 Rosa Campopiano1
Rosa Campopiano1 Rosangela Ferese1
Rosangela Ferese1 Fabio Buttari1Cinzia Femiano1Diego Centonze1,2Francesco Fornai1,3
Fabio Buttari1Cinzia Femiano1Diego Centonze1,2Francesco Fornai1,3 Francesca Biagioni1Maria Antonietta Chiaravalloti1Mauro Magnani4Emiliano Giardina5,6Anna Ruzzo4
Francesca Biagioni1Maria Antonietta Chiaravalloti1Mauro Magnani4Emiliano Giardina5,6Anna Ruzzo4 Stefano Gambardella1,4*
Stefano Gambardella1,4*- 1IRCCS Neuromed, Pozzilli, Italy
- 2Dipartimento di Medicina dei Sistemi, Università di Roma Tor Vergata, Rome, Italy
- 3Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
- 4Department of Biomolecular Sciences, University of Urbino “Carlo Bo”, Urbino, Italy
- 5Department of Biomedicine and Prevention, University of Rome “Tor Vergata”, Rome, Italy
- 6Molecular Genetics Laboratory UILDM, Santa Lucia Foundation, Rome, Italy
Ataxia with oculomotor apraxia (AOA) is a clinical syndrome featuring a group of genetic diseases including at least four separate autosomal-recessive cerebellar ataxias. All these disorders are due to altered genes involved in DNA repair. AOA type 4 (AOA4) is caused by mutations in DNA repair factor polynucleotide kinase phosphatase (PNKP), which encodes for a DNA processing enzyme also involved in other syndromes featured by microcephaly or neurodegeneration. To date, only a few AOA4 patients have been reported worldwide. All these patients are homozygous or compound heterozygous carriers for mutations in the kinase domain of PNKP. In this report, we describe a 56 years old patient affected by AOA4 characterized by ataxia, polyneuropathy, oculomotor apraxia, and cognitive impairment with the absence of dystonia. The disease is characterized by a very late onset (50 years) when compared with other AOA4 patients described so far (median age of onset at 4 years). In this proband, Clinical Exome Analysis through Next Generation Sequencing (NGS) consisting of 4,800 genes, identified the PNKP homozygous mutation p.Gln50Glu. This variant, classified as a likely pathogenic variant according to American College of Medical Genetics (ACMG) guidelines, does not involve the kinase domain but falls in the fork-head-associated (FHA) domain. So far, mutations in such a domain were reported to associate only with a pure seizure syndrome without the classic AOA4 features. Therefore, this is the first report of patients carrying a mutation of the FHA domain within the PNKP gene which expresses the clinical phenotype known as the AOA4 syndrome and the lack of any seizure activity. Further studies are required to investigate specifically the significance of various mutations within the FHA domain, and it would be worth to correlate these variants with the age of onset of the AOA4 syndrome.
Introduction
Mutations in the DNA repair factor polynucleotide kinase phosphatase (PNKP) have been linked to multiple distinct human inherited syndromes. These include (i) microcephaly with seizures (MCSZ, MIM 613402, also known as early infantile epileptic encephalopathy-10), which is characterized by microcephaly, early-onset, intractable seizures, and developmental delay (1), (ii) progressive cerebellar atrophy and polyneuropathy (2), and (iii) ataxia with oculomotor apraxia type 4 (AOA4) (3, 4).
Ataxia with oculomotor apraxia (AOA), is a group of at least four autosomal-recessive cerebellar ataxia syndromes. They are related to genes which provide instructions for making proteins that are involved in repairing damaged DNA: (i) APTX (aprataxin), responsible for AOA1 (MIM 208920), a progressive syndrome associated with hypoalbuminemia and elevated levels of cholesterol (5–7); (ii) SETX (senataxin), responsible for AOA2 or SCAR1 (MIM 606002), a progressive ataxia occurring later than AOA1, characterized by increased alpha-fetoprotein levels (8, 9), (iii) PIK3R5 (phosphoinositide-3-kinase, regulatory subunit 5), responsible for AOA3 (MIM 615217), with clinical features similar to AOA2; (iv) PNKP (polynucleotide kinase 3′-phosphatase) responsible for AOA4 (MIM 616267) characterized by ataxia, oculomotor apraxia, peripheral neuropathy, and dystonia (3).
