 Sanskriti Sasikumar1
Sanskriti Sasikumar1 Antonio P. Strafella1,2,3,4*
Antonio P. Strafella1,2,3,4*- 1Division of Neurology, University of Toronto, Toronto, ON, Canada
- 2Morton and Gloria Shulman Movement Disorder Unit & E. J. Safra Parkinson Disease Program, Neurology Division, Department of Medicine, Toronto Western Hospital, UHN, University of Toronto, Toronto, ON, Canada
- 3Research Imaging Centre, Centre for Addiction and Mental Health, Campbell Family Mental Health Research Institute, University of Toronto, Toronto, ON, Canada
- 4Division of Brain, Imaging and Behaviour – Systems Neuroscience, Krembil Research Institute, UHN, University of Toronto, Toronto, ON, Canada
Cognitive dysfunction is a significant non-motor feature of Parkinson's disease, with the risk of dementia increasing with prolonged disease duration. Multiple cognitive domains are affected, and the pathophysiology cannot be explained by dopaminergic loss alone. Sophisticated neuroimaging techniques can detect the nature and extent of extra-nigral involvement by targeting neurotransmitters, abnormal protein aggregates and tissue metabolism. This review identifies the functional and anatomical imaging characteristics that predict cognitive impairment in PD, the limitations that challenge this process, and the avenues of potential research.
Introduction
Parkinson's Disease (PD) is a neurodegenerative disorder that is often characterized by motor symptoms such as resting tremor, rigidity, bradykinesia, and postural instability. However, non-motor features such as cognitive impairment play a significant role in this progressive, multisystem condition, and often precede the onset of motor symptoms. In fact, mild cognitive impairment (MCI) is present in 42.5% of patients at the time of diagnosis, and progresses to dementia (PDD) in 70–80% of cases within 15–20 years (1, 2). The prevalence of dementia in PD is 3 times higher than the general population (3, 4), and leads to caregiver burden, early institutionalization and death (5).
The clinical syndrome in PD reflects the progressive degeneration of nigrostriatal dopamine neurons and accumulation of intranuclear misfolded alpha-synuclein, referred to as Lewy bodies. Executive dysfunction, that is a prominent feature of the cognitive dysfunction in PD, can be attributed to dopaminergic fronto-striatal impairment. However, other symptoms like alertness and visuo-perceptual dysfunction, cannot fully be explained by dopaminergic loss, and is suspected to involve several other neurotransmitter systems such as serotonergic, noradrenergic and cholinergic circuits. However, the role of extra-dopaminergic pathways in the cognitive symptoms in PD has not been fully elucidated and remains poorly understood.
The advent of sophisticated neuroimaging techniques has facilitated the detection of disease beyond the limitations of the clinical examination. Imaging biomarkers can be used as a potential tool to detect these extra-dopaminergic pathways, by specifically targeting neurotransmitters, abnormal protein aggregates and tissue metabolism (Table 1). They can also clarify the pathological burden that results in cognitive impairment, and can prognosticate the risk factors that determine the progression to dementia. This review highlights the imaging modalities that facilitate the detection of cognitive impairment and dementia in PD, and identify prognostic factors and molecular mechanisms that result in its pathology.
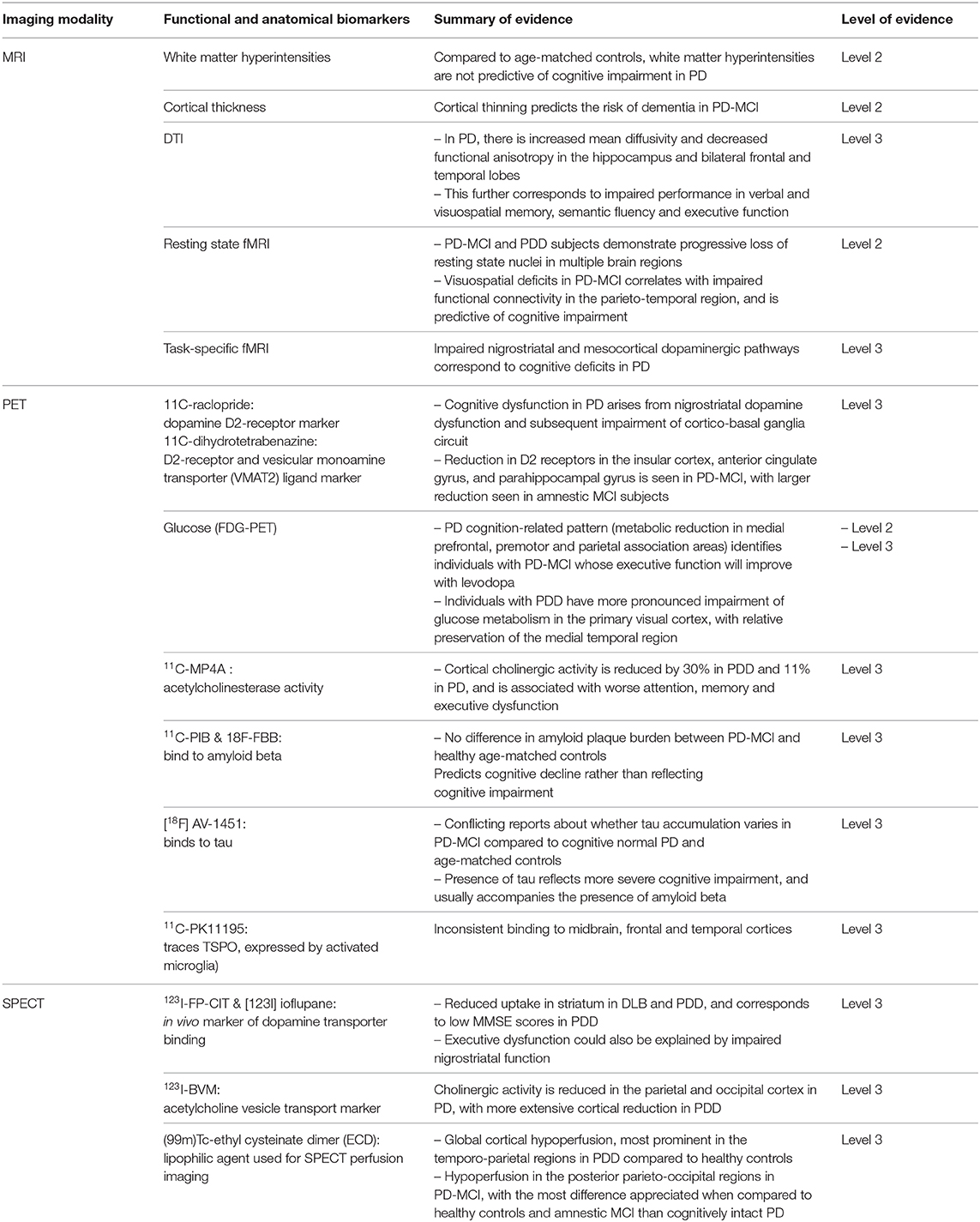
Table 1. Summary of imaging biomarkers in cognitive impairment and dementia in Parkinson's Disease, with their respective grades of evidence.
