 Xiao Yang
Xiao Yang Jing Chen*
Jing Chen* BiXia Zheng
BiXia Zheng Xianyu Liu
Xianyu Liu- Department of Neurology, Children's Hospital of Nanjing Medical University, Nanjing, China
Mutations in PCDH19 are associated with epilepsy, intellectual disability and behavioral disturbances, mostly related to females. The unique X-linked pattern of inheritance affects females predominantly, while usually is transmitted through asymptomatic males. Recently, new research has demonstrated that males with a mosaic pattern of inheritance could also be affected. As yet, PCDH19 mutations have been reported in hundreds of females; however, only 15 mosaic males were reported to exhibit epileptic seizures with the onset ranges between 6 and 31 months. These patients were usually reported to carry various mutations in the PCDH19. Here we describe a non-sense variant at the PCDH19 (c.498C>G; p.Y166*) in the Chinese male that exhibited early developmental delay and frequent seizures starting from the age of 5 months. We aim that this case report, focusing on studying clinical seizures, therapeutic approaches, and the patient's prognosis, will contribute to the cumulative knowledge of this rare and complex genetic disorder.
Introduction
In 1971, Juberg reported a unique inherited epilepsy in a family from North American, in which 15 females seizured at different ages (range from 6 to 18 months) and no male patient was diagnosed (1). Later in 1997, Ryan proposed a X-linked theory to be responsible for the female-related heredity (2). Then the PCDH19 mutation was first reported to be associated with epilepsy and intellectual disability limited to the females (EFMR) in a family in 2008 and proposed as the cause of epilepsy by Dibbens et al. (3).
The gene is located on the long arm of the X-chromosome (Xq22.1) and encodes for protocadherin-19. The location of the gene assumes an unusual X-linked female exclusive inheritance (4). Most of the known mutations are located in the first exon that encodes the extracellular domain of the protein (5, 6). In 2012, Depienne and LeGuern reported the synergetic effect the protocadherin 19 may have with N-cadherin (NCAD) during anterior neurulation in zebrafish. This study found that the protocadherin 19 forms a cis-complex with NCAD, which was shown to play an important role in the regulation of cell motility during brain development. Within the complex, protocadherin 19 plays a dominant part, while NCAD is mostly acting as an essential cis-cofactor (5). It has been proposed that the mutations in PCDH19 has resulted in the loss of the protein function lead to the disruption of the cell-cell adhesion, which could be the underlying reason for the seizures (7). Other than the cellular interference hypothesis, Higurashi proposed the dysfunction of blood-brain barrier (BBB) could be another cause of PCDH19-related epilepsy (8). They found seizures resulting from PCDH19 mutations commonly originate in the limbic region, where it is more closer to the periventricular regions without the BBB. Furthermore, deficiency of some hormones or metabolites caused by these abnormal genes is considered to induce seizures (9). Decrease of allopregnanolone and neuroactive steroids in female patients with PCDH19-related epilepsy can provide strong evidence and further studies of other neurosteroids are needed (10).
Until recently, Cellular Interference was considered to have been associated with the inheritance pattern in which heterozygous females are affected while hemizygous males are healthy (11). However, recent findings show that mosaic male carriers of the mutated PCDH19 may also be affected (12–14). To date, more than 160 mutation sites on PCDH19 in nearly 400 female patients have been detected whereas male patients are significantly rare (11, 12). In this article, we report one male patient with a non-sense variant of the PCDH19 (c.498C>G; p.Y166*) and summarized other reported male cases for further study on clinical seizures, onset ages, drug treatment and disease prognosis.
