 Juan Huang
Juan Huang Huan-yu Meng3
Huan-yu Meng3 Hong-Wei Xu
Hong-Wei Xu Ya-fang Zhou
Ya-fang Zhou Lin Zhou
Lin Zhou- 1Department of Geriatric Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Neurology, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 4Department of Neurology, Hunan Aerospace Hospital, Changsha, China
- 5Department of Neurology, Xiangya Changde Hospital, Changde, China
Stiff limb syndrome (SLS) is a rare autoimmune-related central nervous system disorder, resulting in stiffness and spasms of limbs since onset with rare involvement of the truncal muscles. However, SLS patients will gain notable effects by appropriate therapy focusing on symptomatic treatment and immunotherapy. We reported on a 55-year-old female who showed typical painful spasms in both lower limbs and abduction of the right eyeball that partially responded to low-dose diazepam and had high-titer anti–glutamic acid decarboxylase (anti-GAD) antibody. Electromyography (EMG) only showed continuous motor unit activity (CMUA) in the anterior tibialis and right triceps. Eventually, our patient was diagnosed with SLS and treated with intravenous immunoglobulin (IVIG) and glucocorticoid combined simultaneously. She obtained notable effects. We also review and summarize the current literature on clinical characteristics, coexisting disease, treatment, and outcome of 40 patients with SLS. We hope that this report will provide a basis for further understanding of SLS and promote the formation of more advanced diagnosis and treatment processes.
Introduction
Stiff limb syndrome, a variant of stiff-person syndrome (SPS), is a rare autoimmune-related central nervous system disorder (1–3). SLS is characterized by stiffness and spasms limited to the limbs since onset with rare involvement of the truncal muscles. In 1956, Moersch and Woltman reported on 14 patients with fluctuating truncal and limb muscle rigidity and spasms and first defined a newly discovered disease, “stiff man syndrome” (4). Although some progress has been made in the etiology of SLS, the exact mechanism remains controversial. Previous studies claimed that pathogenic autoantibodies impairing γ-aminobutyric acid (GABA) pathways in the brain and spinal cord could be the reason for the clinical manifestations (2). The incidence of SPS is reported to be approximately one in a million (5), while SLS occurs in 13% of SPS patients (6). The prognosis of SLS is variable and largely depends on the underlying autoimmune response, as antibody-positive patients usually have worse clinical outcomes than antibody-negative patients. We recommend that antibody-positive patients receive both long-term immunotherapy and symptomatic treatment, especially for those with chronic symptoms. For antibody-negative patients, symptomatic treatment can be given in the early stage. Whether to give the immunotherapy depends on the severity of symptoms. In this article, we reported on an anti–glutamic acid decarboxylase (anti-GAD) antibody-positive patient with SLS complicating diabetes mellitus (DM). Treatments with intravenous immunoglobulin (IVIG) and glucocorticoid combined simultaneously, instead of sequentially, obtained significant improvement.
Case Presentation
A 55-year-old female complained that she had experienced episodic bilateral lower limb spasms and pains since November 2017. In September 2018, she felt intense lower lumbar pain after lifting a heavy weight. Magnetic resonance imaging of the spinal cord demonstrated lumbar hyperlordosis and spinal stenosis. To reduce the compression of the lumbar spinal canal and nerve root canal, the patient underwent a lumbar discectomy + lumbar fusion + internal fixation operation. Although lumbar pain was largely relieved, she noticed that the frequency and duration of lower limb spasms were significantly aggravated. At the third month post-operation, she was bedridden and had to maintain lower limb flexion due to severe spasms and pains (Figure 1A).
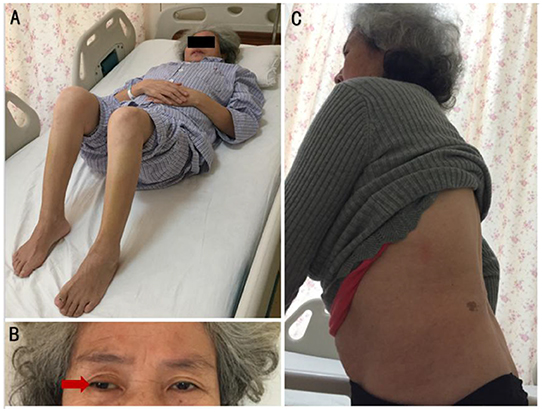
Figure 1. (A) Compulsion position. Lower limb flexion due to severe spasms and pains, with painful spasms triggered by slight movements of the lower limbs. (B) When gazing forward, the right eyeball (red arrow) was abducted relative to the center of the left eyeball. (C) Hyperlordosis of the lumbar spine, without rigidity of the anterior abdominal and lumbar muscles.