Search Strategies and Selection Criteria
A search strategy was conducted using the PubMed database (www.ncbi.nlm.nih.gov/pubmed). The search criteria consisted of all English language articles published until September 23rd 2019, that contained combinations of “Parkinson,” “cognitive impairment,” “dementia,” “imaging,” “amyloid,” “tau,” “microglia,” “MRI,” and “PET,” The abstracts were screened for relevance, and read in their entirety if they were suitable.
Magnetic Resonance Imaging (MRI)
White Matter Hyperintensities
White matter hyperintensities (WMH) can be best visualized on the T2-weighted, fluid-attenuated inversion recovery (FLAIR) and proton density-weighted sequences, and their presence has been associated with worse cognition and postural stability in PD (6). However, previous studies have been inconsistent with the association of cognitive impairment and WMH.
White matter hyperintensities arise due to ischemic injury from either hemodynamic compromise, lipohyalinosis, or arteriosclerosis (7, 8). They are increasingly present with age, and are distributed in the deep subcortical white matter and periventricular regions (9, 10). Risk factors for their formation include hypertension, fluctuations in blood pressure and loss of cerebral vascular autoregulation (11). Because of their prevalence, it can be challenging to attribute pathology to their presence. A recent study by Pozorski et al. used an automated lesion segmentation tool to compare the WMH burden in PD to age matched controls over an 18-month period. Their findings reveal that PD patients do not experience more WMH than the average population, but the concurrent presence of WMH can exacerbate the cognitive and motor symptoms in the condition (12).
Volumetric Assessment of Cortical Thickness
Atrophy in PD with mild cognitive impairment (PD-MCI) occurs predominantly over the frontal, temporal and parietal regions, as well as the basal forebrain, and it becomes more pronounced with progression to dementia (13). MRI analysis of cortical thickness can also be a means of prognosticating conversion to dementia. In a study that followed subjects with PD-MCI and healthy controls, cortical thinning in the frontal, left medial temporal and insular regions was associated with increased likelihood for MCI subjects converting to dementia (14). Loss of tissue volume in these regions is associated with impaired decision-making, executive function, recognition of facial expressions and visual memory (14). Voxel-based assessment of gray matter volume (GMV) corroborates this loss in the frontal-limbic and temporal regions, and suggests that unilateral-to-bilateral loss of GMV is a quantitative predictor for the progression from PD-MCI to PDD (15).
The cortex of PD-MCI converters is thinner than non-converters, despite the degree of cognitive impairment being comparable between the two groups (16). More cortical thinning poses an increased risk of developing dementia (16). Retrospective analysis of structural MRI imaging in cognitively intact individuals with PD demonstrated that cognitive impairment can be predicted based on the region of atrophy. PD-MCI occurred within 6 months of hippocampal atrophy (17), 18 months of temporal cortex atrophy (18), and 2 years of prefrontal cortex, insular and caudate atrophy (19, 20).
Diffusion Tensor Imaging (DTI)
DTI is an MRI-based imaging technique that measures the magnitude (mean diffusivity) and direction (fractional anisotropy) of water molecule flow along the white matter tracts. In PD, there is increased mean diffusivity and decreased fractional anisotropy predominantly in the bilateral frontal and temporal regions and hippocampus, suggesting damage to the white matter tracts in these regions (21–25). These manifest clinically with impaired performance in verbal and visuospatial memory, semantic fluency and executive function (22).
Resting State Functional MRI (fMRI)
Functional MRI measures the blood-oxygen-level dependent (BOLD) signal to reflect the functional connectivity (FC) in the brain. This provides information about the interaction between different cortical regions that make up resting-state networks (RSN). These fMRI images are obtained in the absence of prompted task, and demonstrate the resting connectivity in the brain.
PD subjects demonstrate a progressive loss of RSN in multiple brain regions over the course of a 3-year follow-up (26). Despite controlling for dopaminergic medications, subjects with PD-MCI and PDD demonstrate impaired FC in the fronto-parietal network and its associated RSNs (27, 28). In PD-MCI with visuo-spatial impairment, the FC changes involve the parieto-temporal regions as well, and is predictive of evolution to dementia (26, 29). These findings suggest a dual signature of cognitive impairment: the dopaminergic fronto-striatal pathway and the parieto-temporal pathway (30, 31).
