 Xianjuan Kou
Xianjuan Kou Dandan Chen
Dandan Chen Ning Chen
Ning Chen- Hubei Key Laboratory of Exercise Training and Monitoring, Tianjiu Research and Development Center for Exercise Nutrition and Foods, College of Health Science, Wuhan Sports University, Wuhan, China
MicroRNAs are small non-coding nucleic acids that are responsible for regulating the gene expression by binding to the coding region and 3' and 5' un-translated region of target messenger RNA. Approximately 70% of known microRNAs are expressed in the brain and increasing evidences demonstrate the possible involvement of microRNAs in Alzheimer's disease (AD) according to the statistics. The characteristic symptoms of AD are the progressive loss of memory and cognitive functions due to the deposition of amyloid β (Aβ) peptide, intracellular aggregation of hyperphosphorylated Tau protein, the loss of synapses, and neuroinflammation, as well as dysfunctional autophagy. Therefore, microRNA-mediated regulation for above-mentioned changes may be the potential therapeutic strategies for AD. In this review, the role of specific microRNAs involved in AD and corresponding applications are systematically discussed, including positive effects associated with the reduction of Aβ or Tau protein, the protection of synapses, the inhibition of neuroinflammation, the mitigation of aging, and the induction of autophagy in AD. It will be beneficial to develop effective targets for establishing a cross link between pharmacological intervention and AD in the near future.
Introduction
Alzheimer's disease (AD) is the most common form of late-life dementia, with the characteristics of memory loss, impaired cognitive function, and various neuropsychiatric disturbances. The pathological changes in AD include the deposition of amyloid β (Aβ) peptide as senile plaques (SPs), the aggregation of hyperphosphorylated Tau protein as neurofibrillary tangles (NFTs), and neurodegeneration. With the acceleration of an aging society, the more and more elderly will be suffered from AD in the future. Although the underlying molecular mechanisms of AD are still unknown, the growing evidence indicates that the deposition of Aβ, the abnormal aggregation of Tau protein, and neuroinflammation play major roles in the pathogenesis of AD.
MicroRNAs with the length of 18–25 nucleotides, as the post-transcriptional regulators of gene expression, usually down-regulate the expression of mRNA and protein upon targeting specific mRNAs by binding to (3′-UTR) of the targets. It has been estimated that about 60% of human genes are regulated by microRNAs (1–3), suggesting that these microRNAs play critical roles in a series of biological processes. Data from literature have shown that microRNAs are widely found in the nervous system, some microRNAs exhibit the abundant expression in the brain and participate in neuronal development, synaptic plasticity, neuronal differentiation, and the pathogenesis of neurodegenerative disorders (4). Meanwhile, the expression of some microRNAs is dynamically regulated during the process of brain development, neuronal maturation and neurogenesis (5). Therefore, the slight aberration in the expression and activity of microRNAs may be detrimental to brain functions (6–8).
Although pathological features of AD are very well-documented; unfortunately, the available treatments cannot terminate disease progression, but can slow down it. Recently, growing evidence has demonstrated that the dysfunction of microRNAs within neurons and the altered expression of microRNAs are highly associated with the pathogenesis of neurodegenerative diseases (9–12). Thus, the regulation of microRNAs by exogenous interventions will provide a new perspective to explore the pathogenesis and neuropathology of AD.
microRNAs Involvement in AD
Aβ Regulation Mediated by microRNAs in AD
Excessive accumulation of Aβ may induce significant cytotoxicity in neurons, and is a key pathogenic factor of AD. Increasing evidence suggests that microRNAs can affect Aβ production. Several studies have used profiling strategies to identify the dys-regulation of microRNAs in AD. Some dys-regulated microRNAs involved in the regulation of Aβ deposition have been reported in human brain, mouse models, and cell lines with AD (9, 13). Aβ peptide is produced from amyloid precursor protein (APP) after cleaved by beta-site APP cleaving enzyme 1 (BACE1). Moreover, presenilin 1 (PS1) mutation with the function to produce Aβ from its precursor beta APP can enhance p53 activity in human embryonic kidney (HEK)-293 cells and p53 expression in familial AD (FAD)-affected brains (14). Some specific microRNAs have been reported to be either up-regulated or down-regulated in AD. For example, miR-9, miR-29, miR-29a/b-1, miR-124, miR-101, miR-107, miR-298, and miR-328 contribute to the increase of Aβ production, all of them can exhibit the reduced expression in patients or model animals with AD by regulating the expression of BACE1 and/or APP (15–18). Data from clinical studies have demonstrated that miR-29a/101 in peripheral whole blood from AD patients is markedly down-regulated (19). Certain microRNAs also participate in physiological regulation of APP levels. For example, the overexpression of miR-106a and miR-520c can result in the significant reduction of APP level in HEK-293 cells (20). Moreover, one study has demonstrated that the reduced expression of miR-16 can potentially cause the accumulation of APP protein in the embryo of spontaneous senescence-accelerated mouse P8 (SAMP8) model mice with AD; in contrast, the overexpression of miR-16 also can cause the decreased expression of APP protein in vitro and in vivo (17). Thus, the exogenous overexpression of these microRNAs may play a critical role in the generation of Aβ. The overexpression of miR-29 in humans and transgenic mice can cause the decrease of endogenous BACE1 and the increase of Aβ production (9). Meanwhile, the decreased expression of miR-17, miR-101 and miR-16 is accompanied with high APP level (21), suggesting that the overexpression of miR-17, miR-101 and miR-16 suppresses APP. Another class of microRNAs down-regulated in 12-month-old SAMP8 mice is miR-195 when compared with SAMR1 mice (22). The overexpression of miR-195 in N2a/APP695 cells presents the decreased Aβ level, while the inhibition of miR-195 leads to the increase of Aβ. The reduced expression of these microRNAs may result in the elevated expression and function of BACE1, thus causing aberrant Aβ production as the characteristics of the brains from humans and mice with AD. In addition, overexpressed miR-186 in neuronal cells can result in reduced Aβ level by suppressing BACE1 expression; however, the down-regulated endogenous miR-186 can cause the increased BACE1 level (23). These findings provide the molecular mechanisms associated with BACE1, APP and Aβ deregulation in AD and new perspectives for the etiology of this disease. However, it remains unclear whether the reduced microRNAs play a primary role in the induction of AD. Besides, other microRNAs increase Aβ levels; for example, miR-128 is involved in the development and progression of AD. The levels of miR-128 and Aβ are significantly increased in the cerebral cortex of 3xTg-AD mice when compared with wild type mice; in contrast, miR-128 knockout mice reveal the improvement of cognitive capacity when compared with 3xTg-AD mice. In another study (24), the inhibition of miR-126 has been found to be neuroprotective against Aβ42 toxicity, suggesting that both miR-128 and miR-126 may be the important mechanistic link with AD progression (25).
