 Walter J. Lukiw1,2,3,4*
Walter J. Lukiw1,2,3,4*- 1Bollinger Professor of Alzheimer's Disease, Louisiana State University School of Medicine, New Orleans, LA, United States
- 2LSU Neuroscience Center, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 3Department of Ophthalmology, LSUHSC, New Orleans, LA, United States
- 4Department of Neurology, Louisiana State University Health Sciences Center, New Orleans, LA, United States
The mouse- and human-brain-resident, nuclear factor kappa B (NF-κB)-regulated, micro RNA-146a-5p (miRNA-146a-5p) is an inducible, 22-nucleotide, single-stranded non-coding RNA (sncRNA) easily detected in several brain and immunological cell types, and an important epigenetic modulator of inflammatory signaling and the innate-immune response in several neurological disorders. Among all studied microRNAs, miRNA-146a-5p (typically referred to as just miRNA-146a) has been well characterized and its pathological function in progressive, age-related, and lethal human inflammatory neurodegenerative disease states is well documented. This communication will review our current understanding of miRNA-146a, its induction by the NF-kB-stimulating actions of inflammatory mediators, including the secretory products of certain microbial species such as viral vectors, and Gram-negative bacteria (such as Bacteroides fragilis) that are normal residents of the human gastrointestinal (GI) tract microbiome, and how miRNA-146a appears to contribute to neuro-pathological, neuro-inflammatory, and altered neuro-immunological aspects of both Alzheimer's disease (AD) and prion disease (PrD).
Overview
One of the most important advances in modern neurobiology over the last fifteen years has been the discovery of microRNA (miRNA) in the mammalian central nervous system (CNS), and the characterization of the multiple roles of certain of these CNS-enriched miRNA signals in aging, development, longevity, and neurological health and disease (1–10). As small, soluble, amphipathic regulatory molecules that are important posttranscriptional and epigenetic regulators of messenger RNA (mRNA) abundance, speciation, and complexity, miRNAs (i) exist as 18- to 25-ribonucleotide (nt), single-stranded non-coding RNAs (sncRNAs) whose sequences are both unique and highly selected over evolution; (ii) represent the smallest information-carrying ribonucleic acids yet defined; and (iii) have been repeatedly shown to play critical and determinant roles in the onset and propagation of many human CNS disorders including progressive, incapacitating, and terminal neurological syndromes. Genome-wide analysis of all known human miRNAs, which currently number about ~2,650 unique species, has indicated that only certain miRNAs, which probably number only about ~25–30 miRNA types, (i) are abundant in the cytoplasm of human brain, retinal, and other CNS cells; (ii) are inducible by pathological factors such as pro-inflammatory cytokines and chemokines, by many different types of viral gene-encoded products and secreted bacterial exudates including the Gram-negative bacteria-derived lipopolysaccharide (LPS) and pathogenic enterotoxins (such as fragilysin) of many microbial species resident of the human gastrointestinal (GI) tract microbiome. Of these human brain-resident miRNA species are a selective group of inducible miRNAs that are all under transcriptional control by the pro-inflammatory transcription factor NF-kB (p50/p65). This well-characterized subset of “rapidly activateable” miRNAs defines a sub-family of miRNA all containing one-to-several NF-kB (p50/p65) recognition features in the immediate 5' upstream sequence of their proximal promoter regulatory regions (9–12) (Figure 1). One of these miRNAs is miRNA-146a, normally only moderately abundant in the human brain neocortex and hippocampus, but inducible to many times its basal level by pathogenic agents associated with stress, pro-inflammatory glycolipids (such as LPS), local levels of reactive oxygen species (ROS), and the abundance of Aβ42 peptides and prion amyloids whose accumulation are characteristic, respectively, of the human disorders Alzheimer's disease (AD) and prion disease (PrD) (11, 12, 14, 24–32). For example, in one study, Genechip- and microfluidic fluorescent array-based and/or LED-Northern dot blot miRNA analysis that interrogates the entire 2,650 miRNAs yet characterized in human brain tissues revealed a selective upregulation of miRNA-146a in 36/36 short post-mortem interval (PMI) human superior temporal lobe neocortical tissue specimens analyzed, compared to 30- age-, gender-, and PMI-matched controls (all PMIs < 3.1 h) (10–12, 14, 24, 25). miRNA-146a levels were found to range from 1.7- to 3.6-fold greater in AD than in age-, gender-, and PMI-matched controls in the same anatomical regions (11, 12, 14, 24, 25). Similarly, multiple independent laboratories have reported that miRNA-146a is significantly upregulated in all human prion diseases (PrD) so far examined including sporadic Creutzfeldt-Jakob disease (sCJD), Gerstmann-Straussler-Scheinker (GSS) syndrome, and fatal familial insomnia (FFI) of humans, bovine spongiform encephalopathy (BSE) of cattle, chronic wasting diseases of the family Cervidae, and murine and other rodent models of these prion disorders (14–20; unpublished observations). Most naturally occurring human forms of PrD represent a group of extremely rare (incidence 1–10 per 100 million) fatal, progressive, transmissible neurodegenerative encephalopathies that appear to arise following the misfolding of the cellular prion protein (PrPC) into an immunogenic disease-associated conformation (PrPSc), or for related lethal neurodegenerations such as AD and accumulation of extremely insoluble, highly pro-inflammatory Aβ42 peptide-containing amyloids. Interestingly, as is further discussed below, there is what appears to be a strong microglial cell-mediated inflammatory and immunological component to both AD and PrD. It has been suggested that miRNA-146a may be both fundamental and integral to innate-immune or inflammatory brain cell responses in AD and PrD-mediated infections, and in other terminal neurological disorders that involve (i) the progressive and irreversible inflammatory neurodegeneration of both diseased murine and human brain tissues and cells; (ii) end-stage proteolipid accumulation; and (iii) the many altered neurochemical and neuropathological signaling processes associated with them (26–36).