Case Report
A two-and-half-year-old male was reported to suffer from repetitive seizures starting the age of 5 months. He was born at full-term via normal vaginal delivery without birth-related distress or dysmorphia. Familial and personal neurologic antecedents were negative. Perinatal history and pregnancy were both unremarkable. Except for mildly laryngeal cartilage dysplasia, no other serious congenital diseases of the neonatal period were reported prior to the appearance of the first seizure at the age of 5 months. The first seizure appeared as a tonic seizure following by screaming and a frightened face, including loss of contact, stiff limbs, purple lips, and chewing movements. These onsets usually started during sleep and continued for about 20–30 s with or without a high fever. A cluster of focal seizures appeared with slight fever after 4 months' seizure-free and frequent attacks, this made the patients seek help from the hospital. EEG showed sharp waves, and an MRI scan suggested a widened bilateral frontotemporal gap. Valproate (VPA) treatment started, then tonic seizures still existed from time to time. Levetiracetam (LEV) was added in his treatment after 6 month of continuous seizures, and later of topiramate (TPM). TPM was later replaced by phenobarbital (PB) due to the serious side effect of hypohidrosis. The patient experienced reduced attack frequency after the regimen change; however, the damage to the liver functions forced the withdrawal of VPA starting from August 2019. On August 30, 2019, the patient had another cluster of seizures, which led to the hospitalization and an addition to the regimen of dexamethasone (DXMS) and clonazepam (CZP). The latest reported onset occurred in September 2019. The appearance of epilepsy seizures accompanied by a distinct developmental delay and intellectual disability(ID). No scales had been used for accurately assessing patient's intelligence level and mental disorder whether before, or after his onset. But during the follow-up period, his parents found increased improvement in language communication and physical activity.
We conducted genetic testing (exon sequence) of the patient and his parent's DNA, extracted from the peripheral blood leukocytes, with the cooperation of Running Gene, Beijing, China. Gene analysis revealed a suspected mosaic mutation (c.498C>G) in the exon region (NM_001184880, exon1) of the PCDH19 of the patient but not of his parents (Figures 1A–C). This non-sense mutation results in the early termination of protein translation and significantly affects protein function. According to the guide of The American College of Medical Genetics and Genomics (ACMG), this gene mutation could receive a pathogenic rating.
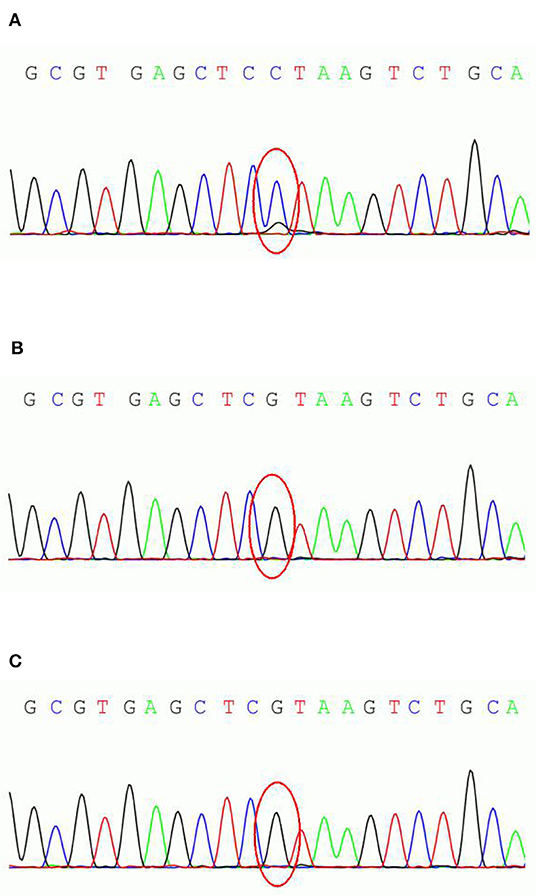
Figure 1. PCDH19 sequencing results of the parent-proband trio. (A) the male patient; (B) patient's father; (C) patient's mother.
Review of Male Patients With PCDH19 Mutations
We used “PCDH19” and “male” as key words to accomplish our literature retrieval in Pubmed. Further reviewing of the literature has detected 14 male PCDH19-related epilepsy cases from eight reports that were integrated with the current case for comparison analysis of the parameters that included the onset age, clinical seizures, variant positions, and treatment. The purpose of this study was to investigate whether PCDH19-related epilepsy has characteristic similarities among the affected males. The study results are summarized in Table 1 (11, 15–18).