Her vital signs were normal. Neurological examinations revealed abduction of the right eyeball when she gazed forward (Figure 1B). In addition, slight lumbar hyperlordosis was found (Figure 1C). Her muscle tone was significantly increased in both lower limbs. Muscle tone was normal in the upper limbs. Deep tendon reflexes were mildly brisk. The Babinski sign was spontaneously positive in both lower limbs. The results from the remainder of the neurological assessments (mental status, cognitive functions, affect, cranial nerves, muscle bulk, and strength sensory examination and coordination) were normal.
Needle electromyography (EMG) revealed continuous motor unit activity (CMUA) only in the anterior tibialis and right triceps (Figure 2). She was found to be positive (++ 1:32) for anti-GAD IgG antibody with an indirect immunofluorescence test (IIFT), strongly positive (+++) for anti-GAD65 IgG antibody by western blot, and negative for anti-amphiphysin IgG antibody (Table 1) with IIFT and western blot. Other laboratory tests after admission showed a moderately increased erythrocyte sedimentation rate [64 mm/h (normal 0–15)] and D-lactate dehydrogenase [288.9 U/L (normal 120–250)], creatine kinase [323.6 U/L (normal 40–200)], and myoglobin levels [141.2 μg/L (normal 0–70)]. Random postprandial blood glucose was up to 13.8mmol/L, and glucose was controlled by daily injections of exogenous insulin. In addition, computed tomography (CT) of the chest, abdomen, pelvis, and brain and 24 h electroencephalography revealed normal results. Laboratory testing results, including a complete blood cell count; a comprehensive metabolic panel including liver, kidney, and thyroid function tests; and tests for levels of electrolytes, C-reactive protein, parathyroid hormone (PTH), HBV-DNA, tumor markers (CEA, AFP, CA15-3, CA125, NSE, CA 72-4, PGI, PGII, and PGR), ANA, ANCA, ACCP, anti-GBM antibody, anti-MPO, anti-PR3, immune factors, rheumatoid factors, and vasculitis factors, were negative. According to the medical history and auxiliary examination results, she was diagnosed with SLS supported by the clinical diagnosis criteria for SPS proposed by Dalakas et al. (2).
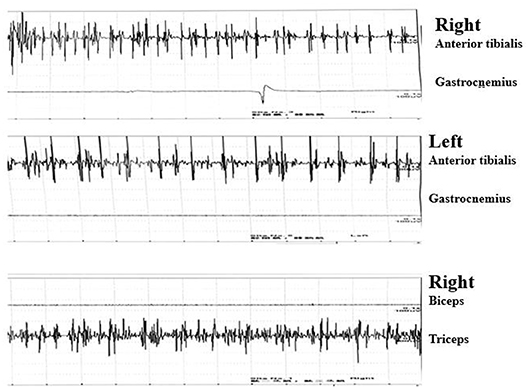
Figure 2. Continuous motor unit activity (CMUA) was found only in the anterior tibialis and right triceps.
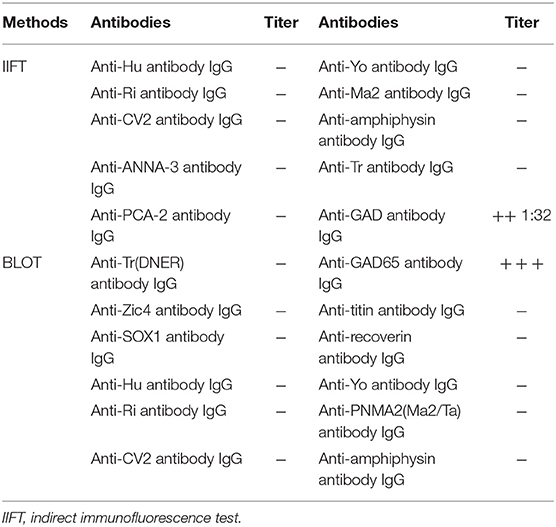
Table 1. Paraneoplastic neurological antibody results.