These syndromes impact cerebellar function and result in a profound loss of motor control characterized by cerebellar degeneration, apraxia, and oculomotor apraxia (abnormal saccadic eye movement). Very often these symptoms occur in the presence of dystonia and peripheral neuropathy (5, 10).
AOA4 is characterized by prominent dystonia which spontaneously attenuates during the course of the disease. Muscle wasting in the hands and feet and neuropathy are also common and lead to tetraplegia and short atrophic hands and feet. Usually, cognitive function is not affected, although some people may present intellectual disability. AOA diseases are characterized by an early onset which occurs in the first decade; strikingly, AOA4 is characterized by the earliest disease onset among AOAs which occurs at about 4 years of age (11).
PNKP is a DNA processing enzyme in which the C-terminal catalytic domain contains a fused bimodal phosphatase and kinase domain, with a fork-head-associated (FHA) domain at its N-terminus (12).
The FHA domain of PNKP is important for interaction with either the XRCC1 or XRCC4 scaffold proteins, which are required for assembling single-strand break repair (SSBR) or dominant pathway for DNA double-strand break repair (DSBR) (NHEJ, non-homologous end-joining) components respectively (13–15). In fact, PNKP is important for both the SSBR/Base excision repair (BER) and NHEJ pathways (12).
To date, only a few patients with mutations in PNKP have been reported worldwide. Substantial variation exists among affected individuals with PNKP mutations, as individuals diagnosed with MCSZ show no neurodegeneration, while individuals with AOA4 show pronounced neurodegeneration.
Among AO4 patients, the different phenotypes associated with mutations in PNKP do not seem to relate to either the type or the location of the mutation (3). So far, all PNKP mutations related to AOA4 have been described in the kinase domain.
In this report, we describe a patient affected by AOA4 related to a homozygous mutation in PNKP, with very late onset at 50 years. Despite the occurrence of AOA4 phenotype, this variant falls within the FHA domain, which was never reported before in AOA4 patients.
Materials and Methods
Genetic Testing
The pedigree of the proband's family is shown in Figure 1A. After genetic counseling, written informed consent was obtained, and clinical exome sequencing (4,800 human genes) was performed including 100 genes related to Ataxia and or Spastic Paraplegia (Clinical Exome Solution, SOPHiA genetics) on MiSeq platform (Illumina) (16).