The microRNA-Medicated Hyperphosphorylation of Tau Protein in AD
In addition to Aβ, the accumulation of intracellular insoluble hyperphosphorylated Tau protein is another pathological feature in AD. The detrimental effects of altered microRNAs in AD neurons may not be restricted to Aβ production and deposition. MicroRNA is also closely related to the phosphorylation and pathological aggregation of Tau protein. For example, miR-132 has a strong regulatory effect on the central nervous system. According to the studies on miR-132/-212 double knockout mouse model, double knockout mice exhibit significant cognitive deficits in recognition, new object recognition and spatial memory (26). In addition, miR-132/-212 has been reported as the down-regulation in the frontal cortex of the AD subjects with mild cognitive decline (27), thus confirming that miR-132/-212 plays a critical regulatory role in cognitive capacity. On the other hand, miR-101b mimic can rescue Tau pathology, dendritic abnormality, and memory deficits in AD mice (28). MiR-137 level has been shown to be a regulator of neuronal development and cognitive function; and clinically to be decreased in the serum of patients with AD so that it could be used as a marker for early diagnosis (29). Similarly, the level of miR-137 also exhibits a decrease in APP/PS1 transgenic mice; however, miR-137 mimics can inhibit p-Tau (Ser202, Ser396, and Ser404) induced by Aβ1-42 in SH-SY5Y cells. In addition, miR-15a, as one of the members in miR-15 family, is frequently down-regulated in AD (30). Moreover, miR-15a can target extracellular signal-regulated kinase 1 (ERK1) for the involvement of Tau hyperphosphorylation (9). The decreased miR-15 can participate neuronal Tau hyperphosphorylation. Data from clinical trials indicate that miR-106b is down-regulated in sporadic AD patients and SH-SY5Y cells (31), and can inhibit Aβ42-induced Tau phosphorylation at the site of Tyr18. Similarly, the expression of miR-512 from Tau protein-rich brains of the patients with advanced AD is significantly reduced, indicating that miR-512 can negatively regulate Tau protein through targeting Fas-related death domain protein (32). Furthermore, miR-153 from the frontal cortex of AD patients is reduced when compared with age-matched control (33). Of course, there are some abnormally elevated microRNAs involved in the hyperphosphorylation of Tau protein, and miR-125b is markedly elevated in animal models with AD. In C57BL/6 wild-type mice, the injection with miR-125b can cause increased phosphorylation of Tau protein and impaired learning and memory capacity (34). Similarly, in primary hippocampal neurons, the overexpression of miR-125b can lead to Tau hyperphosphorylation, affect synaptic morphology, and accelerate apoptosis (34, 35). Conversely, the suppression of miR-125b in primary neurons can reduce Tau phosphorylation and kinase expression/activity. There is inconsistent with the protective role of miR-125b in AD. In IL-1β-induced primary co-culture of human neuronal-glial cells, miR-146a is significantly up-regulated. The expression of miR-146a correlated with senile plaque density and synaptic pathology in the Tg2576 and 5xFAD TG mouse models (36). In China, based on the study involved in 52 patients with mild and moderate AD, the treatment with modified Shuyu Pill could effectively improve the cognitive function of the patients with mild and moderate AD and the underlying mechanism may be related to inhibiting the expressions of IL-1β/NF-κB/miR-146a in peripheral blood (37). In addition, the up-regulation of miR-26b in temporal cortex of the AD models from the early prodromal stage, and the elevated level of miR-26b in postmitotic rodent and human neurons can contribute to the phosphorylation of Tau and apoptotic cell death (38). Another study has shown that compared with the normal elderly group, miR-34a in blood mononuclear cells of AD patients is significantly down-regulated for regulating the phosphorylation of Tau (39), suggesting that miR-34a could be used as a non-invasive biomarker for AD. Similarly, the co-aggregation of Tau could be associated with specific mutations of PSEN1 and/or PSEN2 genes in sporadic and dominantly inherited AD (40).
microRNA-Mediated Synaptic Dysfunction in AD
The alteration in synaptic plasticity is one of the important features for patients with AD. The genome-wide transcriptome studies indicate that many key genes for synapse activity are down-regulated in AD (41). The recovery of cognitive function can be achieved by restoring the reduced microRNAs acting at the synaptic level. The abnormal down-regulation of miR-188-5p is reported in the cerebral cortices and hippocampus of AD patients when compared with age-matched control subjects (42). Dendritic spine and synapse loss are well-documented in AD. However, the overexpression of miR-188-5p alleviates the decrease in dendritic spine density in rat primary hippocampal neuron cultures with the exposure of Aβ. Long-term potentiation (LTP) is believed to be a synaptic mechanism underlying the storage of long-term memory in the brain. The replenishment of miR-188-5p can improve behavioral outcomes and enhance synaptic activity, importantly, and restore cognitive function in AD mouse models such as 5XFAD mice (42, 43). However, some microRNAs are abnormally elevated in AD models and could have negative effects on neurons. Thus, they could need to be down-regulated by exogenous means. In the study of AD patients, miR-34a/p73 expression is found to be remarkably increased in hippocampal tissues, which participates in modulating synaptic activity by lessening synaptotagmin-1 expression (44). Through microRNA microarray screening analysis, the significant up-regulation of miR-30b in the brain of AD patients and transgenic mice is observed (45). The overexpression of miR-30b in hippocampal tissues can jeopardize synaptic structure and function of hippocampal neurons; in turn, can cause the deficits in cognitive function in normal wild type animals. In contrast, the knockdown of miR-30b in transgenic mice prevents synaptic and cognitive decline. These findings suggest that memory deficits in AD may be caused by microRNA alterations. Additionally, the up-regulation of miR-181 and SIRT1 and the decreased c-Fos protein level are observed in the dorsal and ventral hippocampal tissues of 3xTg-AD mice. SIRT1 and c-Fos transcription factor are involved in memory consolidation as the potential targets of miR-181 (46). Another study has reported that Aβ induces the up-regulation of miR-124 in the brain of Tg2576 mice (47). PTPN1 has been implicated in the formation of hippocampal synapses and learning capacity. Importantly, miR-124 directly targets the 3′-UTR of PTPN1 to suppress its translation, thus disrupting synaptic transmission, plasticity and memory. Consistent with these findings, the up-regulation of miR-574 in APP/PSEN1 mice has been reported and miR-574-5p can influence the expression of neuritin (NRN1) involved in synaptic plasticity (48). The results from HT22 hippocampal neuronal cells have shown that miR-574 inhibitors significantly promote NRN1 expression. Additionally, the treatment with Aβ42 can cause the increase of miR-142-5p in SH-SY5Y neuronal cells. In contrast, the inhibition of miR-142-5p can rescue Aβ42-mediated synaptic dysfunction (49). These findings suggest that the reversal of dys-regulated miR-30b, miR-124, miR-574-5p, and miR-142-5p in the brain may prevent or slow cognitive decline in AD. BDNF as a neurotrophic factor plays a pivotal role in synaptic plasticity and cognition. Previous studies have demonstrated that a reduction in BDNF within the prefrontal cortex and hippocampus is highly related to cognitive deficits in animal models with AD (50, 51). Recent study has demonstrated that miR-10a is also a negative regulator in synapse remodeling as a result of the reduction in BDNF-TrkB signals in AD rats. Similarly, the up-regulation of miR-206 in hippocampal tissue, cerebrospinal fluid, and plasma of APP/PSEN1 transgenic mice is also observed, and the alteration of miR-206 contributes to the pathology of AD through down-regulating BDNF (52).
