc.2006.11.032
16. Riccioni G. Aliskiren in the treatment of hypertension and organ damage. Cardiovasc Ther. (2011) 29:77–87. doi: 10.1111/j.1755-5922.2010.00230.x
17. Wang W, Qiu L, Howard A, Solis N, Li C, Wang X, et al. Protective effects of aliskiren and valsartan in mice with diabetic nephropathy. J Renin Angiotensin Aldosterone Syst. (2014) 15:384–95. doi: 10.1177/1470320313507123
18. Yarows SA. Aliskiren/valsartan combination for the treatment of cardiovascular and renal diseases. Expert Rev Cardiovasc Ther. (2010) 8:19–33. doi: 10.1586/erc.09.143
19. Chen Y, Meng G, Bai W, Ma Y, Xie L, Altaf N, et al. Aliskiren protects against myocardial ischaemia-reperfusion injury via an endothelial nitric oxide synthase dependent manner. Clin Exp Pharmacol Physiol. (2017) 44:266–74. doi: 10.1111/1440-1681.12692
20. Koid SS, Ziogas J, Campbell DJ. Aliskiren reduces myocardial ischemia-reperfusion injury by a bradykinin B2 receptor- and angiotensin AT2 receptor-mediated mechanism. Hypertension. (2014) 63:768–73. doi: 10.1161/HYPERTENSIONAHA.113.02902
21. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. (2010) 8:e1000412. doi: 10.1371/journal.pbio.1000412
22. Kataoka H, Kim SW, Plesnila N. Leukocyte-endothelium interactions during permanent focal cerebral ischemia in mice. J Cereb Blood Flow Metab. (2004) 24:668–76. doi: 10.1097/01.WCB.0000117812.35136.5B
23. Plesnila N, Zinkel S, Le D, Amin-Hanjani S, Wu Y, Qui J, et al. BID mediates neuronal cell death after oxygen/ glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA. (2001) 98:15318–23. doi: 10.1073/pnas.261323298
24. Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab. (2004) 24:458–66. doi: 10.1097/00004647-200404000-00011
25. Terpolilli NA, Kim SW, Thal SC, Kuebler WM, Plesnila N. Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. (2013) 33:311–8. doi: 10.1038/jcbfm.2012.176
26. Terpolilli NA, Kim SW, Thal SC, Kataoka H, Zeisig V, Nitzsche B, et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circ Res. (2012) 110:727–38. doi: 10.1161/CIRCRESAHA.111.253419
27. Llovera G, Hofmann K, Roth S, Salas-Perdomo A, Ferrer-Ferrer M, Perego C, et al. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci Transl Med. (2015) 7:299ra121. doi: 10.1126/scitranslmed.aaa9853
28. Miao J, Wang L, Zhang X, Zhu C, Cui L, Ji H, et al. Protective effect of aliskiren in experimental ischemic stroke: up-regulated p-PI3K, p-AKT, Bcl-2 expression, attenuated bax expression. Neurochem Res. (2016) 41:2300–10. doi: 10.1007/s11064-016-1944-7
29. Schmerbach K, Pfab T, Zhao Y, Culman J, Mueller S, Villringer A, et al. Effects of aliskiren on stroke in rats expressing human renin and angiotensinogen genes. PLoS ONE. (2010) 5:e15052. doi: 10.1371/journal.pone.0015052
30. Zhang W, Han Y, Meng G, Bai W, Xie L, Lu H, et al. Direct renin inhibition with aliskiren protects against myocardial ischemia/reperfusion injury by activating nitric oxide synthase signaling in spontaneously hypertensive rats. J Am Heart Assoc. (2014) 3:e000606. doi: 10.1161/JAHA.113.000606
31. Waldmeier F, Glaenzel U, Wirz B, Oberer L, Schmid D, Seiberling M, et al. Absorption, distribution, metabolism, and elimination of the direct renin inhibitor aliskiren in healthy volunteers. Drug Metab Dispos. (2007) 35:1418–28. doi: 10.1124/dmd.106.013797
32. Buczko W, Hermanowicz JM. Pharmacokinetics and pharmacodynamics of aliskiren, an oral direct renin inhibitor. Pharmacol Rep. (2008) 60:623–31. Available online at: http://www.if-pan.krakow.pl/pjp/pdf/2008/5_623.pdf
33. Allikmets K. Aliskiren–an orally active renin inhibitor. Review of pharmacology, pharmacodynamics, kinetics, and clinical potential in the treatment of hypertension. Vasc Health Risk Manag. (2007) 3:809–15.