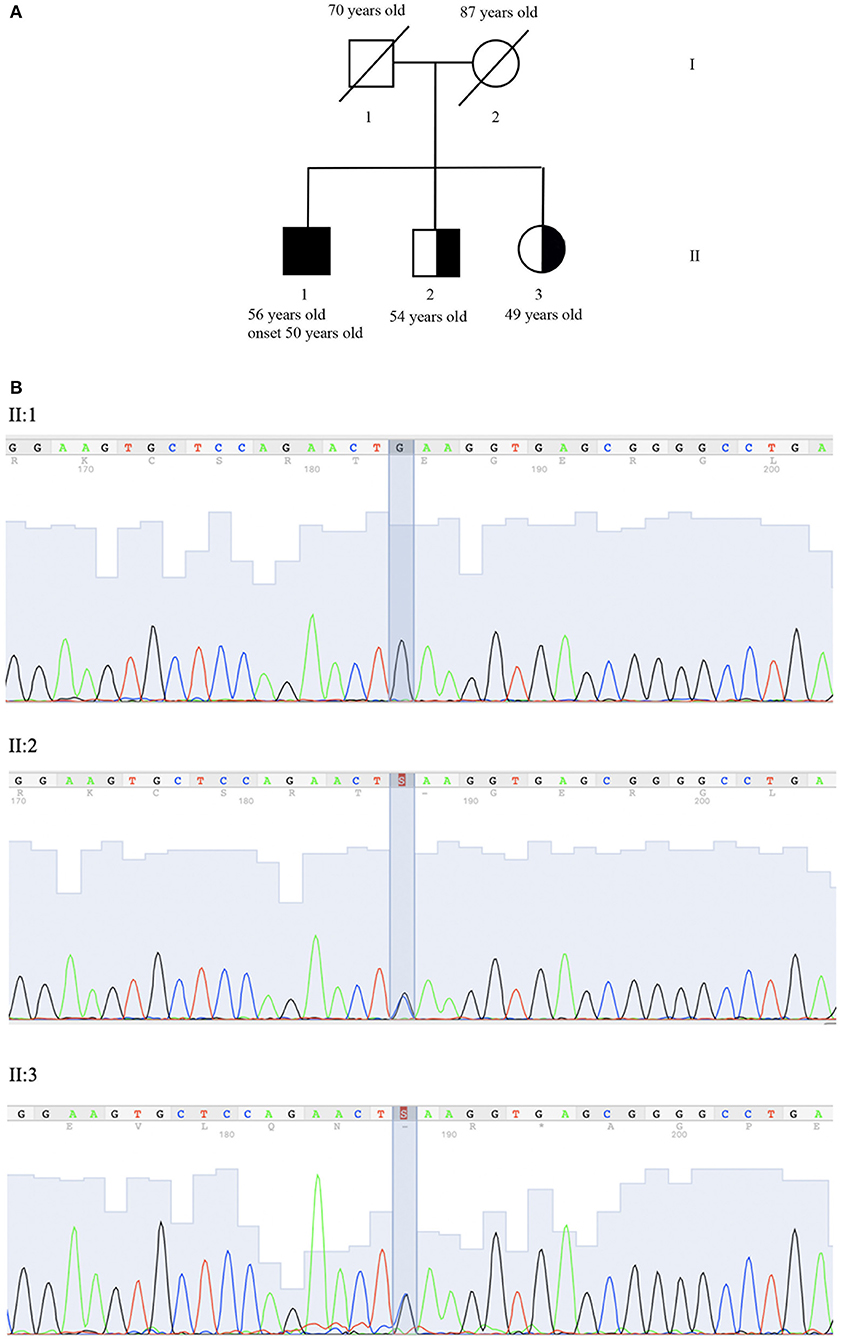
Figure 1. Genetic evaluation. (A) Pedigree: II:1 corresponds to proband, II:2 and II:3 to the asymptomatic carrier brother and sister. (B) Co-segregation analysis of NM_007254.3:c.[148C>G]; NP_009185.2: p. (Gln50Glu) (rs756746191:C>G) (Clin Var Accession Number SUB6349616) in exon 2 of gene PNKP (OMIM #605610). Sequence analysis is shown for proband (II:1) who is homozygous for variant and brother (II:2) and his sister (II:3) who is heterozygous carrier of the same variant.
SOPHiA DDM (SOPHiA genetics) was used for annotation and characterization of variants. Sequence analysis identified the homozygous mutation NM_007254.3:c.[148C>G]; NP_009185.2: p. (Gln50Glu) (rs756746191:C>G).
In the FHA domain PNKP (OMIM #605610), resulting in the replacement of glutamine with glutamic acid in the protein at position 50. The variant was confirmed by Sanger sequencing (ABI 3130xl Genetic Analyzer, Applied Biosystem) (Figure 1B). No mutations in other ataxia-related genes were identified. The variant has been submitted to Clinical Variant (ClinVar Accession Number SUB6349616).
The frequency of the variant is not known either in the ExAC database (http://evs.gs.washington.edu/EVS) or in the 1,000 Genomes database (http://browser.1000genomes.org). The prediction analysis in silico reveals a damaging effect in three out of four tools used [Mutation taster (http://www.mutationtaster.org), SIFT (http://sift.jcvi.org), and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2)]. The PROVEAN (http://provean.jcvi.org/index.php) tool alone predicts it as neutral. The analysis using the PhyloP (http://compgen.bscb.cornell.edu/phast/) and GERP (http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html) tools gave respectively a positive score (1) and an neutral rate (NR) equal to 5.4 indicating that both the wild type nucleotide and the amino acid are conserved in mammals.
The presence of p.Gln50Glu was evaluated in II:2 (54 years old brother) and II:3 (49 years old sister), who are asymptomatic heterozygous carriers (Figure 1A).
According to ACMG guidelines, which consider data on familial segregation, bioinformatic analysis, and allele frequencies, p. (Gln50Glu) has been classified as a likely pathogenic variant.