Treatment with diazepam 2.5 mg q8h po., tizanidine 2 mg tid po., and oxcarbazepine 150 mg bid po. for 7 days only slightly reduced the frequency of lower limb spasms and pain attacks, but she had to stay bedridden to alleviate her remaining symptoms. Another frustrating fact was that she became drowsy with diazepam dosages >10 mg daily. Thus, immunosuppressive therapy was applied. Subsequently, lower limb spasms and pains rapidly disappeared after IVIG (0.4 g/kg/day for 5 days) simultaneously combined with methylprednisolone sodium succinate for injection (500 mg for 3 days and 250 mg for 3 days, subsequently). After the previously mentioned treatment, she could walk with assistance without painful spasms. Post-treatment levels of D-lactate dehydrogenase, creatine kinase, and myoglobin were normal. After she was discharged from the hospital, the dosage of methylprednisolone tablets 48 mg daily was reduced 4 mg per week until 8 mg daily in the long term. Diazepam was maintained at 2.5 mg q8h po. At 8 months following treatment, she could walk with little assistance without painful spasms.
Discussion
Although our patient showed typical painful spasms in both lower limbs and abduction of the right eyeball that partially responded to low-dose diazepam and had high-titer anti-GAD antibody, EMG only showed CMUA in the anterior tibialis and right triceps. CMUA was not specific for SPS, as noted in recent literature (7). In addition, there was not enough clinical or neurophysiological evidence of other neurological disorders resulting in pains and spasms. Therefore, according to the clinical manifestations, this patient was diagnosed with SLS (8).
The age, history, sex, affected limbs, laboratory tests, coexisting diseases, treatments, and outcomes of 40 patients with SLS are summarized in Table 2. The median age at diagnosis was 51 years (range, 11–71 years), and the median time from symptom onset to diagnosis was 2.5 years (range, 20 days −11 years). The ratio of males to females was 1:2.3. Stiffness and spasms were usually asymmetric, more often present in one leg than both legs, and rarely involved the upper limbs. SLS presented with increased probabilities of coexisting autoimmune diseases, such as diabetes (15%) and thyroid disease (17.5%), and a close relationship with breast cancer (12.5%), regardless of antibody-positive or antibody-negative status. With the exception of 13 patients having no baseline data, out of 40 total patients, the remainder (27 patients with SLS) included 19 patients (70.4%) who have autoantibodies against inhibitory synaptic proteins (16 with anti-GAD65 antibody, 2 with anti-glycine receptor (anti-GlyR) antibody, 1 with anti-amphiphysin antibody), 2 patients (7.4%) having another autoantibody (anti-islet and anti-axonal antibody), and 6 patients (22.2%) who were seronegative for antibody. Out of 21 antibody-positive patients, 8 patients (38.1%) gained significant improvements by symptomatic treatment and immunotherapy, and 6 patients (28.6%) only using immunotherapy also showed significant improvements. Moreover, out of six antibody-negative patients, five patients (83.3%) gained significant improvements after symptomatic treatment. We can infer further that the majority (14/21, 66.7%) of antibody-positive patients gained significant improvements by immunotherapy, while most antibody-negative patients (5/6, 83.3%) showed significant improvements with symptomatic treatment. We recommend that antibody-positive patients receive both long-term immunotherapy and symptomatic treatment, especially for those with chronic symptoms. For antibody-negative patients, symptomatic treatment can be given in the early stage. Whether to give the immunotherapy depends on the severity of symptoms.