Task-Based fMRI
In individuals with PD, task-based MRI performed during attentional set-shifting tasks show reduced fronto-striatal activity (32–34). PD-MCI off dopaminergic medication further show diminished activation between the prefrontal cortex and caudate nucleus when performing set-shifting tasks (34). Some studies indicate that the medial temporal lobe compensates for this diminished functioning, and attempts to preserve cognitive function in PD (34–36). Task-based MRI studies demonstrate that the nigrostriatal and mesocortical dopaminergic pathways are involved in the cognitive deficits seen in PD (14).
Positron Emission Tomography (PET)
Dopamine
Dopaminergic molecular imaging has been useful in identifying the subcortical regions of dopamine hypoactivity that contributes to cognitive impairment in PD. By using a marker for dopamine D2-receptor availability and synaptic dopamine function (11C-raclopride, or RAC), Sawamoto et al. studied subjects with early symptomatic PD and age-matched controls during a spatial working memory task and visuomotor task to determine whether the cognitive impairment in PD results from frontal lobe or striatal dopaminergic dysfunction (37). They found that dopamine release in the dorsal caudate was significantly reduced in PD, but preserved in the medial prefrontal cortex, suggesting that cognitive deficits in early patients with PD arises from nigrostriatal dopamine dysfunction and subsequent impairment of the cortico-basal ganglia circuit (37).
Contrary to prior studies that implicated frontal dopamine depletion in cognitive impairment, there was relative preservation of mesocortical dopamine transmission (37). However, the studies that demonstrated diminished activity in the mesolimbic and mesocortical regions (38, 39), were done in PDD instead of early PD with MCI. So, it is possible that with disease progression, the cortical dopaminergic activity becomes more impaired.
By using a D2-receptor and vesicular monoamine transporter (VMAT2) ligands as a marker for dopamine receptor integrity in the cortex and striatum respectively, Christopher et al. demonstrated that cognitive impairment in PD is associated with D2 receptor reduction in the insular cortex, anterior cingulate gyrus and parahippocampal gyrus compared to cognitively intact PD controls. This reduction was also larger in amnestic compared to non-amnestic MCI subjects. This suggests that the medial temporal lobe contributes to the memory impairment and executive dysfunction in PD (40).
Glucose
FDG-PET findings in PDD manifest in a similar pattern to Alzheimer's Disease, where the regional glucose cerebral metabolism affects the posterior cingulate gyrus first, then progresses to the temporal and parietal association cortex, and finally affects the prefrontal cortex (41, 42). Studies on PD-MCI also reveal diminished glucose metabolism in the parietal, temporal, cingulate, and frontal cortices (43, 44).
Despite similar findings in AD and PDD, the degree of regional cerebral hypometabolism can distinguish between the two conditions when matched for age, sex, and dementia severity. Individuals with PDD have pronounced impairment of glucose metabolism in the primary visual cortex, with relative preservation of the medial temporal region (45). However, in AD, there is only mild reduction in glucose metabolism in these regions (45).
Dementia with Lewy Bodies (DLB) is another neurodegenerative condition that shares similar PET imaging features with PDD. Compared to PD, PDD and DLB demonstrate relatively reduced metabolism in the bilateral inferior and medial frontal lobes, as well as the right parietal lobe (46). The only distinguishing feature is that glucose metabolism is more diminished in the anterior cingulate gyrus in DLB compared to PDD (46).
Spatial covariance analysis of FDG-PET images in PD reveals metabolic reductions in the medial prefrontal, premotor and parietal association areas in a pattern that is referred to as PD cognition-related pattern (PDCP). PDCP is associated with severe cognitive impairment, and can identify the population of patients with MCI whose executive function will improve with levodopa; those who do not demonstrate this pattern, do not respond to therapy (47).
Acetylcholinesterase Activity
Cortical cholinergic deficiency plays a critical role in the cognitive decline seen in PD. Cholinergic denervation occurs early in PD, and is more pronounced in individuals with cognitive impairment and dementia (48–50). Lower cortical cholinergic activity is associated with worse cognitive performance scores for attention, memory, and executive function, and is sensitive enough to detect subclinical MCI (51). This justifies the use of cholinergic inhibitors for the symptomatic treatment of cognitive impairment in PDD (51).
While cortical cholinergic deficiency does not correlate with motor symptoms in PD, it appears to be the primary etiology associated with significant cognitive decline (48–50). Cortical binding of 11C-MP4A, a marker of acetylcholinesterase activity, is globally reduced by 30% in PDD, compared to 11% in PD (42). This pattern of cortical hypometabolism is also seen in DLB and AD, with AD demonstrating additional involvement of the thalamus (49, 51). Cholinergic PET also correlates with loss of forebrain cholinergic neurons seen on post-mortem pathology (48–50). 18F-dopa studies indicate that diminished dopaminergic uptake in the frontal and temporo-parietal regions correlates with cholinergic deficiency, suggesting that both systems reduce in conjunction in PD and PDD (48).
Amyloid Beta
The role of amyloid beta in the development of cognitive impairment in PD is debated. Using the 11C-PIB marker, amyloid beta plaques can be identified in the cortex (52–54), but the degree is significantly less in PD compared to cognitively normal elderly people (55, 56). However, there is a higher plaque burden observed in the cortex and striatum of individuals with PDD (15–20%) and DLB (52, 57).
The presence of amyloid beta plaques is not presumed to be a major player in the pathogenesis of cognitive impairment in PD. There was no difference in amyloid beta burden between PD-MCI and healthy age-matched controls, but long-term follow up indicates that both groups experience cognitive decline at 2.5 years (58). This suggests that the presence of plaques might predict cognitive decline rather than reflect the severity of cognitive impairment (58). Additionally, the presence of amyloid plaque in the striatum is associated with worse cognitive decline compared to its presence in the cortex alone (59).