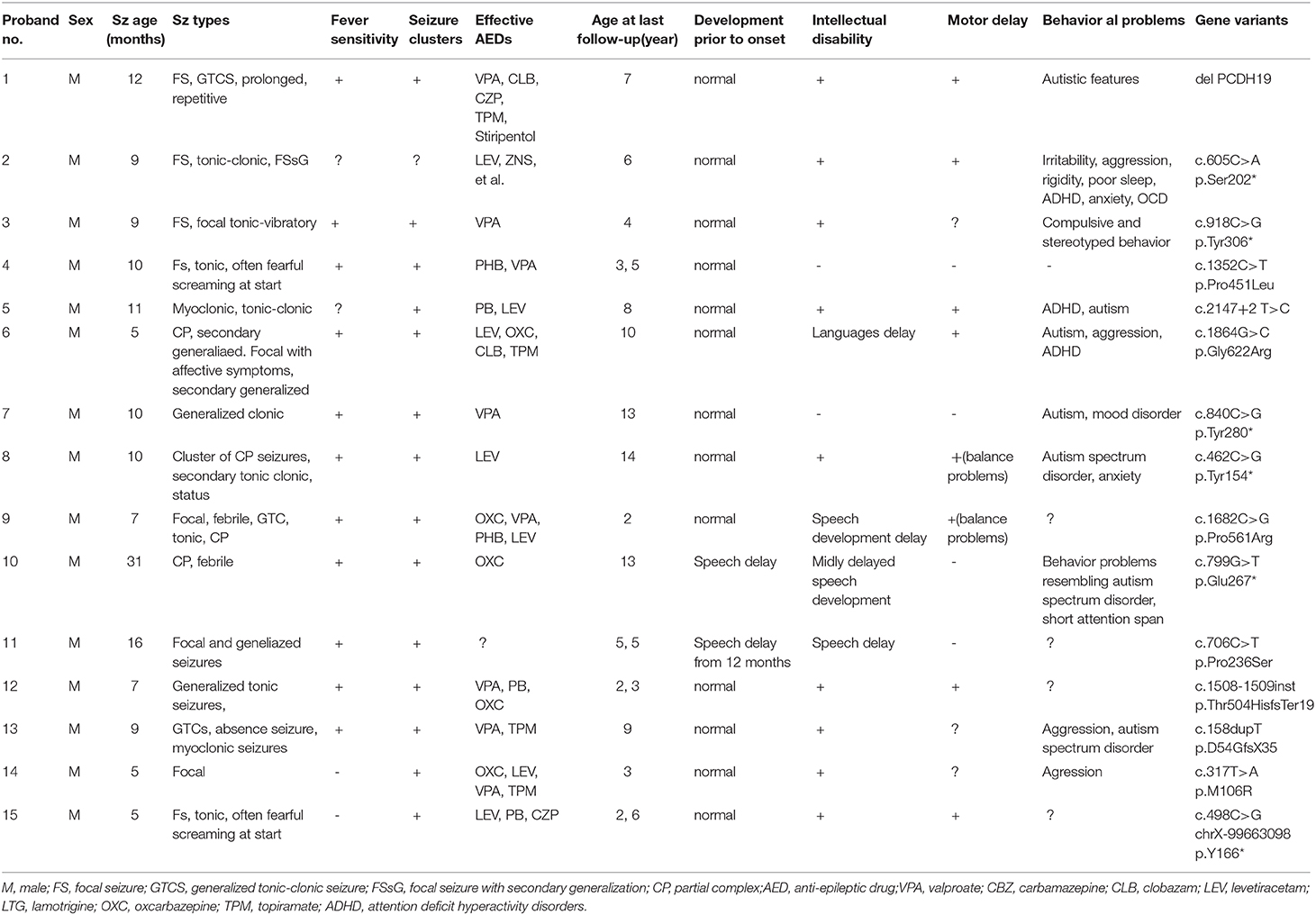
Table 1. Clinical manifestation of 15 male patients with PCDH19 gene variants.