The Modulation Role of Neuroinflammation in AD
Neuroinflammation in brain tissues of AD models is primarily mediated by microglia and astrocytes. It is a high risk of AD and involved in the pathological process of AD. This is substantiated by increased levels of pro-inflammatory cytokines including TNF-α and/or IL-6 in serum and brain tissue of AD patients when compared with the controls (53, 54). Another evidence comes from the presence of microglial cells surrounding amyloid plaques in AD cerebral cortex, the presence of Aβ deposition in T-cells can activate microglia and reactive astrocytes in the brains of AD patients (55). Moreover, the up-regulation of APP is also associated with neuroinflammation. Inflammatory responses are strongly associated with the altered expression of microRNAs in the AD brain. In order to elucidate which microRNA is important in the production of pro-inflammatory cytokines and proteolytic enzymes in AD, mRNA targets and specific roles in brain need to be identified and established.
Several research groups have investigated the effects of microRNAs on LPS-induced neuroinflammation and Toll-like receptor 4 (TLR4)-mediated inflammation. The miR-132 is involved in multiple physiological and pathological mechanisms, such as neuronal cell development (56), synaptic plasticity (57, 58) and inflammation (59, 60). Recent studies indicate that miR-132 participates in the regulation of inflammation and is a negative regulator of the inflammatory response in PC12 (61). Interestingly, resveratrol treatment could ameliorate inflammatory response in PC-12 cells via up-regulating miR-132. Moreover, based on this report, IL-1β, IL-6, and TNF-α are proposed as the targets of miR-132. Similarly, miR-132 is down-regulated in LPS-induced inflammatory injury in neuron HT-22 cells and the overexpression of miR-132 attenuates the inflammatory response (62). TNF receptor associated factor 6 (TRAF6) linked to promote inflammation may be a direct target of miR-132. In addition, miR-206 can enhance LPS-induced inflammation and promote the release of Aβ in microglia by binding to the 3'-UTR of insulin-like growth factor 1 (IGF-1) so that IGF-1 exposure can mitigate miR-206-induced inflammation in microglia, indicating that the miR-206/IGF-1 signaling pathway may be associated with microglial inflammation in AD (63).
Persistent microglial activation is able to initiate inflammatory activity, results in neuronal damage and eventually causes AD. MiR-155 is one of the most well-studied microRNAs in AD-related neuroinflammatory events. In 3xTg AD animal model, there is a high expression level of miR-155. The up-regulation of miR-155 is simultaneously accompanied with an enhanced activation of microglia and astrocytes, thus triggering the production of inflammatory mediators. Moreover, miR-155 can also contribute to the regulation of AD through activating different T cell functions during inflammation (64). Clinical data from human AD brains indicate that miR-125b and miR-146 levels are elevated to aggravate neuroinflammation and reduce complement factor H, which is associated with the neuronal release of mR-146a and miR-155 and inflammatory spreading in the AD brain (65, 66). During investigating the significance of microRNA release in the AD brain, let-7 family has also gained extensive attention. Let-7 has been reported to be critical for maintaining microglial function in inflammation-mediated injury (67). In the studies on let-7a in LPS-treated microglial BV2 cells, let-7a level is found to be remarkably decreased; however, let-7a overexpression can reduce the production of inducible nitric oxide synthase (iNOS) and IL-6, while promoting anti-inflammatory genes at the same time in microglia (68). Consistent with this finding, let-7a can strongly inhibit the expression of inflammatory cytokines by controlling the activation of apoptosis signal-regulating kinase 1 (ASK1), thus activating anti-inflammatory cytokines such as IL-10 and Mycs in microglia (69). Meanwhile, let-7 could act as a regulator of microglial function during inflammation and be a novel target for enhancing the beneficial function of microglia in CNS disorders. In addition, the released let-7b activates the Toll-like receptor 7, thus resulting in neuronal degeneration. Besides, miR-32-5p knockdown also can ameliorate the production of inflammatory cytokines in LPS-treated microglia and dual-specificity phosphatase 5 (Dusp5) is a direct target of miR-32 (70). Similar with this, miR-204 inhibition could repress inflammation process in LPS-induced mouse microglial cell lines (N9 and BV2) via regulating Sirt1 level (71). One of previous studies has found that the loss of miR-29a disrupts the activity of neuronal navigator 3 that is involved in guidance, and is enriched in degenerating pyramidal neurons in AD (72). Thus, above microRNAs may provide potential therapeutic strategies for neuroinflammation. The pharmacological modulation of microRNAs in anti-inflammatory response can be achieved. For example, klotho at different concentrations (0.5, 1 and 2 nM) or linagliptin (50 μM) can inhibit the expression of TNF-α and then alleviate the inflammation in human peripheral blood mononuclear cells (PBMCs) of AD patients, probably by suppressing inflammatory cytokines and up-regulating miR-29a (73). Therefore, microRNAs may have the therapeutic potential of AD through attenuating neuroinflammation.
microRNA-Mediated Aging in AD
Aging is accompanied with behavioral impairments at different degrees, including impaired learning and memory capacity. Increasing evidence suggests that numerous microRNAs are largely implicated in aging and cellular changes associated with aging. Thus, it is very important to evaluate microRNAs that affect these aging events in order to determine the roles of microRNAs in aging. Previous studies have demonstrated that the majority of microRNAs such as miR-151a-3p, miR-181a-5p, miR-1248, miR-103, miR-107, miR-128, miR-130a, miR-155, miR-24, miR-221, miR-496, and miR-1538 in serum of human are down-regulated as the extension of age (74). Interestingly, miR-1248 and miR-181a are negatively associated with the expression of IL-6 and TNF-α and positively correlated with anti-inflammatory cytokines including TGF-β and IL-10, suggesting that circulating microRNAs could be the biological markers of aging. Data from peripheral blood of AD subjects have demonstrated that miR-34a expression is remarkably up-regulated when compared to normal elderly controls (39, 75). In addition, compared with age-matched healthy control, the increased expression of miR-34a in the brain is closely associated with the severity of AD (76). Also, human and mouse SIRT1 mRNA are the targets of miR-34a, so that miR-34a expression is closely related with human longevity (77). Our research team has also found that swimming intervention with a period of 8 weeks can attenuate brain aging in D-galactose-induced AD rats. Mechanically, swimming training down-regulates miR-34a expression in AD rats, which is also confirmed by miR-34a inhibitor in SH-SY5Y cells (78). In normally aged mice, resveratrol treatment can improve learning and memory capacity through down-regulating miR-124/-134, in turn, activating CREB-BDNF signal pathway, which suggests that a resveratrol-rich diet may be beneficial for preserving cognitive function in aged individuals (79). Up to date, the contribution of microRNAs to age and/or senescence-related changes in gene expression is not completely clear. For example, miR-17, miR-19b, miR-20a, and miR-106a are significantly down-regulated in various human aging model systems (80). Meanwhile, the decrease in these microRNAs is correlated with the increased transcript levels of CDK inhibitor p21/CDKN1A. Similar to this, a series of microRNAs such as miR-9, miR-19a, miR-135a, miR-15b, miR-16, miR-214, and miR-141 are reported to be associated with aging (81, 82). These findings indicate that microRNAs can be the novel markers of cell aging in humans.