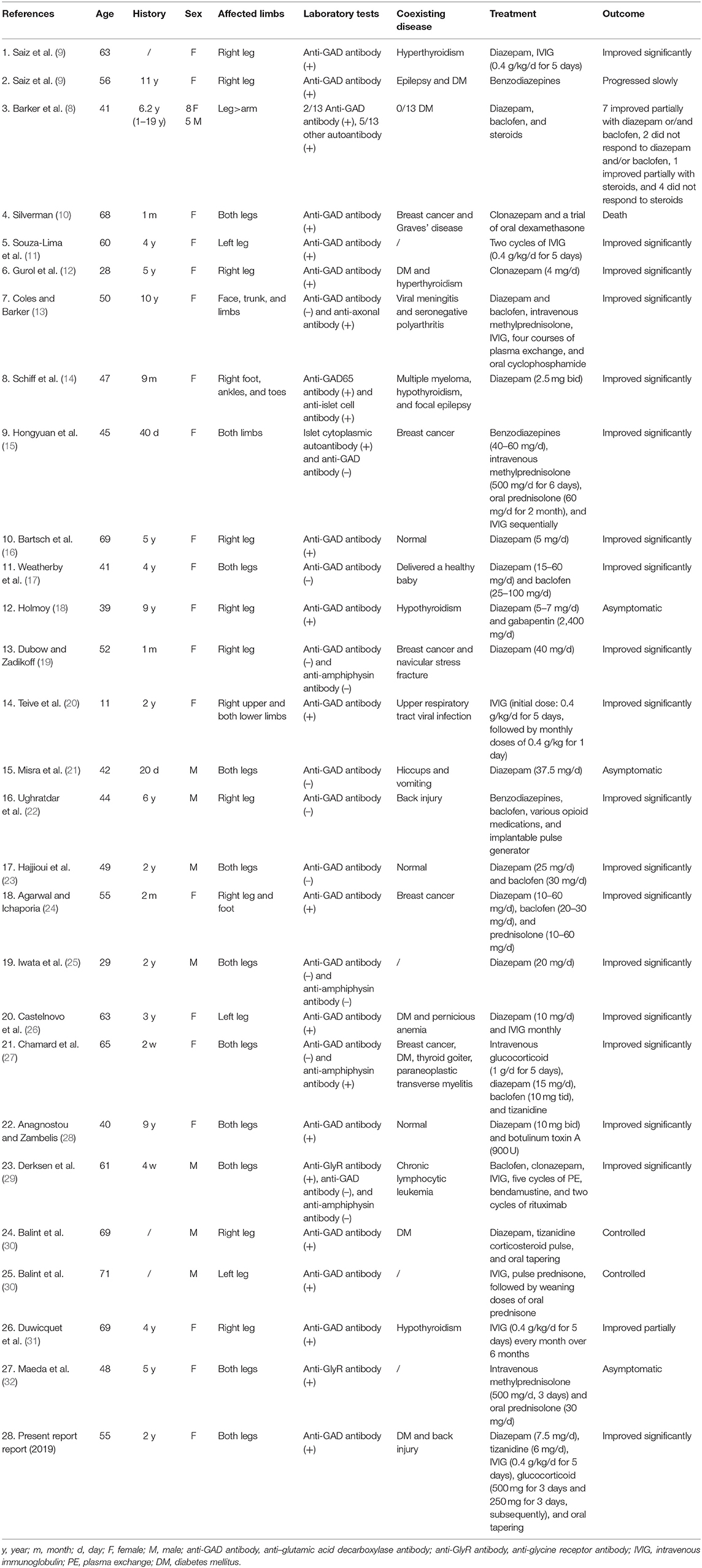
Table 2. Reported cases of stiff limb syndrome.
In 1988, Solimena et al. first reported autoantibody in serum to human brain GAD (anti-GAD) in a patient with SPS, epilepsy, and type I DM (33). Anti-GAD antibody exists in diseases such as type I DM, SPS, cerebellar ataxia, and other neurological disorders (epilepsy, idiopathic limbic encephalitis, and myasthenia gravis) (34). GAD is a pyridoxal 59-phosphate-dependent enzyme that converts glutamate into GABA (35–37). GABA acts as a major inhibitory neurotransmitter in the brain and spinal cord by triggering chloride channels to open, causing the inward movement of chloride into the cell, resulting in membrane hyperpolarization. Anti-GAD antibodies are produced intrathecally to inhibit GAD bioactivity and downregulate GABA expression, resulting in low level of GABA in the brain and cerebrospinal fluid and eventually leading to various clinical manifestations (36).