All patients included in this table were males with median onset age of 9.5 months (range 5–31 months). PCDH19 mutation positions contain c.158dupT(p.D54GfsX35), c.317T>A(p.M106R), c.462C>G (p.Tyr154*), c.498C>G(p.Y166*), c.605C>A(p.Ser202*), c.799G>T(p.Glu267*), c.706C>T(p.Pro236Ser), c.840C>G (p.Tyr280*), c.918C>G (p.Tyr306*), c.1352C>T (p.Pro451Leu), c.1508-1509insT(p.Thr504HisfsTer19), c.1682C>G (p.Pro561Arg), c.1864G>C(p.Gly622Arg), c.2147+2T>C. Case 15 refers to the current study at which the patient possesses the same mutation position that was reported in a female patient by Smith L (16). Smith showed that the seizures onset age of the female patient was 10 months, while in our case 15, the disease onset had occurred significantly earlier (5 months). Repeated and uncontrolled seizures of the female patient still existed while variety of antiepileptic drug combination therapy had been used. Long-term follow-up found clear development disability and sleep disorders which had become a serious situation. In the female patient, a focal seizure was the only seizure type, while the male case included complex seizure types such as tonic, tonic-clonic, myoclonic, and generalized tonic-clonic seizures (GTCS). Nearly all of the cases showed more than one seizure type during the disease progression, while only one patient has been free of seizures (case 13). In both cases 4 and 15, a unique fearful screaming was present at the beginning of seizures. Fever sensitivity and seizure clusters were prevalent in 73% of male cases (11 of 15). Following the latest evaluation, in several cases, the patients were treated with more than three medicaments; however, the management of symptoms remained poor-controlled.
In one patient (case 8), treatment with levetiracetam (LEV) was reported as profoundly effective. In other patients, treatment with oral polypharmacy, clonazepam (CZP) and phenobarbital (PB) also was reported as effective. Most cases (13 of 15) were reported as having normal development before seizures' onset, however almost all of the patients exhibited ID or motor delay (delayed expression and balance disorder) in the later stage (except cases 4 and 7). Many patients reported to have a broad spectrum of behavior disturbances in high incidence. These include autism (7 of 15), aggression (4 of 15), and attention deficit hyperactivity disorder (ADHD) (2 of 15).
Discussion
Molecular genetic screening has become an effective path for defining diagnosis or searching for the cause of some epilepsy and epilepsy syndromes, especially in the identification of epilepsy syndromes with similar clinical manifestation. In previous reports, some fever-related epilepsy patients were found SCN1A-negative and were diagnosed as DS-like. Targeted gene detection on PCDH19 should be reconfirmed in patients with the following features: age at onset before 3 years, seizures with earlier fearful screaming, cluster occurrence and cognitive and behavior disturbances (19). These hallmarks of PCDH19-related epilepsy both happened in female and male patients and there is no characteristic differences between two genders. The patient present here has a reported mutation site while his onset age was much earlier than the female case. For male patients, mosaic condition should be considered as possible genetic cause and further functional analyses are needed to prove the pathogenicity in male cases. Depienne used according to the suggested use, could possibly lower the percentage of mosaicism in male patients and this has led to a milder phenotype (5). This assumption cannot be confirmed due to the limited clinical evidence.
Disorders on cognitive development or psychiatric symptoms has become a common situation among patients with PCDH19-related epilepsy. Rarely cases show developmental delay before seizure onsets. Their impairment usually appears after the age of two and is not known to have a connection with seizure severity. Our case had no developmental issues before seizure onset and therefore the typical cognitive delay and motor disability could be observed after seizure onset. Although original antiepileptic drug-treatment was not working well, his developmental delay still improved progressively over time. This feature could also be noticed in other typical developing patients (20). Detailed cognitive assessment should be used to estimate cognitive level of patients, such as Griffiths Mental Developmental Scales and Wechsler scales. Autism is the most common psychiatric symptom accompanied with PCDH19-related epilepsy, accounted for ~20% of patients described by a systematic review (21). The other symptoms include anxiety, obsessive-compulsive disorders, and oppositional defiant disorders. One-fifth of them were defined with multiple psychiatric comorbidities. High incidence of cognitive delay and psychiatric symptoms guided crux of early psychological assessment and followed behavioral therapy.