Dysfunctional Autophagy and Autolysosomal Proteolysis Mediated by microRNAs in AD
Autophagy involved in the degradation of long-lived proteins, cytosolic components, or damaged organelles is essential for the survival of mature neurons (83–85). Multiple studies have demonstrated that the induction of autophagy plays a neuroprotective role; on the contrary, deficient autophagy or impaired autophagic flux can result in neurological damage in most neurological disorders (86–88). The regulation of Aβ and Tau is critically affected by autophagy. For example, phosphorylated Tau in neurons is mainly removed by normal autophagy, and autophagy activation or enhancement can effectively promote the clearance of Tau (89). In addition, loss-of-function mutations in several genes with autophagy-related function such as Becn1/VPS30/ATG6 (90), Atg7 (91), and Atg5 (92) can result in dysfunctional autophagy and increased accumulation of disordered and aggregated proteins such as Aβ and Tau in AD, indicating that autophagy failure has become an important therapeutic target for AD and regulating autophagy by exogenous means may become a new strategy for AD treatment. Beclin1 plays a significant role in autophagy. The studies from cultured neurons and transgenic mice have verified that the deficiency of Beclin1 can provoke the deposition of Aβ; however, Beclin1 overexpression can mitigate the accumulation of Aβ (93). Moreover, the induction of autophagy via the administration of a lentiviral vector expressing Beclin1 can decrease both intracellular and extracellular Aβ pathology in APP transgenic mice (90). Rapamycin, as an autophagy inducer, can attenuate Aβ accumulation and inhibit Tau phosphorylation in AD mouse models (94). Similarly, autophagy-related genes including LC3-II/LC3-I, Beclin1, Atg7, and autophagic influx are markedly decreased in D-galactose-induced AD rat models when compared with the control; however, 8-week swimming training (autophagy mimics) alleviates cognitive function defects via restoring autophagy in an AD rat model (78). Above findings reveal the reasonable proposition that the induction of autophagy has potential therapeutic benefits in AD. However, considering the functional status of autophagy in AD is context-dependent and complex, controversial data about the applicability of inducing autophagy as a general treatment strategy for AD are also exist. For example, autophagy inhibition has been reported to mitigate Aβ42-induced cell death (95, 96), thus, it appears that the time of intervention for inducing autophagy during the progression of these neurodegenerative diseases should be considered during implementing autophagy induction as a therapeutic approach. As illustrated from one of previous studies, increasing induction of autophagy prior to the development of AD-like pathology in 3×Tg-AD mice can reduce the levels of soluble Aβ, Tau and amyloid plaques, whereas the induction after the formation of mature plaques and tangles has no effect on AD-like pathology or cognitive deficits (97).
In addition to the defects of autophagy at the early stages, autolysosomal proteolysis is significantly impaired in AD and its defect is one of the key pathogenic factors in AD (98, 99); thus, selectively enhancing lysosomal activity by genetic ablation of cystatin B to enhance the clearance of autophagic substrates and ameliorate amyloid pathology and memory deficits in TgCRND8 AD mouse models (100). The recovery of autophagic flux is crucial for reversing spatial learning and cognitive deficit. It has been reported that autophagic flux of AD patients is impaired, and autophagic sequestration is stimulated in AD patients at the early stage, while lysosomal clearance is progressively declined and autophagic flux is gradually hindered due to the lack of the substrate clearance (101). Similarly, increased autophagic flux with daily intra-peritoneal injection of pimozide in AD mice decreases the aggregation of Tau through the mTOR-independent AMPK-ULK1 axis (102). Additionally, the treatment with rapamycin and an anti-epileptic drug carbamazepine can alleviate cognitive impairment and Aβ neuropathology in APP/PS1 transgenic mouse model through restoring normal autophagy (103). These observations support the disruption of substrate proteolysis within autolysosomes as the principal mechanism of dysfunctional autophagy in AD.
In recent years, accumulating evidence suggests that microRNAs play an important role for the regulation of autophagy in brain tissue of AD patients. In addition to the correlation with aging, miR-34 is also linked to autophagy and longevity in several species. The decreased miR-34 level is detected in long-lived dietary-restricted mice. In human cells, miR-34 can target Bcl-2, thereby directly inhibiting autophagy-related BECN1/VPS30 complex (104). According to the reports (105), miR-214-3p is also down-regulated in hippocampal neurons of SAMP8 mice and cerebrospinal fluid from sporadic Alzheimer's disease. The treatment with miR-214-3p for SAMP8 mice improves behavioral performance and attenuates neuronal apoptosis. Another study (106) has also demonstrated that miR-214-3 is down-regulated in patients and model animals with AD. It negatively regulates autophagy, thus exerting its neuroprotective effects and Atg12 is a direct target of miR-214-3p in neurons. MiR-299-5p is also a potent autophagy regulator, and the decreased level of miR-299-5p is also reported in hippocampal tissue of APPswe/PS1dE9 mice and cerebrospinal fluid of AD patients. Strikingly, the overexpression of miR-299-5p promotes cognitive impairment of APPswe/PS1dE9 mice via modulating autophagy and apoptosis by targeting Atg5 (107). Similarly, miR-376a, miR-376b, and miR-181a can prevent starvation-induced autophagy in human cell lines by blocking the expression of Beclin1, Atg4c, or Atg5 (108). In addition, the expression of miR-132/212 in AD brain is also observable, and miR-132/212 is down-regulated in AD (109). Mechanistically, Atg9a and Atg5-12 are its targets. Besides, recent studies have found that autophagy is also regulated by miR-30d and miR-101 through inhibiting Beclin1 and Atg4d expression, which may be a new mechanism for AD (110, 111). The induction of autophagy by pharmacological administration such as resveratrol, osthole and ampelopsin has been proved to effectively reduce neuronal aggregates and alleviate the progression of neurological symptoms in several mouse models with AD through activating microRNA-mediated autophagy (112, 113).