Results
A 56-year-old Italian male (II:1) was referred to the Neurology Unit of the Scientific Institute for Research and Healthcare Neuromed in Pozzilli (IS) Italy, with a 6 years history of distal weakness and tactile hypoesthesia of the legs and arms, cramps, urinary urgency, dysarthria, and mild dysphagia. He was born at term by unrelated parents that died late in life. His comorbidity consists of allergic bronchial asthma which was routinely treated with beta2-agonists and corticosteroids (Figure 1A).
On neurological examination, he had cerebellar dysarthria, gait ataxia (the patient was able to walk without support but with moderate gait instability), mild bilateral dysmetria, oculomotor apraxia, distal weakness of the arms and legs, distal hypoesthesia, absent tendon reflexes. The International Cooperative Ataxia Rating Scale (ICARS) total score was 40/100. Electroneuromyography (ENMG) indicated the presence of a sensory-motor axonal polyneuropathy. Motor evoked potentials (MEPs) were characterized by increased central motor conduction time. Somatosensory evoked potentials (SEP) were not measurable due to a lack of reproducibility of peripheral responses. Brain Magnetic Resonance Imaging (MRI) at the age of 55 showed marked subtentorial atrophy without spinal cord alteration. In detail, Axial T1-weighted showed cerebellar and brainstem atrophy, Sagittal MRI scan showed global cerebellar atrophy, and Axial FLAIR MRI sequences showed no dentate nucleus hyperintensity (Figure 2). The neuropsychological assessment showed impairment of short-term memory, attention and constructive apraxia. Nothing relevant was found on body Computer Tomography (CT) scan. There were no alterations in the liquor analyzed after lumbar puncture. A second neurological examination performed after 6 months showed no significant deterioration, suggesting a slow progression of the disease.
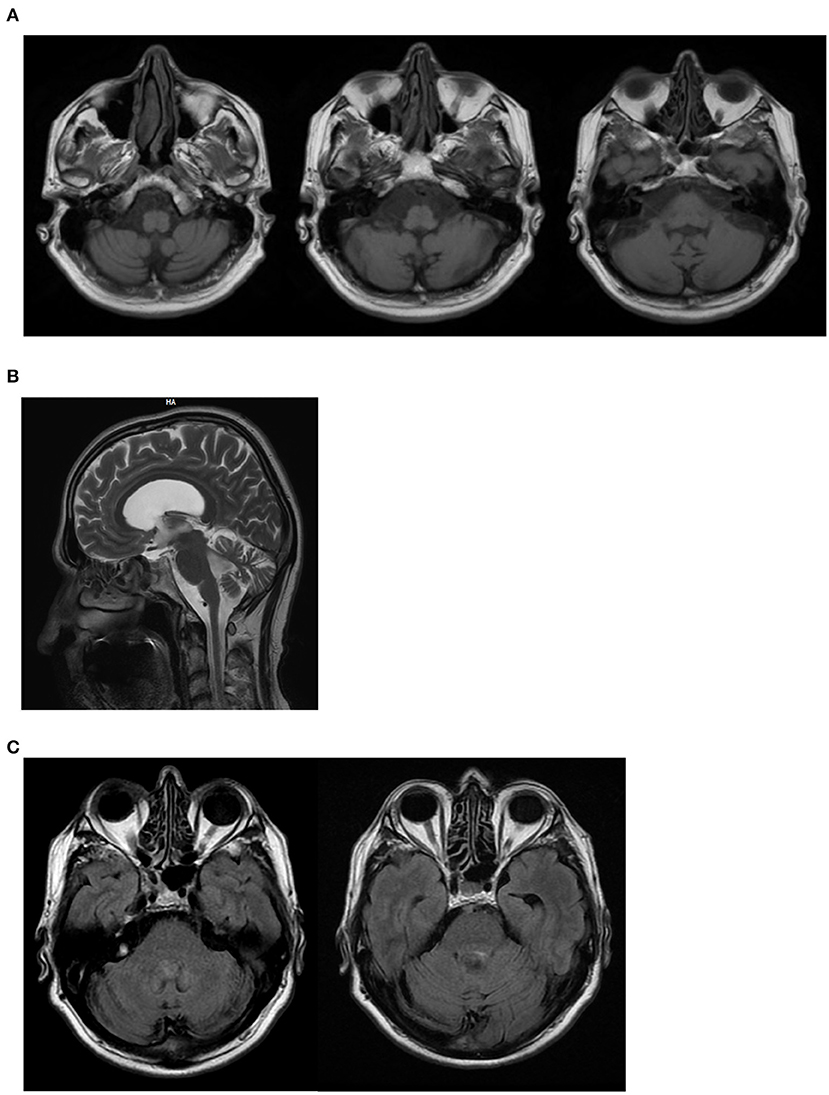
Figure 2. II:1 Brain MRI. (A) Axial T1-weighted shows cerebellar and brainstem atrophy; (B) Sagittal MRI scan shows global cerebellar atrophy; (C) Axial FLAIR MRI sequences shows no dentate nucleus hyperintensity.