Just as our summary showed above, most PCDH19-related epilepsy patients needed polytherapy with various combinations and none have proved definitively superior. Several patients with brain lesions had seizure control improved after surgery while most of all is refractory to antiepileptic treatment (22). Jan et al. reported a retrospective multicenter study of antiepileptic therapy in 58 female patients with PCDH19 mutations (11). After 3 months of treatment, bromide and clobazam were affirmed the most effective drugs with a responder rate of 68% and 67%. After 1-year of treatment, bromide and clobazam, respectively, with the responder rate of 50 and 43%. Three quarters of the patients became seizure-free for at least 3 months, and half of them for at least 1 year. At the same time, positive effective of sodium channel blockers would help us to understand the mechanism of epileptogenesis in PCDH19 mutations (23). Further studies should put more attention on precision therapies which can target underlying pathogenesis, in order to improve epilepsy seizure as well as neuropsychiatric symptoms in these patients.
Conclusion
Here we reported a Chinese male patient with a non-sense variant on PCDH19(c.498C>G; p.Y166*). He had a much earlier onset age with typical clinical features and obvious cognitive developmental disorders, which would usually be seen in female patients reported by other scholars. Early onset age of epilepsy seizure might confuse clinical doctors to a wrong diagnosis. While we discover typical features about PCDH19-related epilepsy, molecular genetic screening can further clarify possible epilepsy syndrome. Early psychological assessment along with antiepileptic drugs treatment and followed by behavioral therapy would be essential for recovering social functions. Several adjustments of oral drugs in our case reminded awareness of side effects. Although epilepsy in PCDH19 mutations is often cognitive pharmacoresistant, bromide and clobazam are expected to be effective in long-term treatment of antiepileptic drugs. Until the latest follow-up, combination therapy of DXMS and clonazepam had effect seizure control and longer observation is needed to estimate long-term effectiveness and potential side effects.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XY wrote the article with guidance of JC, while BZ perfected the contents of genetics. XL, ZC, and XW had collected relevant case information.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Juberg RC, Hellman CD. A new familial form of cunvulsivedisorder and mental ratardation limited to females. J Pediatr. (1971) 79:726–32. doi: 10.1016/S0022-3476(71)80382-7
2. Ryan SG, Chance PF, Zou CH, Spinner NB, Golden JA, Smietana, et al. Epilepsy and mental retardation limited to females: an X-linked dominant disorder with male sparing. Nat Genet. (1997) 17:92–5. doi: 10.1038/ng0997-92
3. Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. (2008) 40:776–81. doi: 10.1038/ng.149
4. Gerosa L, Francolini M, Bassani S, Passafaro M. The role of protocadherin 19 (PCDH19) in neurodevelopment and in the pathophysiology of early infantile epileptic encephalopathy-9 (EIEE9). Dev Neurobiol. (2019) 79:75–84. doi: 10.1002/dneu.22654
5. Depienne C, Leguern E. PCDH19-related infantile epileptic encephalopathy: an unusual X-linked inheritance disorder. Hum Mutat. (2012) 33:627–34. doi: 10.1002/humu.22029
6. Niazi R, Fanning EA. Depienne C, Sarmady M, Abou TAN. A mutation update for the PCDH19 gene causing early-onset epilepsy in females with an unusual expression pattern. Hum Mutat. (2019) 40:243–57. doi: 10.1002/humu.23701
7. Cooper SR, Jontes JD, Sotomayor M. Structural determinants of adhesion by Protocadherin-19 and implications for its role in epilepsy. Elife. (2016) 26:e18529. doi: 10.7554/eLife.18529