Remarks and Future Directions
The deposition of Aβ, intracellular aggregation of hyperphosphorylated Tau protein, the loss of synapses, neuroinflamamiton and autophagic dysfunction, as well as aging reveal the critical roles in the pathogenesis of AD, which is associated with the dysfunctional regulation of a series of microRNAs (Figure 1). Given the large number of microRNAs involved in AD, the analysis of microRNAs in body fluids is a relatively simple procedure when compared with structural magnetic resonance imaging (MRI) and molecular neuroimaging ith positron emission tomography (PET), and microRNAs appear to be promising. A more complete understanding of the regulatory roles of specific microRNAs in AD will be helpful for the development of therapeutic strategies. Therefore, microRNAs as diagnostic and therapeutic agents in AD should be extensively explored and applied in the future. However, possible limitations of microRNAs including the induction of autophagy at suitable stages need to be further explored and clarified.
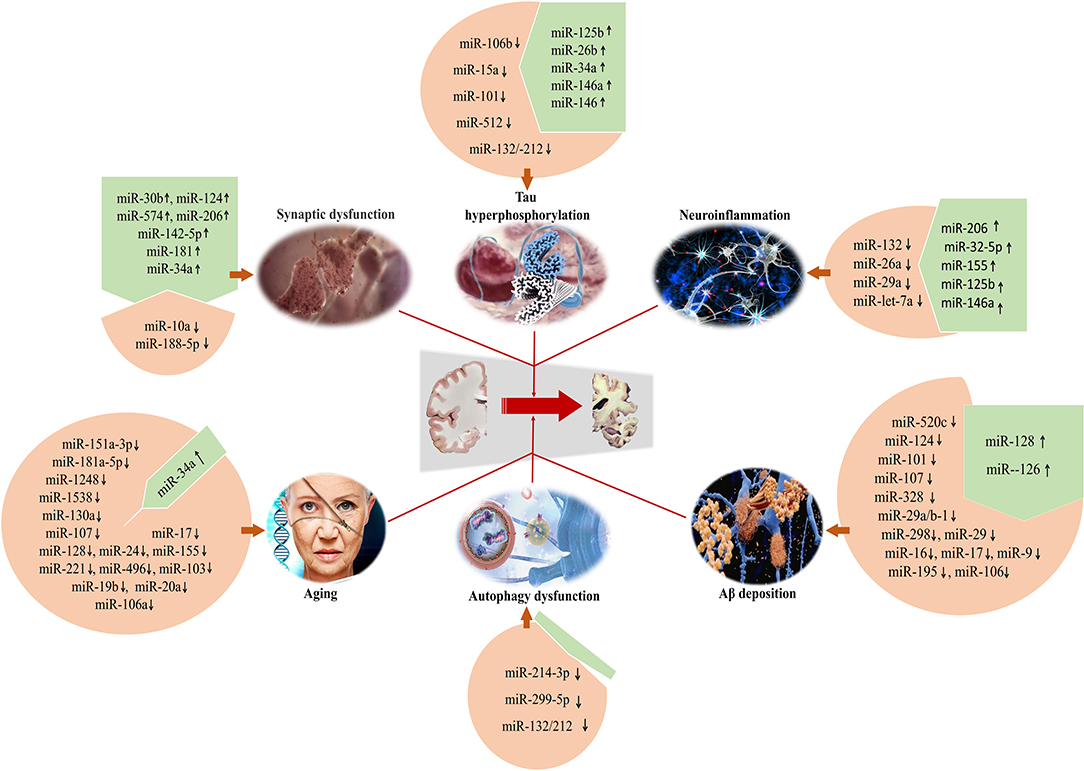
Figure 1. Specific microRNAs involved in the development of AD for regulating Aβ deposition, Tau hyperphosphorylation, synaptic dysfunction, neuroinflammation, and autophagic dysfunction. Meanwhile, microRNAs can be considered as the preventive and therapeutic targets to develop novel and effective intervention strategies for AD.
Author Contributions
NC and XK have designed the project and executed the manuscript writting. XK and DC have participated the literature collection and draft writting. NC have conducted the editing and final reviewing of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 81601228), Natural Science Foundation of Hubei province (2019CFB761), Scientific Research Project of Hubei Ministry of Education (B2019200) and Donghu Scholar Program from Wuhan Sports University to XK; Outstanding Youth Scientific and Research Team (No. T201624) from Hubei Provincial Department of Education, Hubei Superior Discipline Group of Physical Education and Health Promotion and Chutian Scholar Program and Innovative Start-Up Foundation from Wuhan Sports University to NC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. (2004) 5:396–400. doi: 10.1038/nrg1328
2. Bates DJ, Liang R, Li N, Wang E. The impact of noncoding RNA on the biochemical and molecular mechanisms of aging. Biochim Biophys Acta. (2009) 1790:970–9. doi: 10.1016/j.bbagen.2009.03.028
3. Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. (2009) 4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222
4. Müller M, Perrone G, Kuiperij HB, Verbeek MM. Expression of five miRNA targets in hippocampus and cerebrospinal fluid in Alzheimer's disease. Alzheimers Dementia J Alzheimers Association. (2012) 8:P273. doi: 10.1016/j.jalz.2012.05.731
5. Kapsimali M, Kloosterman WP, Bruijn ED, Rosa F, Plasterk RH, Wilson SW. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. (2007) 8:R173. doi: 10.1186/gb-2007-8-8-r173
6. Beveridge NJ, Cairns MJ. MicroRNA dysregulation in schizophrenia. Neurobiol Dis. (2012) 46:263–71. doi: 10.1016/j.nbd.2011.12.029
7. Im HI, Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. (2012) 35:325–34. doi: 10.1016/j.tins.2012.01.004
8. Wong J, Duncan CE, Beveridge NJ, Webster MJ, Cairns MJ, Weickert CS. Expression of NPAS3 in the human cortex and evidence of its posttranscriptional regulation by miR-17 during development, with implications for schizophrenia. Schizophr Bull. (2013) 39:396–406. doi: 10.1093/schbul/sbr177
9. Delay C, Mandemakers W, Hebert SS. MicroRNAs in Alzheimer's disease. Neurobiol Dis. (2012) 46:285–90. doi: 10.1016/j.nbd.2012.01.003
10. Satoh J. Molecular network of microRNA targets in Alzheimer's disease brains. Exp Neurol. (2012) 235:436–46. doi: 10.1016/j.expneurol.2011.09.003
11. Schonrock N, Gotz J. Decoding the non-coding RNAs in Alzheimer's disease. Cell Mol Life Sci. (2012) 69:3543–59. doi: 10.1007/s00018-012-1125-z
12. Iranifar E, Seresht BM, Momeni F, Fadaei E, Mehr MH, Ebrahimi Z, et al. Exosomes and microRNAs: new potential therapeutic candidates in Alzheimer disease therapy. J Cell Physiol. (2019) 234:2296–305. doi: 10.1002/jcp.27214
13. Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem. (2010) 285:18344–51. doi: 10.1074/jbc.M110.112664
14. Alves Da Costa C, Sunyach C, Pardossi-Piquard R, Sevalle J, Vincent B, Boyer N, et al. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer's disease. J Neurosci. (2006) 26:6377–85. doi: 10.1523/JNEUROSCI.0651-06.2006
15. Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci USA. (2008) 105:6415–20. doi: 10.1073/pnas.0710263105
16. Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. (2009) 284:1971–81. doi: 10.1074/jbc.M807530200
17. Hebert SS, Horre K, Nicolai L, Bergmans B, Papadopoulou AS, Delacourte A, et al. MicroRNA regulation of Alzheimer's Amyloid precursor protein expression. Neurobiol Dis. (2009) 33:422–8. doi: 10.1016/j.nbd.2008.11.009
18. Long JM, Lahiri DK. MicroRNA-101 downregulates Alzheimer's amyloid-beta precursor protein levels in human cell cultures and is differentially expressed. Biochem Biophys Res Commun. (2011) 404:889–95. doi: 10.1016/j.bbrc.2010.12.053
19. Ma T, Sun X, Sun S, Guo R, Ma X. The study of peripheral blood miR-29a/101 in the diagnosis of Alzheimer's disease. Chin J Behav Med Brain Sci. (2016) 11:1010–4. doi: 10.3760/cma.j.issn.1674-6554.2016.11.011
20. Patel N, Hoang D, Miller N, Ansaloni S, Huang Q, Rogers JT, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener. (2008) 3:10. doi: 10.1186/1750-1326-3-10
21. Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, et al. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer's-associated pathogenesis in SAMP8 mice. Neurobiol Aging. (2012) 33:522–34. doi: 10.1016/j.neurobiolaging.2010.04.034
22. Zhu HC, Wang LM, Wang M, Song B, Tan S, Teng JF, et al. MicroRNA-195 downregulates Alzheimer's disease amyloid-beta production by targeting BACE1. Brain Res Bull. (2012) 88:596–601. doi: 10.1016/j.brainresbull.2012.05.018
23. Kim J, Yoon H, Chung DE, Brown JL, Belmonte KC, Kim J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem. (2016) 137:436–45. doi: 10.1111/jnc.13507
24. Kim W, Lee Y, Mckenna ND, Yi M, Simunovic F, Wang Y, et al. miR-126 contributes to Parkinson's disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol Aging. (2014) 35:1712–21. doi: 10.1016/j.neurobiolaging.2014.01.021
25. Tiribuzi R, Crispoltoni L, Porcellati S, Di Lullo M, Florenzano F, Pirro M, et al. miR128 up-regulation correlates with impaired amyloid beta(1-42) degradation in monocytes from patients with sporadic Alzheimer's disease. Neurobiol Aging. (2014) 35:345–56. doi: 10.1016/j.neurobiolaging.2013.08.003
26. Hansen KF, Sakamoto K, Aten S, Snider KH, Loeser J, Hesse AM, et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn Mem. (2016) 23:61–71. doi: 10.1101/lm.039578.115
27. Weinberg RB, Mufson EJ, Counts SE. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front Neurosci. (2015) 9:430. doi: 10.3389/fnins.2015.00430
28. Liu D, Tang H, Li XY, Deng MF, Wei N, Wang X, et al. Targeting the HDAC2/HNF-4A/miR-101b/AMPK pathway rescues tauopathy and dendritic abnormalities in Alzheimer's disease. Mol Ther. (2017) 25:752–64. doi: 10.1016/j.ymthe.2017.01.018
29. Jiang Y, Xu B, Chen J, Sui Y, Ren L, Li J, et al. Micro-RNA-137 inhibits Tau hyperphosphorylation in Alzheimer's disease and targets the CACNA1C gene in transgenic mice and human neuroblastoma SH-SY5Y cells. Med Sci Monit. (2018) 24:5635–44. doi: 10.12659/MSM.908765
30. Hebert SS, Papadopoulou AS, Smith P, Galas MC, Planel E, Silahtaroglu AN, et al. Genetic ablation of dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum Mol Genet. (2010) 19:3959–69. doi: 10.1093/hmg/ddq311
31. Reddy PH, Tonk S, Kumar S, Vijayan M, Kandimalla R, Kuruva CS, et al. A critical evaluation of neuroprotective and neurodegenerative MicroRNAs in Alzheimer's disease. Biochem Biophys Res Commun. (2017) 483:1156–65. doi: 10.1016/j.bbrc.2016.08.067
32. Dickson JR, Kruse C, Montagna DR, Finsen B, Wolfe MS. Alternative polyadenylation and miR-34 family members regulate tau expression. J Neurochem. (2013) 127:739–49. doi: 10.1111/jnc.12437
33. Long JM, Ray B, Lahiri DK. MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J Biol Chem. (2012) 287:31298–310. doi: 10.1074/jbc.M112.366336
34. Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H, et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer's disease. EMBO J. (2014) 33:1667–80. doi: 10.15252/embj.201387576
35. Ma X, Liu L, Meng J. MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer's disease. Neurosci Lett. (2017) 661:57–62. doi: 10.1016/j.neulet.2017.09.043
36. Li YY, Cui JG, Hill JM, Bhattacharjee S, Zhao Y, Lukiw WJ. Increased expression of miRNA-146a in Alzheimer's disease transgenic mouse models. Neurosci Lett. (2011) 487:94–8. doi: 10.1016/j.neulet.2010.09.079
37. Xie W, Tan Z, Chen Y, Li X, Liu H. Clinical efficacy of modified shuyu pill for mild and moderate Alzheimer's disease and its effects on expression of IL-1β/NF-κB/miR-146a in peripheral blood. Chin J Integr Tradition Chin Western Med. (2019) 39:668–75.