Blood tests showed mild hyper-CKemia (624 UI/l), hypercholesterolemia (263 mg/dl), hight alfa-fetoprotein (10.85 ng/ml), and low albumin level (3.4 g/dl). BMI index was 24.22. LDL increased level (173 mg/dl, nv < 130) confirmed data by Scheiss et al. (4), who reported increased LDL and IgE levels in AOA4 patients. The immunoglobulin dosage was not performed. High-density lipoprotein (HDL), triglycerides, thyroid hormones, vitamins, autoimmune screen, anti-ganglioside, and anti-neuronal antibodies were normal.
Discussion
A variety of human neurologic diseases are caused by inherited defects in DNA repair, underscoring the critical requirement for genome stability in this tissue (17). An example is provided by the defective enzymatic activity of the genes responsible for ataxia with AOA, a subgroup involving cerebellar ataxia, axonal neuropathy, oculomotor apraxia, and extrapyramidal features. Genes responsible for this condition provide instructions for making proteins that are involved in repairing damaged DNA. DNA damage that is not repaired makes the cell unstable and can lead to cell death, producing a severe effect in the cerebellum, as witnessed by the occurrence of ataxia with oculomotor apraxia (18).
Recently described in a Portuguese family (19), AOA4 is a complex, progressive movement disorder characterized by phenotypic heterogeneity like other AOAs. In most patients, the first symptom is dystonia, which spontaneously attenuates during the course of the disease. Other features include cerebellar ataxia, oculomotor apraxia, and polyneuropathy. Cognitive impairment is rare, while a few patients at later stages of the disease present abnormal values of alfa-fetoprotein, albumin, and cholesterol.
In this report, we describe the later onset of AOA4 in a patient that did not experience dystonia but a classic cerebellar syndrome with gait ataxia, mild dysmetria, oculomotor apraxia, and cerebellar dysarthria and dysphagia. Sensorial-motor axonal polyneuropathy was also present. Cerebellar ataxia was compatible with cerebellar atrophy detected at brain MRI (20) (Figure 2). Dysphagia could be explained by the involvement of the nucleus ambiguous in the brainstem atrophy detected at MRI. However, considering the concomitance of cerebellar dysarthria, the presence of dysphagia is likely to be due to the cerebellar involvement. Similarly, brainstem atrophy could be further investigated concerning the potential involvement of pre-cerebellar nuclei. Mild hyper-CKemia, although non-specific, is likely to be due to the motor component of polyneuropathy.
The AO4 patient here described is unique considering his age of onset at 50. In fact, age of onset of AO4 patients ranges from 1 to 9 years (with a mean of 4.3). So far, only a German woman was described with a delayed onset although she was much younger (23-year-old), compared with the present case report. Additionally, this 23 year old diagnosed patient possessed an AOA phenotype which was combined with a cerebellar pilocytic astrocytoma (21).
The present paper reports a patient who is a carrier of the homozygous mutation NM_007254.3:c.[148C>G]; NP_009185.2: p. (Gln50Glu) (rs756746191:C>G) in the FHA domain PNKP. To date, multiple distinct human syndromes have been linked to mutations in PNKP, leading to microcephaly or neurodegeneration (17). PNKP is a multifunctional DNA repair enzyme important for both the SSBR/BER and NHEJ pathways (12), and this dual functionality potentially explains the presence of both microcephaly and neurodegeneration in patients with certain inherited PNKP mutations (22).