8. Higurashi N, Takahashi Y, Kashimada A, Sugawara Y, Sakuma H, Tomonoh Y, et al. Immediate suppression of seizure clusters by corticosteroids in PCDH19 female epilepsy. Seizure. (2015) 27:1–5. doi: 10.1016/j.seizure.2015.02.006
9. Trivisano M, Lucchi C, Rustichelli C, Terracciano A, Cusmai R, Ubertini GM, et al. Reduced steroidogenesis in patients with PCDH19-female limited epilepsy. Epilepsia. (2017) 58:e91–5. doi: 10.1111/epi.13772
10. Tan C, Shard C, Ranieri E, Hynes K, Pham DH, Leach D, et al. Mutations of protocadherin 19 in female epilepsy (PCDH19-FE) lead to allopregnanolone deficiency. Hum Mol Genet. (2015) 24:5250–9. doi: 10.1093/hmg/ddv245
11. Lyons S, Marnane M, Reavey E, Williams N, Costello D. PCDH19-related epilepsy: a rare but recognisable clinical syndrome in females. Practical Neurol. (2017) 17:314–7. doi: 10.1136/practneurol-2016-001521
12. Terracciano A, Trivisano M, Cusmai R, Palma LD, Fusco L, Compagnucci, et al. PCDH19-related epilepsy in two mosaic male patients. Epilepsia. (2016) 57: e51-5. doi: 10.1111/epi.13295
13. Thiffault I, Farrow E, Smith L, Lowry J, Zellmer L, Black B, et al. PCDH19-related epileptic encephalopathy in a male mosaic for a truncating variant. Am J Med Genet A. (2016) 170:1585–9. doi: 10.1002/ajmg.a.37617
14. Liu A, Yang X, Yang X, Wu Q, Zhang J, Sun, et al. Mosaicism and incomplete penetrance of PCDH19 mutations. J Med Genet. (2018) 56:81–8. doi: 10.1136/jmedgenet-2017-105235
15. Lange, I.M.D, Rump P, Neuteboom RF, Augustijn. Male patients affected by mosaic PCDH19 mutations: five new cases. Neurogenetics. (2017) 18:147–53. doi: 10.1007/s10048-017-0517-5
16. Romasko EJ, DeChene ET, Balciuniene J, Akgumus GT, Helbig I, Tarpinian JM. PCDH19-related epilepsy in a male with Klinefelter syndrome: additional evidence supporting PCDH19 cellular interference disease mechanism. Epilepsy Res. (2018) 145:89–92. doi: 10.1016/j.eplepsyres.2018.06.008
17. Tan Y, Hou M, Ma S, Liu P, Xia S, Wang Y, et al. Chinese cases of early infantile epileptic encephalopathy: a novel mutation in the PCDH19 gene was proved in a mosaic male- case report. BMC Med Genet. (2018) 19:92. doi: 10.1186/s12881-018-0621-x
18. Perez D, Hsieh DT, Rohena L. Somatic Mosaicism of PCDH19 in a male with early infantile epileptic encephalopathy and review of the literature. Am J Med Genet A. (2017) 173:1625–30. doi: 10.1002/ajmg.a.38233
19. Trivisano M, Specchio N. The role of PCDH19 in refractory status epilepticus. Epilepsy Behav. (2019) 101:106539. doi: 10.1016/j.yebeh.2019.106539
20. Cappelletti S, Specchio N, Moavero R, Terracciano A, Trivisano M, Pontrelli G, et al. Cognitive development in females with PCDH19 gene-related epilepsy. Epilepsy Behav. (2015) 42:36e40. doi: 10.1016/j.yebeh.2014.10.019
21. Kolc KL, Sadleir LG, Scheffer IE, Ivabcevic A, Roberts R, Pham DH, et al. A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Mol Psychiatry. (2019) 2019:241–51. doi: 10.1038/s41380-018-0066-9
22. Kurian M, Korff CM, Ranza E, Bernasconi A, Lubbig A, Nangia S, et al. Focal cortical malformations in children with early infantile epilepsy and PCDH19 mutations: case report. Dev Med Child Neurol. (2018) 60:100–5. doi: 10.1111/dmcn.13595
Keywords: PCDH19, gene mutation, epilepsy, male, neurology
Citation: Yang X, Chen J, Zheng B, Liu X, Cao Z and Wang X (2020) PCDH19-Related Epilepsy in Early Onset of Chinese Male Patient: Case Report and Literature Review. Front. Neurol. 11:311. doi: 10.3389/fneur.2020.00311
Received: 29 December 2019; Accepted: 31 March 2020;
Published: 30 April 2020.
Edited by:
Eliane Kobayashi, McGill University, CanadaReviewed by:
Marina Trivisano, Bambino Gesù Children Hospital (IRCCS), ItalyKette D. Valente, University of São Paulo, Brazil
Copyright © 2020 Yang, Chen, Zheng, Liu, Cao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Chen, Y2hlbmppbmc1NjQwMDQyQDE2My5jb20=