The in vivo amyloid beta PET imaging studies have several limitations; particularly, low sample size, low power, lack of cognitive characterization and no standardization for age (60). A recently published cross-sectional study by Melzer et al. (60) looked at 115 subjects divided into PD-CN, PD-MCI and PDD. Using 18F-Florbetaben (FBB) amyloid PET and structural MRI, the study determined that while FBB binding was higher in PDD population, it was not significant when adjusted to findings in age-matched controls, and is much lower than the deposition seen in AD (54, 61–63). This further suggests that amyloid beta is not the primary pathology that constitutes the cognitive impairment seen in PD.
Tau
Tau deposits using tracer [18F] AV-1451 show increased binding in precuneus and inferior temporal gyrus in PDD and DLB, compared to age-matched controls (64, 65). However, in a recent cross-sectional study by Winer et al., tau accumulation did not differ between PD-CN and PD-MCI compared to age-matched controls (66). This suggests that cortical tau aggregates are present with more progressive disease only. While it is uncertain whether they contribute to cognitive dysfunction in PD, their presence certainly reflects more advanced cognitive impairment (51). Additionally, tau accumulation increases with amyloid beta, regardless of cognitive status and age (66).
Microglia
Microglia are resident macrophages of the central nervous system that are activated during neuronal injury. They have been implicated in the pathogenesis of PD because post-mortem studies demonstrate increased microglia in the substantia nigra, putamen, hippocampus, cingulate and temporal cortex (67). As such, identifying microglial activity could serve as a biomarker of inflammation and potentially, PD.
Upon activation, microglia express TSPO (18 kDA translocator protein) in the outer mitochondrial membrane (68). However, PET studies using the 11C-PK11195 tracer show inconsistent binding to TSPO in the midbrain (69), frontal and temporal cortices (70). This variability might be explained by the limitations of the tracer itself, as 11C-PK11195 has non-specific binding, low blood-brain barrier penetration, and high plasma protein binding. It is also challenging to prepare, and these demonstrate different affinities because of polymorphisms in TPSO (71, 72).
Increased cortical microglial activation is associated with glucose hypometabolism in the posterior cingulate, frontal, temporal, parietal, and occipital regions in PD-MCI and PDD, suggesting that neuronal disconnections occur both locally and remotely (73). The degree of microglial activation also correlates with the cortical load of amyloid beta, so neuroinflammation may be an early event in dementia and likely initiates neuronal dysfunction (73).
Single-Proton Emission Computed Tomography (SPECT)
Dopamine
Similar to PET imaging, SPECT employs radiotracers to study the basal ganglia and its synaptic projections. 123I-FP-CIT SPECT, which is an in vivo marker of dopamine transporter (DAT) binding, shows reduced striatal uptake in individuals with DLB and PDD (74). In comparison, cognitively intact individuals with PD demonstrate more selective DAT binding to the putamen (75). Moreover, 123I-FP-CIT SPECT binding corresponds to MMSE scores in PDD patients, suggesting that striatal dopaminergic loss contributes to cognitive impairment in PD (74). SPECT studies using [123I] ioflupane suggest that executive dysfunction in PD could also be explained by impaired nigrostriatal function (76, 77).
Acetylcholine
123I-BVM is a marker of acetylcholine vesicle transport, and reflects cholinergic deficiency in PD. Cognitively intact individuals with PD have reduced 123I-BVM binding in the parietal and occipital cortex, whereas those with PDD show extensive reduction in cortical binding (78).
Perfusion
There is limited data on the role of SPECT perfusion imaging in PD-related cognitive impairment, but the reported literature suggests its potential in reflecting the severity of underlying cognitive impairment. A study of 22 patients with PDD using the SPECT radiotracer (99m)Tc-ethyl cysteinate dimer (ECD) reveals global cortical hypoperfusion, most prominent in the temporo-parietal regions, compared to healthy, age-matched controls (79). In individuals with PD-MCI, the hypoperfusion pattern is limited to the posterior parieto-occipital regions, with the most difference seen in comparison to healthy controls and amnestic MCI than to those with cognitively intact PD (80).
Alpha Synuclein
The pathology of alpha-synuclein's invasion into the nervous system is unknown. It is theorized to originate in the enteric or peripheral nervous system, and enter the central nervous system through retrograde vagal nerve transport. However, not all PD subjects demonstrate pathology in the dorsal motor nucleus of the vagus to support this theory (81). Borghammer et al. hypothesize that there are two types of disease processes: (i) peripheral nervous system first, which manifests clinically with REM sleep behavioral disorder (RSBD) and (ii) central nervous system first, with nigrostriatal dopamine involvement prior to autonomic symptoms (81).
They support this theory using in vivo imaging of RSBD positive (idiopathic RSBD, who eventually develop PD or DLB) and RSBD negative (de novo H&Y Stage I PD patients) subjects. This revealed that RSBD positive subjects demonstrate severely reduced sympathetic cardiac innervation (using 123 I -MIBG scintigraphy) but normal striatal dopamine storage capacity (using 18 F-DOPA PET). RSBD negative subjects, who represent de novo PD, show normal cardiac innervation and reduced striatal dopamine capacity. These findings suggest that distinguishing RSBD reflects the initial location of the alpha synuclein pathology (81). Whether the disease starts in the enteric and peripheral nervous system vs. brainstem and limbic system might be determined by different patterns of cellular vulnerability (81). Unfortunately, there is no nucleotide tracer for alpha-synuclein, so much of its role in PD remains theoretical, as does its role in the progression of PD-MCI to PDD.