38. Absalon S, Kochanek DM, Raghavan V, Krichevsky AM. MiR-26b, upregulated in Alzheimer's disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J Neurosci. (2013) 33:14645–59. doi: 10.1523/JNEUROSCI.1327-13.2013
39. Schipper HM, Maes OC, Chertkow HM, Wang E. MicroRNA expression in Alzheimer blood mononuclear cells. Gene Regul Syst Bio. (2007) 1:263–74. doi: 10.4137/GRSB.S361
40. Gondim DD, Oblak A, Murrell JR, Richardson R, Epperson F, Ross OA, et al. Diffuse lewy body disease and Alzheimer disease: neuropathologic phenotype associated with the PSEN1 p.A396T mutation. J Neuropathol Exp Neurol. (2019) 78:585–94. doi: 10.1093/jnen/nlz039
41. Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, et al. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA. (2008) 105:4441–6. doi: 10.1073/pnas.0709259105
42. Lee K, Kim H, An K, Kwon OB, Park S, Cha JH, et al. Replenishment of microRNA-188-5p restores the synaptic and cognitive deficits in 5XFAD mouse model of Alzheimer's disease. Sci Rep. (2016) 6:34433. doi: 10.1038/srep34433
43. Zhang J, Hu M, Teng Z, Tang YP, Chen C. Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer's disease. J Neurosci. (2014) 34:14919–33. doi: 10.1523/JNEUROSCI.1165-14.2014
44. Agostini M, Tucci P, Killick R, Candi E, Sayan BS, Rivetti Di Val Cervo P, et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc Natl Acad Sci USA. (2011) 108:21093–8. doi: 10.1073/pnas.1112061109
45. Song Y, Hu M, Zhang J, Teng ZQ, Chen C. A novel mechanism of synaptic and cognitive impairments mediated via microRNA-30b in Alzheimer's disease. EBioMedicine. (2019) 39:409–21. doi: 10.1016/j.ebiom.2018.11.059
46. Rodriguez-Ortiz CJ, Baglietto-Vargas D, Martinez-Coria H, Laferla FM, Kitazawa M. Upregulation of miR-181 decreases c-Fos and SIRT-1 in the hippocampus of 3xTg-AD mice. J Alzheimers Dis. (2014) 42:1229–38. doi: 10.3233/JAD-140204
47. Wang X, Liu D, Huang HZ, Wang ZH, Hou TY, Yang X, et al. A novel microRNA-124/PTPN1 signal pathway mediates synaptic and memory deficits in Alzheimer's disease. Biol Psychiatr. (2018) 83:395–405. doi: 10.1016/j.biopsych.2017.07.023
48. Li F, Wei G, Bai Y, Li Y, Huang F, Lin J, et al. MicroRNA-574 is involved in cognitive impairment in 5-month-old APP/PS1 mice through regulation of neuritin. Brain Res. (2015) 1627:177–88. doi: 10.1016/j.brainres.2015.09.022
49. Song J, Kim YK. Identification of the role of miR-142-5p in Alzheimer's disease by comparative bioinformatics and cellular analysis. Front Mol Neurosci. (2017) 10:227. doi: 10.3389/fnmol.2017.00227
50. Arancibia S, Silhol M, Mouliere F, Meffre J, Hollinger I, Maurice T, et al. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. (2008) 31:316–26. doi: 10.1016/j.nbd.2008.05.012
51. Ramser EM, Gan KJ, Decker H, Fan EY, Suzuki MM, Ferreira ST, et al. Amyloid-β oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol Biol Cell. (2013) 24:2494–505. doi: 10.1091/mbc.e12-12-0858
52. Silvestro S, Bramanti P, Mazzon E. Role of miRNAs in Alzheimer's disease and possible fields of application. Int J Mol Sci. (2019) 20:E3979. doi: 10.3390/ijms20163979
53. Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, et al. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. (1991) 129:318–20. doi: 10.1016/0304-3940(91)90490-K
54. Strauss S, Bauer J, Ganter U, Jonas U, Berger M, Volk B. Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer's disease patients. Lab Invest. (1992) 66:223–30.
55. Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int J Dev Neurosci. (2006) 24:167–76. doi: 10.1016/j.ijdevneu.2005.11.014
56. Tognini P, Pizzorusso T. MicroRNA212/132 family: molecular transducer of neuronal function and plasticity. Int J Biochem Cell Biol. (2012) 44:6–10. doi: 10.1016/j.biocel.2011.10.015
57. Impey S, Davare M, Lesiak A, Fortin D, Ando H, Varlamova O, et al. An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol Cell Neurosci. (2010) 43:146–56. doi: 10.1016/j.mcn.2009.10.005
58. Lambert TJ, Storm DR, Sullivan JM. MicroRNA132 modulates short-term synaptic plasticity but not basal release probability in hippocampal neurons. PLoS ONE. (2010) 5:e15182. doi: 10.1371/journal.pone.0015182
59. Luikart BW, Bensen AL, Washburn EK, Perederiy JV, Su KG, Li Y, et al. miR-132 mediates the integration of newborn neurons into the adult dentate gyrus. PLoS ONE. (2011) 6:e19077. doi: 10.1371/journal.pone.0019077
60. Soreq H, Wolf Y. NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol Med. (2011) 17:548–55. doi: 10.1016/j.molmed.2011.06.009
61. Zhang G, Liu Y, Xu L, Sha C, Zhang H, Xu W. Resveratrol alleviates lipopolysaccharide-induced inflammation in PC-12 cells and in rat model. BMC Biotechnol. (2019) 19:10. doi: 10.1186/s12896-019-0502-1
62. Ji YF, Wang D, Liu YR, Ma XR, Lu H, Zhang BA. MicroRNA-132 attenuates LPS-induced inflammatory injury by targeting TRAF6 in neuronal cell line HT-22. J Cell Biochem. (2018) 119:5528–37. doi: 10.1002/jcb.26720
63. Xing H, Guo S, Zhang Y, Zheng Z, Wang H. Upregulation of microRNA-206 enhances lipopolysaccharide-induced inflammation and release of amyloid-β by targeting insulin-like growth factor 1 in microglia. Mol Med Rep. (2016) 14:1357–64. doi: 10.3892/mmr.2016.5369
64. Song J, Lee JE. miR-155 is involved in Alzheimer's disease by regulating T lymphocyte function. Front Aging Neurosci. (2015) 7:61. doi: 10.3389/fnagi.2015.00061
65. Lukiw WJ, Alexandrov PN. Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer's disease (AD) brain. Mol Neurobiol. (2012) 46:11–9. doi: 10.1007/s12035-012-8234-4
66. Redis RS, Calin S, Yang Y, You MJ, Calin GA. Cell-to-cell miRNA transfer: from body homeostasis to therapy. Pharmacol Ther. (2012) 136:169–74. doi: 10.1016/j.pharmthera.2012.08.003
67. Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. (2008) 18:505–16. doi: 10.1016/j.tcb.2008.07.007
68. Cho KJ, Song J, Oh Y, Lee JE. MicroRNA-Let-7a regulates the function of microglia in inflammation. Mol Cell Neurosci. (2015) 68:167–76. doi: 10.1016/j.mcn.2015.07.004
69. Song J, Lee JE. ASK1 modulates the expression of microRNA Let7A in microglia under high glucose in vitro condition. Front Cell Neurosci. (2015) 9:198. doi: 10.3389/fncel.2015.00198
70. Yan T, Zhang F, Sun C, Sun J, Wang Y, Xu X, et al. miR-32-5p-mediated Dusp5 downregulation contributes to neuropathic pain. Biochem Biophys Res Commun. (2018) 495:506–11. doi: 10.1016/j.bbrc.2017.11.013
71. Li L, Sun Q, Li Y, Yang Y, Yang Y, Chang T, et al. Overexpression of SIRT1 induced by resveratrol and inhibitor of miR-204 suppresses activation and proliferation of microglia. J Mol Neurosci. (2015) 56:858–67. doi: 10.1007/s12031-015-0526-5
72. Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, et al. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. (2010) 36:320–30. doi: 10.1111/j.1365-2990.2010.01076.x