In support of this, it's well-known that different domains of the protein are linked to different diseases. The kinase domain of PNKP is associated with neurodegeneration associated with AOA4, as witnessed by the variants p.G375W, p.T408del, p.R439fs, p.T442fs, p.Thr424fs*49, and p.Tyr515* (21). Mutations responsible for MCSZ are mainly located in the phosphatase domain (p.L176F and p.E326) but spread along the whole gene. In detail, an MCSZ patient carries the compound heterozygote genotype p.G292R/p.A55S, falling respectively in the phosphatase and FHA domains; while another MCSZ patient is homozygous for p.T424GfsX48 in the kinase domain (a variant which was also reported in a compound heterozygous AOA4 patient) (2, 23).
Remarkably, so far variants in the FHA domain have never been associated with AOA4 and even with neurodegeneration or microcephaly, as they have been solely reported in individuals who manifest only seizures (p.P20S, p.A55S) (11).
The genetic analysis performed in this paper has limitations. In fact, although the sequencing analysis has been carried out on 4,800 human genes including 100 genes related to Ataxia and Spastic paraplegia, these can't rule out the involvement of other genes related to clinical phenotype. Probably, a more extensive approach like whole exome or whole genome sequencing would guarantee more informative data. Moreover, segregation analysis is not fully informative. In fact, the age of the brother and the sister of the proband, both asymptomatic heterozygous carriers, can't rule out the appearance of clinical symptoms in the next years.
The present paper describes the first variant in the FHA domain leading to neurodegeneration and appearing with an AOA4 phenotype. Further studies are required to establish the role of mutations in different PNKP domains with a special emphasis of site-specificity within the FHA domain, and to identify and understand the role of modifier genes (24). This is required to explain the molecular mechanisms leading to the onset of AOA4 phenotype and to understand whether a late disease onset is a typical feature of AOA4 when this is produced by FHA domain specific mutations.
Author Contributions
SG and RF interpretation of data and drafting of manuscript. FF and DC acquisition of data, analysis and interpretation of data, and critical revision. RC, RF, FBi, and MC acquisition of data, analysis and interpretation of data. FBu and CF neurological history and interpretation of clinical data. MM, EG, and AR analysis and interpretation of data and critical revision. SG study conception and design, analysis and interpretation of data, and drafting of manuscript.
Funding
This work was supported by the Italian Ministry of Health (Current Research 2019-2023: Identification of New Variants and/or New Genes Responsible for Ataxia and Spastic Paraplegia).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients and their family for participating in the study.
References
1. Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. (2010) 42:245–9. doi: 10.1038/ng.526
2. Poulton C, Oegema R, Heijsman D, Hoogeboom J, Schot R, Stroink H, et al. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics. (2013) 14:43–51. doi: 10.1007/s10048-012-0351-8
3. Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T, et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet. (2015) 96:474–9. doi: 10.1016/j.ajhg.2015.01.005
4. Schiess N, Zee DS, Siddiqui KA, Szolics M, El-Hattab AW. Novel PNKP mutation in siblings with ataxia-oculomotor apraxia type 4. J. Neurogenet. (2017) 31:23–5. doi: 10.1080/01677063.2017.1322079
5. Barbot C, Coutinho P, Chorão R, Ferreira C, Barros J, Fineza I, et al. Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch Neurol. (2001) 58:201–5. doi: 10.1001/archneur.58.2.201
6. Moreira MC, Barbot C, Tachi N, Kozuka N, Mendonça P, Barros J, et al. Homozygosity mapping of Portuguese and Japanese forms of ataxia-oculomotor apraxia to 9p13, and evidence for genetic heterogeneity. Am J Hum Genet. (2001) 68:501–8. doi: 10.1086/318191
7. Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. (2001) 29:189–93. doi: 10.1038/ng1001-189
8. Le Ber I, Bouslam N, Rivaud-Péchoux S, Guimarães J, Benomar A, Chamayou C, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. (2004) 127:759–67. doi: 10.1093/brain/awh080
9. Moreira MC, Klur S, Watanabe M, Németh AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. (2004) 36:225–7. doi: 10.1038/ng1303
10. Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. (2012) 366:636–46. doi: 10.1056/NEJMra1006610
11. Paucar M, Malmgren H, Taylor M, Reynolds JJ, Svenningsson P, Press R, et al. Expanding the ataxia with oculomotor apraxia type 4 phenotype. Neurol. Genet. (2016) 2:e49. doi: 10.1212/NXG.0000000000000049
12. Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci. (2011) 36:262–71. doi: 10.1016/j.tibs.2011.01.006
13. Ali AA, Jukes RM, Pearl LH, Oliver AW. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. (2009) 37:1701–12. doi: 10.1093/nar/gkn1086
14. Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Karimi-Busheri F, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol Cell. (2005) 17:657–70. doi: 10.1016/j.molcel.2005.02.012
15. Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. (2004) 117:17–28. doi: 10.1016/S0092-8674(04)00206-5
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Dumitrache LC, McKinnon PJ. Polynucleotide kinase-phosphatase (PNKP) mutations and neurologic disease. Mech Ageing Dev. (2017) (Pt A):121–9. doi: 10.1016/j.mad.2016.04.009
18. Gueven N, Becherel OJ, Howe O, Chen P, Haince JF, Ouellet ME, et al. A novel form of ataxia oculomotor apraxia characterized by oxidative stress and apoptosis resistance. Cell Death Differ. (2007) 14:1149–61. doi: 10.1038/sj.cdd.4402116
19. Rudenskaya GE, Marakhonov AV, Shchagina OA, Lozier ER, Dadali EL, Akimova IA, et al. Ataxia with oculomotor apraxia type 4 with PNKP common “Portuguese” and novel mutations in two Belarusian families. J Pediatr Genet. (2019) 8:58–62. doi: 10.1055//s-0039-1684008
20. Ronsin S, Hannoun S, Thobois S, Petiot P, Vighetto A, Cotton F, et al. A new MRI marker of ataxia with oculomotor apraxia. Eur J Radiol. (2019) 110:187–92. doi: 10.1016/j.ejrad.2018.11.035
21. Scholz C, Golas MM, Weber RG, Hartmann C, Lehmann U, Sahm F, et al. Rare compound heterozygous variants in PNKP identified by whole exome sequencing in a German patient with ataxia-oculomotor apraxia 4 and pilocytic astrocytoma. Clin Genet. (2018) 94:185–6. doi: 10.1111/cge.13216.
22. Hoch NC, Hanzlikova H, Rulten SL, Tétreault M, Komulainen E, Ju L, et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. (2017) 541:87–91. doi: 10.1038/nature20790.
23. Nakashima M, Takano K, Osaka H, Aida N, Tsurusaki Y, Miyake N, et al. Causative novel PNKP mutations and concomitant PCDH15 mutations in a patient with microcephaly with early-onset seizures and developmental delay syndrome and hearing loss. J Hum Genet. (2014) 59:471–4. doi: 10.1038/jhg.2014.51
Keywords: ataxia with oculomotor apraxia, PNKP gene, clinical exome, late onset, neurogenetics
Citation: Campopiano R, Ferese R, Buttari F, Femiano C, Centonze D, Fornai F, Biagioni F, Chiaravalloti MA, Magnani M, Giardina E, Ruzzo A and Gambardella S (2020) A Novel Homozygous Variant in the Fork-Head-Associated Domain of Polynucleotide Kinase Phosphatase in a Patient Affected by Late-Onset Ataxia With Oculomotor Apraxia Type 4. Front. Neurol. 10:1331. doi: 10.3389/fneur.2019.01331
Received: 07 June 2019; Accepted: 02 December 2019;
Published: 15 January 2020.
Edited by:
Matthew James Farrer, University of Florida, United StatesReviewed by:
Mario Reynaldo Cornejo-Olivas, National Institute of Neurological Sciences, PeruLuca Marsili, University of Cincinnati, United States
Copyright © 2020 Campopiano, Ferese, Buttari, Femiano, Centonze, Fornai, Biagioni, Chiaravalloti, Magnani, Giardina, Ruzzo and Gambardella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Gambardella, c3RlZmFuby5nYW1iYXJkZWxsYUBuZXVyb21lZC5pdA==