Discussion
Emerging imaging techniques have facilitated a broader understanding of cognitive impairment, where extra-nigral pathways have been identified to play a key role in the pathogenesis of neurodegeneration in PD. Several mechanisms have been identified, including location of cortical atrophy as means of predicting risk of progression to dementia (17–20), meso-cortical dopaminergic dysfunction in advanced dementia, glucose hypometabolism in the posterior cingulate gyrus and temporo-parietal association cortices, and diffusely diminished cholinergic activity. Certain features like cortical deposition of amyloid beta and tau, as well as white matter burden, are associated with advanced cognitive impairment but do not participate in the primary pathology of cognitive impairment in PD. These processes act synergistically, and have poor prognostic implications for individuals with the disease (58, 82).
Cognitive impairment in PD starts in the temporo-parietal cortices before involving the remainder of the cortex and subcortical gray matter, and predicts the progression to PDD. There is diminished cholinergic activity in this region, as well as in the basal nucleus and occipital cortex, which justifies the use of cholinergic inhibitors for symptomatic treatment of attention and visuospatial disorientation in PD.
There are several limitations to the value of imaging to detect cognitive impairment in PD. Co-pathology of synucleinopathy (Lewy Body inclusions) and proteinopathy (amyloid beta and tau) is a feature shared with other dementing conditions like DLB and AD, which makes it challenging to distinguish PD-related cognitive impairment from these co-pathologies (83). It also remains unclear whether amyloid beta and tau influence the onset of cognitive impairment in PD, enhance existing symptoms or are predictive of its rate of progression. Studies are further limited by design and sample size, which challenges the ability to draw conclusions from their results. The average age of the patient population in the reported studies is over 55 years, with a disease duration between 8 and 10 years, so the relevance of these biomarkers to early-onset PD cannot be determined. There is also a need for new ligands with high affinity and better selectivity, that not only assess neurotransmission but also neuroinflammation (84). This will shed light on the series of events that take place in cognitive impairment, and determine the risk factors that drive this process. Identifying anatomical and functional predictors of cognitive impairment can facilitate early detection and target appropriate interventions to optimize patient care.
Author Contributions
SS: execution and write-up of first draft of the manuscript. AS: conception of research subject and reviewing the manuscript.
Funding
This work was supported by Canadian Institutes of Health Research (MOP-136778) to AS. AS was supported by Canada Research Chair Program.
Conflict of Interest
The authors declare that the study was performed in the absence of commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Aarsland D, Bronnick K, Williams-Gray C, Weintraub D, Marder K, Kulisevsky J, et al. Mild cognitive impairment in Parkinson disease: a multicenter pooled analysis. Neurology. (2010) 75:1062–9. doi: 10.1212/WNL.0b013e3181f39d0e
2. Palavra NC, Naismith SL, Lewis SJ. Mild cognitive impairment in Parkinson's disease: a review of current concepts. Neurol Res Int. (2013) 2013:576091. doi: 10.1155/2013/576091
3. Aarsland D, Andersen K, Larsen JP, Lolk A. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol. (2003) 60:387–92. doi: 10.1001/archneur.60.3.387
4. Svenningsson P, Westman E, Ballard C, Aarsland D. Cognitive impairment in patients with Parkinson's disease: diagnosis, biomarkers, and treatment. Lancet Neurol. (2012) 11:697–707. doi: 10.1016/S1474-4422(12)70152-7
5. Jones AJ, Kuijer RG, Livingston L, Myall D, Horne K, MacAskill M, et al. Caregiver burden is increased in Parkinson's disease with mild cognitive impairment (PD-MCI). Transl Neurodegeneration. (2017) 6:17. doi: 10.1186/s40035-017-0085-5
6. Duncan GW, Firbank MJ, O'Brien JT, Burn DJ. Magnetic resonance imaging: a biomarker for cognitive impairment in Parkinson's disease? Mov Disord. (2013) 28:425–38. doi: 10.1002/mds.25352
7. Kim KW, MacFall JR, Payne ME. Classification of white matter lesions on magnetic resonance imaging in elderly persons. Biol Psychiatr. (2008) 64:273–80. doi: 10.1016/j.biopsych.2008.03.024
8. Xiong YY, Mok V. Age-related white matter changes. J Aging Res. (2011) 2011:617927. doi: 10.4061/2011/617927
9. Murray ME, Senjem ML, Petersen RC, Hollman JH, Preboske GM, Weigand SD, et al. Functional impact of white matter hyperintensities in cognitively normal elderly subjects. Arch Neurol. (2010) 67:1379–85. doi: 10.1001/archneurol.2010.280
10. Yamawaki M, Wada-Isoe K, Yamamoto M, Nakashita S, Uemura Y, Takahashi Y, et al. Association of cerebral white matter lesions with cognitive function and mood in Japanese elderly people: a population-based study. Brain Behav. (2015) 5:e00315. doi: 10.1002/brb3.315
11. Mok V, Kim JS. Prevention and management of cerebral small vessel disease. J Stroke. (2015) 17:111. doi: 10.5853/jos.2015.17.2.111
12. Pozorski V, Oh JM, Okonkwo O, Krislov S, Barzgari A, Theisen F, et al. Cross-sectional and longitudinal associations between total and regional white matter hyperintensity volume and cognitive and motor function in Parkinson's disease. NeuroImage Clin. (2019) 23:101870. doi: 10.1016/j.nicl.2019.101870
13. Delgado-Alvarado M, Gago B, Navalpotro-Gomez I, Jiménez-Urbieta H, Rodriguez-Oroz MC. Biomarkers for dementia and mild cognitive impairment in Parkinson's disease. Mov Disord. (2016) 31:861–81. doi: 10.1002/mds.26662
14. Lehericy S, Vaillancourt DE, Seppi K, Monchi O, Rektorova I, Antonini A, et al. The role of high-field magnetic resonance imaging in parkinsonian disorders: pushing the boundaries forward. Mov Disord. (2017) 32:510–25. doi: 10.1002/mds.26968