34. Agrawal V, Gupta JK, Qureshi SS, Vishwakarma VK. Role of cardiac renin angiotensin system in ischemia reperfusion injury and preconditioning of heart. Indian Heart J. (2016) 68:856–61. doi: 10.1016/j.ihj.2016.06.010
35. Ertl G, Hu K. Anti-ischemic potential of drugs related to the renin-angiotensin system. J Cardiovasc Pharmacol. (2001) 37 (Suppl 1), S11–20. doi: 10.1097/00005344-200109011-00003
36. Kohara K, Mikami H, Okuda N, Higaki J, Ogihara T. Angiotensin blockade and the progression of renal damage in the spontaneously hypertensive rat. Hypertension. (1993) 21:975–9. doi: 10.1161/01.HYP.21.6.975
37. Yang T, Xu C. Physiology and pathophysiology of the intrarenal renin-angiotensin system: an update. J Am Soc Nephrol. (2017) 28:1040–9. doi: 10.1681/ASN.2016070734
38. Fouda AY, Artham S, El-Remessy AB, Fagan SC. Renin-angiotensin system as a potential therapeutic target in stroke and retinopathy: experimental and clinical evidence. Clin Sci (Lond). (2016) 130:221–38. doi: 10.1042/CS20150350
39. Sadoshima S, Fujii K, Ooboshi H, Ibayashi S, Fujishima M. Angiotensin converting enzyme inhibitors attenuate ischemic brain metabolism in hypertensive rats. Stroke. (1993) 24:1561–6. doi: 10.1161/01.STR.24.10.1561
40. Werner C, Hoffman WE, Kochs E, Rabito SF, Miletich DJ. Captopril improves neurologic outcome from incomplete cerebral ischemia in rats. Stroke. (1991) 22:910–4. doi: 10.1161/01.STR.22.7.910
41. Panahpour H, Dehghani GA, Bohlooli S. Enalapril attenuates ischaemic brain oedema and protects the blood-brain barrier in rats via an anti-oxidant action. Clin Exp Pharmacol Physiol. (2014) 41:220–6. doi: 10.1111/1440-1681.12210
42. Porritt MJ, Chen M, Rewell SS, Dean RG, Burrell LM, Howells DW. ACE inhibition reduces infarction in normotensive but not hypertensive rats: correlation with cortical ACE activity. J Cereb Blood Flow Metab. (2010) 30:1520–6. doi: 10.1038/jcbfm.2010.57
43. Schrader J, Kulschewski A, Dendorfer A. Inhibition of the renin-angiotensin system and the prevention of stroke. Am J Cardiovasc Drugs. (2007) 7:25–37. doi: 10.2165/00129784-200707010-00003
44. Lee S, Brait VH, Arumugam TV, Evans MA, Kim HA, Widdop RE, et al. Neuroprotective effect of an angiotensin receptor type 2 agonist following cerebral ischemia in vitro and in vivo. Exp Transl Stroke Med. (2012) 4:16. doi: 10.1186/2040-7378-4-16
45. McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. (2009) 40:1482–9. doi: 10.1161/STROKEAHA.108.531509
46. Rashikh A, Ahmad SJ, Pillai KK, Kohli K, Najmi AK. Aliskiren attenuates myocardial apoptosis and oxidative stress in chronic murine model of cardiomyopathy. Biomed Pharmacother. (2012) 66:138–43. doi: 10.1016/j.biopha.2011.11.020
47. Rashikh A, Pillai KK, Ahmad SJ, Akhtar M, Najmi AK. Aliskiren alleviates doxorubicin-induced nephrotoxicity by inhibiting oxidative stress and podocyte injury. J Renin Angiotensin Aldosterone Syst. (2013) 14:14–22. doi: 10.1177/1470320312459980
48. Wang Z, Liu Y, Han Y, Guan W, Kou X, Fu J, et al. Protective effects of aliskiren on ischemia-reperfusion-induced renal injury in rats. Eur J Pharmacol. (2013) 718:160–6. doi: 10.1016/j.ejphar.2013.08.038
49. Heo JH, Han SW, Lee SK. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. (2005) 39:51–70. doi: 10.1016/j.freeradbiomed.2005.03.035
50. Shichiri M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr. (2014) 54:151–60. doi: 10.3164/jcbn.14-10
51. Smith JA, Park S, Krause JS, Banik NL. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem Int. (2013) 62:764–75. doi: 10.1016/j.neuint.2013.02.013
52. Desjarlais M, Dussault S, Dhahri W, Mathieu R, Rivard A. Direct renin inhibition with aliskiren improves ischemia-induced neovascularization: blood pressure-independent effect. Atherosclerosis. (2015) 242:450–60. doi: 10.1016/j.atherosclerosis.2015.08.009
53. Ye Y, Qian J, Castillo AC, Perez-Polo JR, Birnbaum Y. Aliskiren and Valsartan reduce myocardial AT1 receptor expression and limit myocardial infarct size in diabetic mice. Cardiovasc Drugs Ther. (2011) 25:505–15. doi: 10.1007/s10557-011-6339-z
54. Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. (1996) 16:981–7. doi: 10.1097/00004647-199609000-00023
55. Santizo R, Baughman VL, Pelligrino DA. Relative contributions from neuronal and endothelial nitric oxide synthases to regional cerebral blood flow changes during forebrain ischemia in rats. Neuroreport. (2000) 11:1549–53. doi: 10.1097/00001756-200005150-00037
56. Carreau A, Duval D, Poignet H, Scatton B, Vige X, Nowicki JP. Neuroprotective efficacy of N omega-nitro-L-arginine after focal cerebral ischemia in the mouse and inhibition of cortical nitric oxide synthase. Eur J Pharmacol. (1994) 256:241–9. doi: 10.1016/0014-2999(94)90549-5
57. Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. (2005) 36:2251–7. doi: 10.1161/01.STR.0000181077.84981.11
Keywords: brain edema, focal Ischemia, neuroprotection, stroke, acute stroke, hypertension, experimental
Citation: Panahpour H, Terpolilli NA, Schaffert D, Culmsee C and Plesnila N (2019) Central Application of Aliskiren, a Renin Inhibitor, Improves Outcome After Experimental Stroke Independent of Its Blood Pressure Lowering Effect. Front. Neurol. 10:942. doi: 10.3389/fneur.2019.00942
Received: 10 February 2019; Accepted: 14 August 2019;
Published: 04 September 2019.
Edited by:
Anna Rosell, Vall d'Hebron Research Institute (VHIR), SpainReviewed by:
Jesus Miguel Pradillo, Complutense University of Madrid, SpainGiuseppe Faraco, Weill Cornell Medicine, United States
Copyright © 2019 Panahpour, Terpolilli, Schaffert, Culmsee and Plesnila. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolaus Plesnila, bmlrb2xhdXMucGxlc25pbGFAbWVkLnVuaS1tdWVuY2hlbi5kZQ==
†These authors have contributed equally to this work