73. Sedighi M, Baluchnejadmojarad T, Fallah S, Moradi N, Afshin-Majdd S, Roghani M. Klotho ameliorates cellular inflammation via suppression of cytokine release and upregulation of miR-29a in the PBMCs of diagnosed Alzheimer's disease patients. J Mol Neurosci. (2019) 69:157–65. doi: 10.1007/s12031-019-01345-5
74. Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE. (2010) 5:e10724. doi: 10.1371/journal.pone.0010724
75. Chen J, Qi Y, Liu CF, Lu JM, Shi J, Shi Y. MicroRNA expression data analysis to identify key miRNAs associated with Alzheimer's disease. J Gene Med. (2018) 20:e3014. doi: 10.1002/jgm.3014
76. Sarkar S, Jun S, Rellick S, Quintana DD, Cavendish JZ, Simpkins JW. Expression of microRNA-34a in Alzheimer's disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. (2016) 1646:139–51. doi: 10.1016/j.brainres.2016.05.026
77. Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. (2008) 105:13421–6. doi: 10.1073/pnas.0801613105
78. Kou X, Li J, Liu X, Chang J, Zhao Q, Jia S, et al. Swimming attenuates d-galactose-induced brain aging via suppressing miR-34a-mediated autophagy impairment and abnormal mitochondrial dynamics. J Appl Physiol. (2017) 122:1462–9. doi: 10.1152/japplphysiol.00018.2017
79. Zhao YN, Li WF, Li F, Zhang Z, Dai YD, Xu AL, et al. Resveratrol improves learning and memory in normally aged mice through microRNA-CREB pathway. Biochem Biophys Res Commun. (2013) 435:597–602. doi: 10.1016/j.bbrc.2013.05.025
80. Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Muck C, et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell. (2010) 9:291–6. doi: 10.1111/j.1474-9726.2010.00549.x
81. Tacutu R, Budovsky A, Yanai H, Fraifeld VE. Molecular links between cellular senescence, longevity and age-related diseases - a systems biology perspective. Aging. (2011) 3:1178–91. doi: 10.18632/aging.100413
82. Inukai S, De Lencastre A, Turner M, Slack F. Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE. (2012) 7:e40028. doi: 10.1371/journal.pone.0040028
83. Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta. (2009) 1793:1516–23. doi: 10.1016/j.bbamcr.2008.12.013
84. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. (2011) 146:682–95. doi: 10.1016/j.cell.2011.07.030
85. Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med. (2013) 368:1845. doi: 10.1056/NEJMc1303158
86. Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, et al. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. (2005) 171:87–98. doi: 10.1083/jcb.200505082
87. Zemke D, Azhar S, Majid A. The mTOR pathway as a potential target for the development of therapies against neurological disease. Drug News Perspect. (2007) 20:495–9. doi: 10.1358/dnp.2007.20.8.1157618
88. Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. (2013) 19:51–60. doi: 10.1016/j.molmed.2012.11.001
89. Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, Hanger DP. Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging. (2013) 34:2146–57. doi: 10.1016/j.neurobiolaging.2013.03.015
90. Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. (2008) 118:2190–9. doi: 10.1172/JCI33585
91. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. (2006) 441:880–4. doi: 10.1038/nature04723
92. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. (2006) 441:885–9. doi: 10.1038/nature04724
93. Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, et al. Impaired autophagy and APP processing in Alzheimer's disease: the potential role of Beclin 1 interactome. Prog Neurobiol. (2013) 106–107:33–54. doi: 10.1016/j.pneurobio.2013.06.002
94. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. (2010) 285:13107–20. doi: 10.1074/jbc.M110.100420
95. Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE. (2009) 4:e4201. doi: 10.1371/journal.pone.0004201
96. Wang H, Ma J, Tan Y, Wang Z, Sheng C, Chen S, et al. Amyloid-beta1-42 induces reactive oxygen species-mediated autophagic cell death in U87 and SH-SY5Y cells. J Alzheimers Dis. (2010) 21:597–610. doi: 10.3233/JAD-2010-091207
97. Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE. (2011) 6:e25416. doi: 10.1371/journal.pone.0025416
98. Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. (2008) 28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008
99. Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. (2008) 4:590–9. doi: 10.4161/auto.6259
100. Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. (2011) 7:788–9. doi: 10.4161/auto.7.7.15596
101. Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. (2016) 12:2467–83. doi: 10.1080/15548627.2016.1239003
102. Kim YD, Jeong EI, Nah J, Yoo SM, Lee WJ, Kim Y, et al. Pimozide reduces toxic forms of tau in TauC3 mice via 5' adenosine monophosphate-activated protein kinase-mediated autophagy. J Neurochem. (2017) 142:734–46. doi: 10.1111/jnc.14109
103. Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer's disease. Curr Alzheimer Res. (2013) 10:433–41. doi: 10.2174/1567205011310040008
104. Yang F, Li QJ, Gong ZB, Zhou L, You N, Wang S, et al. MicroRNA-34a targets Bcl-2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol Cancer Res Treat. (2014) 13:77–86. doi: 10.7785/tcrt.2012.500364
105. Zhang Y, Li Q, Liu C, Gao S, Ping H, Wang J, et al. MiR-214-3p attenuates cognition defects via the inhibition of autophagy in SAMP8 mouse model of sporadic Alzheimer's disease. Neurotoxicology. (2016) 56:139–49. doi: 10.1016/j.neuro.2016.07.004
106. Du X, Huo X, Yang Y, Hu Z, Botchway BOA, Jiang Y, et al. miR-124 downregulates BACE 1 and alters autophagy in APP/PS1 transgenic mice. Toxicol Lett. (2017) 280:195–205. doi: 10.1016/j.toxlet.2017.08.082
107. Zhang Y, Liu C, Wang J, Li Q, Ping H, Gao S, et al. MiR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci Rep. (2016) 6:24566. doi: 10.1038/srep24566
108. Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy. (2015) 11:867–80. doi: 10.1080/15548627.2015.1034410
109. Smith PY, Hernandez-Rapp J, Jolivette F, Lecours C, Bisht K, Goupil C, et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. (2015) 24:6721–35. doi: 10.1093/hmg/ddv377
110. Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J. (2011) 30:4628–41. doi: 10.1038/emboj.2011.331
111. Xu Y, An Y, Wang Y, Zhang C, Zhang H, Huang C, et al. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep. (2013) 29:2019–24. doi: 10.3892/or.2013.2338
112. Kou X, Liu X, Chen X, Li J, Yang X, Fan J, et al. Ampelopsin attenuates brain aging of D-gal-induced rats through miR-34a-mediated SIRT1/mTOR signal pathway. Oncotarget. (2016) 7:74484–95. doi: 10.18632/oncotarget.12811
Keywords: Alzheimer's disease, Aβ, microRNA, biomarker, autophagy
Citation: Kou X, Chen D and Chen N (2020) The Regulation of microRNAs in Alzheimer's Disease. Front. Neurol. 11:288. doi: 10.3389/fneur.2020.00288
Received: 07 January 2020; Accepted: 26 March 2020;
Published: 17 April 2020.
Edited by:
Y-H. Taguchi, Chuo University, JapanReviewed by:
Manabu Funayama, Juntendo University, JapanLiena Elbaghir Omer Elsayed, University of Khartoum, Sudan
Copyright © 2020 Kou, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Chen, bmNoZW41MTBAZ21haWwuY29t