The treatment of SLS focuses on symptomatic treatment and immunotherapy. Diazepam could increase the opening frequency of the GABAA receptor and lead to hyperpolarization to inhibit excessive neurophysiological activities so that the rigidity and spasms of muscles could be relieved (38, 39). The effective dose ranges from 5 to 60 mg/day for patients with SLS. Most patients with SLS respond to diazepam, but many of them cannot tolerate the high-dose-related adverse effects, including respiratory depression, drowsiness, and dysarthria. The adverse effects of high-dose diazepam have become a major reason why symptomatic treatment fails. Our patient's muscle spasms were partially reduced but were not eliminated due to high doses of diazepam causing drowsiness. The combination of diazepam with other symptomatic treatment, such as baclofen (20–100 mg), clonazepam (4–10 mg/day), tizanidine (6–36 mg/day), and gabapentin (up to 2,400 mg/day), has also been used for relieving SLS symptoms (12, 16–18, 24, 29, 30).
When patients with SLS incompletely respond to diazepam and/or baclofen, IVIG (2 g/kg in 2–5 days) is recommended to improve significant disability in daily activities and is a safe and effective therapy for patients with SLS (9, 20, 30, 31). The effects of IVIG usually last several months, and another IVIG session is started with the return of symptoms (11, 20, 26, 31). Immunoglobulin may suppress the activities of anti-GAD65 antibody by accelerating the rate of IgG catabolism, and the other immunomodulatory effects of immunoglobulin on the neutralization of cytokines and T cells may also play a complementary role (7, 40). Glucocorticoids (e.g., induced as pulse therapy with 500–1,000 mg/day methylprednisolone for 3–5 days and an oral maintenance dose with prednisone 1 mg/kg or 60–80 mg po., oral tapering) could be efficient in treating SLS (24, 27, 30, 32). Plasma exchange may benefit patients with SLS who failed to respond to baclofen and diazepam and cannot tolerate IVIG and glucocorticoids by reducing serum levels of antibodies and other proinflammatory mediators (13, 29). The use of “further immunotherapy” (rituximab, oral cyclophosphamide) is rare in SLS. Interestingly, oral cyclophosphamide was effective in a patient with SLS who was reluctant to accept repeated courses of plasma exchange (13). Another patient with SLS used rituximab and bendamustine (first day, rituximab 350 mg/m2; second and third days, bendamustine 90 mg/m2) to deplete mature B cells, which caused deadly side effects (mydriasis, tachycardia, high blood pressure, hyperthermia, and even cardiac arrest) (29). Therefore, caution should be taken when starting “further immunotherapy” due to a high risk of adverse events, such as severe autonomic instability, opportunistic infections, and neoplasms.
Conclusion
SLS is initially limited to stiffness and spasms of the limbs. Many SLS patients ultimately progress to whole-body stiffness and spasms without appropriate treatment. The etiology in most SLS cases is an autoimmune process, and in rare cases coexisting malignant tumors, especially breast tumors. The treatment of SLS follows the principle of symptomatic treatment and immunotherapy. We recommend that antibody-positive patients receive both long-term immunotherapy and symptomatic treatment, especially for those with chronic symptoms. For antibody-negative patients, symptomatic treatment can be given in the early stage. Whether to give the immunotherapy depends on the severity of symptoms. In summary, we reported on an SLS patient with elevated anti-GAD antibody in serum. Our patient gained notable effects by treating with IVIG and glucocorticoid combined simultaneously. As a result, she resumed normal daily activities after the treatment. However, our patient should maintain long-term low-dose methylprednisolone and diazepam, and tumor monitoring is still necessary. We hope our article provides further understanding of SLS and becomes a reference for future treatment.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JH and HM participated in writing of the paper. XD, WL and H-WX participated in collecting the information of the paper. YZ participated in the clinical data analysis. LZ participated in the revising of the paper.
Funding
This study was supported by a grant from the Key Applied Basic Research Programs of Hunan Province (Grant No. 2016JC2060).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baizabal-Carvallo JF, Jankovic J. Stiff-person syndrome: insights into a complex autoimmune disorder. J Neurol Neurosurg Psychiatry. (2015) 86:840–8. doi: 10.1136/jnnp-2014-309201
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. (2009) 11:102–10. doi: 10.1007/s11940-009-0013-9
3. Martinez-Hernandez E, Ariño H, McKeon A, Iizuka T, Titulaer MJ, Simabukuro MM. Clinical and Immunologic Investigations in Patients With Stiff-Person Spectrum Disorder. JAMA Neurol. (2016) 73:714–20. doi: 10.1001/jamaneurol.2016.0133
4. Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin. (1956) 31:421–7.
5. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. (2011) 11:272–82. doi: 10.1136/practneurol-2011-000071
6. Mckeon A, Robinson MT, Mcevoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. (2012) 69:230–8. doi: 10.1001/archneurol.2011.991
7. Balint B, Meinck HM. Pragmatic Treatment of Stiff Person Spectrum Disorders. Mov Disord Clin Pract. (2018) 5:394–401. doi: 10.1002/mdc3.12629
8. Barker RA, Revesz T, Thom M, Marsden CD, Brown P. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. (1998) 65:633–40. doi: 10.1136/jnnp.65.5.633
9. Saiz A, Graus F, Valldeoriola F, Valls-Sole J, Tolosa E. Stiff-leg syndrome: a focal form of stiff-man syndrome. Ann Neurol. (1998) 43:400–3. doi: 10.1002/ana.410430322
10. Silverman IE. Paraneoplastic stiff limb syndrome. J Neurol Neurosurg Psychiatry. (1999) 67:126–7. doi: 10.1136/jnnp.67.1.126
11. Souza-Lima CF, Ferraz HB, Braz CA, Araujo AM, Manzano GM. Marked improvement in a stiff-limb patient treated with intravenous immunoglobulin. Mov Disord. (2000) 15:358–9. doi: 10.1002/1531-8257(200003)15:2 <358::aid-mds1032>3.0.co;2-l
12. Gurol ME, Ertas M, Hanagasi HA, Sahin HA, Gursoy G, Emre M. Stiff leg syndrome: case report. Mov Disord. (2001) 16:1189–93. doi: 10.1002/mds.1224
13. Coles A, Barker R. A case of stiff limb syndrome responsive to plasma exchange. J Neurol Neurosurg Psychiatry. (2001) 70:407–8. doi: 10.1136/jnnp.70.3.407a
14. Schiff D, Dalmau J, Myers DJ. Anti-GAD antibody positive stiff-limb syndrome in multiple myeloma. J Neurooncol. (2003) 65:173–5. doi: 10.1023/B:NEON.0000003754.34527.f2
15. Hongyuan D, Jie L, Xu Fei. Clinical Analysis of Stiff Limb Syndrome. Stroke and Nervous Disease 10. (2003) 43–45. doi: 10.3969/j.issn.1007-0478.2003.01.012
16. Bartsch T, Herzog J, Baron R, Deuschl G. The stiff limb syndrome–a new case and a literature review. J Neurol. (2003) 250:488–90. doi: 10.1007/s00415-003-1002-7
17. Weatherby SJM, Woolner P, Clarke CE. Pregnancy in stiff-limb syndrome. Mov Disord. (2004) 19:852–854. doi: 10.1002/mds.20094
18. Holmoy T. Long-term effect of gabapentin in stiff limb syndrome: a case report. Eur Neurol. (2007) 58:251–2. doi: 10.1159/000107973
19. Dubow JS, Zadikoff C. Isolated navicular fracture presenting as stiff limb syndrome: a case report. Mov Disord. (2007) 22:2136–7. doi: 10.1002/mds.21704
20. Teive HA, Munhoz RP, Cardoso J, Amaral VC, Werneck LC. Stiff-three limbs syndrome. Mov Disord. (2009) 24:311–2. doi: 10.1002/mds.22344
21. Misra UK, Maurya PK, Kalita J, Gupta RK. Stiff limb syndrome: end of spectrum or a separate entity? Pain Med. (2009) 10:594–7. doi: 10.1111/j.1526-4637.2009.00578.x
22. Ughratdar I, Sivakumar G, Basu S. Spinal cord stimulation to abort painful spasms of atypical stiff limb syndrome. Stereotact Funct Neurosurg. (2010) 88:183–6. doi: 10.1159/000313871
23. Hajjioui A, Benbouazza K, Faris Mel A, Missaoui A, Hassouni NH. Stiff limb syndrome: a case report. Cases J. (2010) 3:60. doi: 10.1186/1757-1626-3-60
24. Agarwal PA, Ichaporia NR. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. (2010) 58:449–51. doi: 10.4103/0028-3886.65704
25. Iwata T, Shigeto H, Ogata K, Hagiwara K, Kanamori Y, Uehara T, et al. Hyperexcitability restricted to the lower limb motor system in a patient with stiff-leg syndrome. J Clin Neurosci. (2011) 18:1720–2. doi: 10.1016/j.jocn.2011.03.021
26. Castelnovo G, Renard D, Bouly S, Labauge P. Isolated hypertrophy of the tibialis anterior muscle in the stiff leg syndrome. Muscle Nerve. (2011) 44:306. doi: 10.1002/mus.22152
27. Chamard L, Magnin E, Berger E, Hagenkotter B, Rumbach L, Bataillard M. Stiff leg syndrome and myelitis with anti-amphiphysin antibodies: a common physiopathology? Eur Neurol. (2011) 66:253–5. doi: 10.1159/000331592
28. Anagnostou E, Zambelis T. Botulinum toxin A in anti-GAD-positive stiff-limb syndrome. Muscle Nerve. (2012) 46:457–8. doi: 10.1002/mus.23416
29. Derksen A, Stettner M, Stocker W, Seitz RJ. Antiglycine receptor-related stiff limb syndrome in a patient with chronic lymphocytic leukaemia. BMJ Case Rep 2013. (2013). doi: 10.1136/bcr-2013-008667. [Epub ahead of print].