15. Xu Y, Yang J, Hu X, Shang H. Voxel-based meta-analysis of gray matter volume reductions associated with cognitive impairment in Parkinson's disease. J Neurol. (2016) 263:1178–87. doi: 10.1007/s00415-016-8122-3
16. Gasca-Salas C, García-Lorenzo D, Garcia-Garcia D, Clavero P, Obeso JA, Lehericy S, et al. Parkinson's disease with mild cognitive impairment: severe cortical thinning antedates dementia. Brain Imaging Behav. (2019) 13:180–8. doi: 10.1007/s11682-017-9751-6
17. Kandiah N, Zainal NH, Narasimhalu K, Chander RJ, Ng A, Mak E, et al. Hippocampal volume and white matter disease in the prediction of dementia in Parkinson's disease. Parkinsonism Relat Disord. (2014) 20:1203–8. doi: 10.1016/j.parkreldis.2014.08.024
18. Mak E, Su L, Williams GB, Firbank MJ, Lawson RA, Yarnall AJ, et al. Baseline and longitudinal grey matter changes in newly diagnosed Parkinson's disease: ICICLE-PD study. Brain. (2015) 138:2974–86. doi: 10.1093/brain/awv211
19. Lee JE, Cho KH, Song SK, Kim HJ, Lee HS, Sohn YH, et al. Exploratory analysis of neuropsychological and neuroanatomical correlates of progressive mild cognitive impairment in Parkinson's disease. J Neurol Neurosurg Psychiatr. (2014) 85:7–16. doi: 10.1136/jnnp-2013-305062
20. Aarsland D, Creese B, Politis M, Chaudhuri KR, Weintraub D, Ballard C. Cognitive decline in Parkinson disease. Nat Rev Neurol. (2017) 13:217. doi: 10.1038/nrneurol.2017.27
21. Koshimori Y, Segura B, Christopher L, Lobaugh N, Duff-Canning S, Mizrahi R, et al. Imaging changes associated with cognitive abnormalities in Parkinson's disease. Brain Struct Funct. (2015) 220:2249–61. doi: 10.1007/s00429-014-0785-x
22. Duncan GW, Firbank MJ, Yarnall AJ, Khoo TK, Brooks DJ, Barker RA, et al. Gray and white matter imaging: a biomarker for cognitive impairment in early P arkinson's disease? Mov Disord. (2016) 31:103–10. doi: 10.1002/mds.26312
23. Carlesimo GA, Piras F, Assogna F, Pontieri FE, Caltagirone C, Spalletta G. Hippocampal abnormalities and memory deficits in Parkinson disease: a multimodal imaging study. Neurology. (2012) 78:1939–45. doi: 10.1212/WNL.0b013e318259e1c5
24. Agosta F, Canu E, Stefanova E, Sarro L, Tomić A, Špica V, et al. Mild cognitive impairment in Parkinson's disease is associated with a distributed pattern of brain white matter damage. Human Brain Mapp. (2014) 35:1921–9. doi: 10.1002/hbm.22302
25. Chen B, Fan GG, Liu H, Wang S. Changes in anatomical and functional connectivity of Parkinson's disease patients according to cognitive status. Eur J Radiol. (2015) 84:1318–24. doi: 10.1016/j.ejrad.2015.04.014
26. Dubbelink KT, Schoonheim MM, Deijen JB, Twisk JW, Barkhof F, Berendse HW. Functional connectivity and cognitive decline over 3 years in Parkinson disease. Neurology. (2014) 83:2046–53. doi: 10.1212/WNL.0000000000001020
27. Rektorova I, Krajcovicova L, Marecek R, Mikl M. Default mode network and extrastriate visual resting state network in patients with Parkinson's disease dementia. Neurodegenerative Dis. (2012) 10:232–7. doi: 10.1159/000334765
28. Borroni B, Premi E, Formenti A, Turrone R, Alberici A, Cottini E, et al. Structural and functional imaging study in dementia with Lewy bodies and Parkinson's disease dementia. Parkinsonism Relat Disord. (2015) 21:1049–55. doi: 10.1016/j.parkreldis.2015.06.013
29. Baggio HC, Segura B, Sala-Llonch R, Marti MJ, Valldeoriola F, Compta Y, et al. Cognitive impairment and resting-state network connectivity in P arkinson's disease. Human Brain Mapp. (2015) 36:199–212. doi: 10.1002/hbm.22622
30. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol. (2010) 9:1200–13. doi: 10.1016/S1474-4422(10)70212-X
31. Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW, et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow-up of the CamPaIGN cohort. Brain. (2009) 132:2958–69. doi: 10.1093/brain/awp245
32. Lewis SJ, Dove A, Robbins TW, Barker RA, Owen AM. Cognitive impairments in early Parkinson's disease are accompanied by reductions in activity in frontostriatal neural circuitry. J Neurosci. (2003) 23:6351–6. doi: 10.1523/JNEUROSCI.23-15-06351.2003
33. Monchi O, Petrides M, Doyon J, Postuma RB, Worsley K, Dagher A. Neural bases of set-shifting deficits in Parkinson's disease. J Neurosci. (2004) 24:702–10. doi: 10.1523/JNEUROSCI.4860-03.2004
34. Nagano-Saito A, Habak C, Mejía-Constaín B, Degroot C, Monetta L, Jubault T, et al. Effect of mild cognitive impairment on the patterns of neural activity in early Parkinson's disease. Neurobiol Aging. (2014) 35:223–31. doi: 10.1016/j.neurobiolaging.2013.06.025
35. Carbon M, Reetz K, Ghilardi MF, Dhawan V, Eidelberg D. Early Parkinson's disease: longitudinal changes in brain activity during sequence learning. Neurobiol Dis. (2010) 37:455–60. doi: 10.1016/j.nbd.2009.10.025
36. Dagher A, Owen AM, Boecker H, Brooks DJ. The role of the striatum and hippocampus in planning: a PET activation study in Parkinson's disease. Brain. (2001) 124:1020–32. doi: 10.1093/brain/124.5.1020
37. Sawamoto N, Piccini P, Hotton G, Pavese N, Thielemans K, Brooks DJ. Cognitive deficits and striato-frontal dopamine release in Parkinson's disease. Brain. (2008) 131:1294–302. doi: 10.1093/brain/awn054
38. Ito K, Nagano-Saito A, Kato T, Arahata Y, Nakamura A, Kawasumi Y, et al. Striatal and extrastriatal dysfunction in Parkinson's disease with dementia: a 6-[18F] fluoro-l-dopa PET study. Brain. (2002) 125:1358–65. doi: 10.1093/brain/awf134
39. Nagano-Saito A, Kato T, Arahata Y, Washimi Y, Nakamura A, Abe Y, et al. Cognitive-and motor-related regions in Parkinson's disease: FDOPA and FDG PET studies. Neuroimage. (2004) 22:553–61. doi: 10.1016/j.neuroimage.2004.01.030
40. Christopher L, Duff-Canning S, Koshimori Y, Segura B, Boileau I, Chen R, et al. Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann Neurol. (2015) 77:269–80. doi: 10.1002/ana.24323
41. Hoffman JM, Welsh-Bohmer KA, Hanson M, Crain B, Hulette C, Earl N, et al. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med. (2000) 41:1920–8.