30. Balint B, Mahant N, Meinck HM, Fung V. Stiff Limb Syndrome Mimicking Corticobasal Syndrome. Mov Disord Clin Pract. (2014) 1:354–356. doi: 10.1002/mdc3.12059
31. Duwicquet C, Biberon J, de Toffol B, Corcia P. Pseudo spastic gait can reveal a Stiff Leg Syndrome (SLS). Clin Neurol Neurosurg. (2016) 147:108–9. doi: 10.1016/j.clineuro.2016.05.026
32. Maeda K, Shimizu F, Sugihara F, Kanazawa N, Iizuka T. A case of anti-glycine receptor antibody-positive stiff-limb syndrome. Clinical neurology. (2019) 59:98–101. doi: 10.5692/clinicalneurol.cn-001239
33. Solimena M, Folli F, Denis-Donini S, Comi GC, Pozza G, De Camilli P, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med. (1988) 318:1012–20. doi: 10.1056/NEJM198804213181602
34. Albert S, Yolanda B, Lidia S, Félix G, Luis B, Roser C, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain A J Neurol. (2008) 10. doi: 10.1093/brain/awn183
35. Balint B, Bhatia KP. Stiff person syndrome and other immune-mediated movement disorders - new insights. Curr Opin Neurol. (2016) 29:496–506. doi: 10.1097/WCO.0000000000000351
36. Ali F, Rowley M, Jayakrishnan B, Teuber S, Gershwin ME, Mackay IR. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: Protean additions to the autoimmune central neuropathies. J Autoimmun. (2011) 37:79–87. doi: 10.1016/j.jaut.2011.05.005
37. Sarva H, Deik A, Ullah A, Severt WL. Clinical spectrum of stiff person syndrome: a review of recent reports. Tremor Other Hyperkinet Mov (N Y). (2016) 6:340. doi: 10.7916/D85M65GD
38. Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology. (2000) 55:1531–5. doi: 10.1212/WNL.55.10.1531
39. Bhatti AB, Gazali ZA. Recent Advances and Review on Treatment of Stiff Person Syndrome in Adults and Pediatric Patients. Cureus. (2015) 7:e427. doi: 10.7759/cureus.427
Keywords: stiff limb syndrome, anti–glutamic acid decarboxylase (anti-GAD) antibody, diazepam, intravenous immunoglobulin, glucocorticoid
Citation: Huang J, Meng H-y, Duan X, Li W-w, Xu H-W, Zhou Y-f and Zhou L (2020) Effectiveness of Combined Immunoglobulin and Glucocorticoid Treatments in a Patient With Stiff Limb Syndrome: Case Report and Review of the Literature. Front. Neurol. 11:284. doi: 10.3389/fneur.2020.00284
Received: 19 July 2019; Accepted: 26 March 2020;
Published: 08 May 2020.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Ricardo Constantino Ginestal, Hospital Clínico San Carlos, SpainBonaventura Casanova, University and Polytechnic Hospital of La Fe, Spain
Copyright © 2020 Huang, Meng, Duan, Li, Xu, Zhou and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-fang Zhou, enlmXzE5ODFAY3N1LmVkdS5jbg==; Lin Zhou, emhvdWxpbjkwMTE3QHNpbmEuY29t