42. Brooks DJ, Pavese N. Imaging non-motor aspects of Parkinson's disease. Prog Brain Res. (2010) 184:205–18. doi: 10.1016/S0079-6123(10)84011-7
43. Gonzalez-Redondo R, García-García D, Clavero P, Gasca-Salas C, García-Eulate R, Zubieta JL, et al. Grey matter hypometabolism and atrophy in Parkinson's disease with cognitive impairment: a two-step process. Brain. (2014) 137:2356–67. doi: 10.1093/brain/awu159
44. Pappatá S, Santangelo G, Aarsland D, Vicidomini C, Longo K, Bronnick K, et al. Mild cognitive impairment in drug-naive patients with PD is associated with cerebral hypometabolism. Neurology. (2011) 77:1357–62. doi: 10.1212/WNL.0b013e3182315259
45. Vander Borght T, Minoshima S, Giordani B, Foster NL, Frey KA, Berent S, et al. Cerebral metabolic differences in Parkinson's and Alzheimer's Diseases matched for dementia severity. J Nucl Med. (1997) 38:797–802.
46. Yong SW, Yoon JK, An YS, Lee PH. A comparison of cerebral glucose metabolism in Parkinson's disease, Parkinson's disease dementia and dementia with Lewy bodies. Eur J Neurol. (2007) 14:1357–62. doi: 10.1111/j.1468-1331.2007.01977.x
47. Hirano S, Shinotoh H, Eidelberg D. Functional brain imaging of cognitive dysfunction in Parkinson's disease. J Neurol Neurosurg Psychiatr. (2012) 83:963–9. doi: 10.1136/jnnp-2011-301818
48. Hilker R, Thomas AV, Klein JC, Weisenbach S, Kalbe E, Burghaus L, et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology. (2005) 65:1716–22. doi: 10.1212/01.wnl.0000191154.78131.f6
49. Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. (2003) 60:1745–8. doi: 10.1001/archneur.60.12.1745
50. Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. (2009) 73:273–8. doi: 10.1212/WNL.0b013e3181ab2b58
51. Kotagal V, Müller ML, Kaufer DI, Koeppe RA, Bohnen NI. Thalamic cholinergic innervation is spared in Alzheimer disease compared to parkinsonian disorders. Neurosci Lett. (2012) 514:169–72. doi: 10.1016/j.neulet.2012.02.083
52. Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R) PK11195-PET and [11C] PIB-PET study. Neurobiol Dis. (2008) 32:412–9. doi: 10.1016/j.nbd.2008.08.001
53. Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging β-amyloid burden in aging and dementia. Neurology. (2007) 68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea
54. Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, et al. Imaging amyloid deposition in Lewy body diseases. Neurology. (2008) 71:903–10. doi: 10.1212/01.wnl.0000326146.60732.d6
55. Mielke MM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, Roberts RO, et al. Indicators of amyloid burden in a population-based study of cognitively normal elderly. Neurology. (2012) 79:1570–7. doi: 10.1212/WNL.0b013e31826e2696
56. Ghadery C, Koshimori Y, Christopher L, Kim J, Rusjan P, Lang AE, et al. The interaction between neuroinflammation and β-amyloid in cognitive decline in Parkinson's Disease. Mol Neurobiol. (2019) 57:492–501. doi: 10.1007/s12035-019-01714-6
57. Maetzler W, Reimold M, Liepelt I, Solbach C, Leyhe T, Schweitzer K, et al. [11C] PIB binding in Parkinson's disease dementia. Neuroimage. (2008) 39:1027–33. doi: 10.1016/j.neuroimage.2007.09.072
58. Gomperts SN, Locascio JJ, Rentz D, Santarlasci A, Marquie M, Johnson KA, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. (2013) 80:85–91. doi: 10.1212/WNL.0b013e31827b1a07
59. Shah N, Frey KA, Müller ML, Petrou M, Kotagal V, Koeppe RA, et al. Striatal and cortical β-amyloidopathy and cognition in Parkinson's Disease. Mov Disord. (2016) 31:111–7. doi: 10.1002/mds.26369
60. Melzer TR, Stark MR, Keenan RJ, Myall DJ, MacAskill MR, Pitcher TL, et al. Beta amyloid deposition is not associated with cognitive impairment in Parkinson's disease. Front Neurol. (2019) 10:391. doi: 10.3389/fneur.2019.00391
61. Petrou M, Bohnen NI, Müller ML, Koeppe RA, Albin RL, Frey KA. Aβ-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology. (2012) 79:1161–7. doi: 10.1212/WNL.0b013e3182698d4a
62. Lee SH, Cho H, Choi JY, Lee JH, Ryu YH, Lee MS, et al. Distinct patterns of amyloid-dependent tau accumulation in Lewy body diseases. Mov Disord. (2018) 33:262–72. doi: 10.1002/mds.27252
63. Donaghy PC, Firbank MJ, Thomas AJ, Lloyd J, Petrides G, Barnett N, et al. Clinical and imaging correlates of amyloid deposition in dementia with Lewy bodies. Mov Disord. (2018) 33:1130–8. doi: 10.1002/mds.27403
64. Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol. (2016) 73:1334–41. doi: 10.1001/jamaneurol.2016.3338
65. Kantarci K, Lowe VJ, Boeve BF, Senjem ML, Tosakulwong N, Lesnick TG, et al. AV-1451 tau and β-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol. (2017) 81:58–67. doi: 10.1002/ana.24825
66. Winer JR, Maass A, Pressman P, Stiver J, Schonhaut DR, Baker SL, et al. Associations between tau, β-amyloid, and cognition in parkinson disease. JAMA Neurol. (2018) 75:227–35. doi: 10.1001/jamaneurol.2017.3713
67. Best L, Ghadery C, Pavese N, Tai YF, Strafella AP. New and old TSPO PET radioligands for imaging brain microglial activation in neurodegenerative disease. Curr Neurol Neurosci Rep. (2019) 19:24. doi: 10.1007/s11910-019-0934-y
68. Stoessl AJ, Lehericy S, Strafella AP. Imaging insights into basal ganglia function, Parkinson's disease, and dystonia. Lancet. (2014) 384:532–44. doi: 10.1016/S0140-6736(14)60041-6
69. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. (2005) 57:168–75. doi: 10.1002/ana.20338
70. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. (2006) 21:404–12. doi: 10.1016/j.nbd.2005.08.002
71. Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN, et al. [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. (2003) 61:686–9. doi: 10.1212/01.WNL.0000078192.95645.E6
72. Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. (2012) 32:1–5. doi: 10.1038/jcbfm.2011.147
73. Fan Z, Aman Y, Ahmed I, Chetelat G, Landeau B, Chaudhuri KR, et al. Influence of microglial activation on neuronal function in Alzheimer's and Parkinson's disease dementia. Alzheimer's Demen. (2015) 11:608–21. doi: 10.1016/j.jalz.2014.06.016
74. O'Brien JT, McKeith IG, Walker Z, Tatsch K, Booij J, Darcourt J, et al. Diagnostic accuracy of 123 I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatr. (2009) 194:34–9. doi: 10.1192/bjp.bp.108.052050
75. Walker Z, Costa DC, Walker RW, Shaw K, Gacinovic S, Stevens T, et al. Differentiation of dementia with Lewy bodies from Alzheimer's disease using a dopaminergic presynaptic ligand. J Neurol Neurosurg Psychiatr. (2002) 73:134–40. doi: 10.1136/jnnp.73.2.134
76. Nobili F, Campus C, Arnaldi D, De Carli F, Cabassi G, Brugnolo A, et al. Cognitive-nigrostriatal relationships in de novo, drug-naïve Parkinson's disease patients: a [I-123] FP-CIT SPECT study. Mov Disord. (2010) 25:35–43. doi: 10.1002/mds.22899
77. Antonelli F, Ray N, Strafella AP. Imaging cognitive and behavioral symptoms in Parkinson's disease. Exp Rev Neurother. (2010) 10:1827–38. doi: 10.1586/ern.10.173
78. Kuhl DE, Minoshima S, Fessler JA, Ficaro EP, Wieland DM, Koeppe RA, et al. In vivo mapping of cholinergic terminals in normal aging, Alzheimer's disease, and Parkinson's disease. Ann Neurol. (1996) 40:399–410. doi: 10.1002/ana.410400309
79. Antonini A, De Notaris R, Benti R, De Gaspari D, Pezzoli G. Perfusion ECD/SPECT in the characterization of cognitive deficits in Parkinson's disease. Neurol Sci. (2001) 22:45–6. doi: 10.1007/s100720170039
80. Nobili F, Abbruzzese G, Morbelli S, Marchese R, Girtler N, Dessi B, et al. Amnestic mild cognitive impairment in Parkinson's disease: a brain perfusion SPECT study. Mov Disord. (2009) 24:414–21. doi: 10.1002/mds.22381
81. Borghammer P, Van Den Berge N. Brain-first versus gut-first Parkinson's Disease: A hypothesis. J Parkinson's Dis. (2019) 9:S281–95. doi: 10.3233/JPD-191721.
82. Braak H, Del Tredici K, Rüb U, De Vos RA, Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
83. Shi M, Huber BR, Zhang J. Biomarkers for cognitive impairment in Parkinson disease. Brain Pathol. (2010) 20:660–71. doi: 10.1111/j.1750-3639.2009.00370.x
Keywords: mild cognitive impairment, dementia, Parkinson's disease, biomarker, imaging
Citation: Sasikumar S and Strafella AP (2020) Imaging Mild Cognitive Impairment and Dementia in Parkinson's Disease. Front. Neurol. 11:47. doi: 10.3389/fneur.2020.00047
Received: 11 November 2019; Accepted: 14 January 2020;
Published: 31 January 2020.
Edited by:
Yu Zhang, VA Palo Alto Health Care System, United StatesReviewed by:
Lin Tan, Qingdao Municipal Hospital, ChinaSoichiro Shimizu, Tokyo Medical University, Japan
Copyright © 2020 Sasikumar and Strafella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio P. Strafella, YW50b25pby5zdHJhZmVsbGFAdWhuLmNh