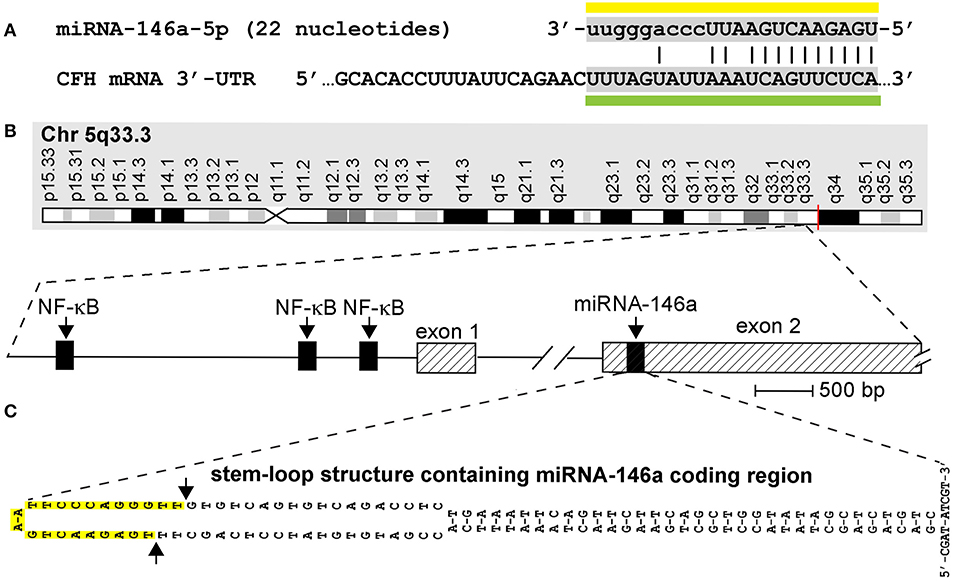
Figure 1. (A) Homo sapiens micro RNA-146a-5p (hsa-miRNA-146a) is a 22-nucleotide (45.5%G+C) single-stranded non-coding RNA (sncRNA) abundant in human brain and immune cells and in the central nervous system (CNS) of mice and humans (miRNA sequence overlined with yellow bar); the homology of the miRNA-146a sequence has been studied in human and several rodent and ape species, and in zebrafish [see Figure 1 in reference (13)] although the actual quenching of specific mRNA targets by miRNA-146a in most of these species still requires further analysis; miRNA-146a has several known mRNA 3′-untranslated region (3′-UTR) targets that include the 3′-UTR of complement factor H (CFH) mRNA (DNA-miRNA recognition sequence underlined with green bar; the miRNA seed sequence is indicated by capitalized ribonucleotides); CFH mRNA encodes a sialic acid-containing 383-amino-acid serum-enriched glycoprotein that plays an essential role in maintaining a homeostatic innate-immune response by modulating complement activation via acting as a soluble inhibitor of complement; CFH is one of the most abundant glycoproteins in the blood serum. CFH has a calculated molecular mass of 43 kDa; however, the apparent molecular mass of CFH in SDS-PAGE gels is approximately 55–60 kDa under reducing conditions due to extensive glycosylation (https://www.genecards.org/cgi-bin/carddisp.pl?gene=CFH; https://www.uniprot.org/uniprot/P08603); miRNA-146a ribonucleotides involved in fully complementary hydrogen bonding are indicated with an (|); the energy of association (EA; predicted energy of association for miRNA–mRNA interaction between hsa-miRNA-146a and CFH is about −24.5 kcal/mol, indicating an extremely stable miRNA–mRNA pairing (14–19); the chromosome location of the human CFH gene is chromosome 1 (chr1q31.3; GenBank accession NC_000001.11); mouse and human miRNA-146a are identical in RNA sequence (3, 15, 18). (B) Structural features of the miRNA-146a encoding DNA locus at chromosome 5q33.3 and details of the NF-κB-sensitive miRNA146a gene showing three upstream (5′) canonical regulatory NF-κB binding sites; as might be expected, miRNA-146a transcription is highly sensitive to induction by NF-κB (2, 3, 14, 15, 20, 21); (C) pre-miRNA-146a transcribed from the miRNA-146a locus has the intrinsic potential to form an extremely stable 35-base-pair stem, 60-nucleotide loop sncRNA structure (stem-loop EA = −49.5 kcal/mol) with miRNA-146a encoded in the distal loop; several other secondary structures of alternate miRNA-146a-stem-loop containing configurations are possible and the 5′ and 3′ ends of pre-miRNA-146a may be significantly extended; in all other high-stability precursor miRNA-146a models, the stem-loop structure containing the mature miRNA-146a sequence is consistently located in the very distal end of the predicted loop [DNA sequence shown; upper case; highlighted in yellow and delineated by arrows on the left side of (C)]; RNA-polymerase II-based transcription and processing by Dicer (RNase III) of the miRNA-146a-5p precursor (pre-miRNA-146a-5p) generated by this unique stem-loop structure yields a mature miRNA-146a-5p [miRNA-146a-5p sequence over-lined with a yellow bar in (A)] (2, 14–16, 23). This figure is an updated and upgraded figure derived in part from a previous publication [(26); Figure 1].
miRNA-146a Attributes in Neurological Disease
In general, the major typical mode of action of miRNAs is to interact via base-pair complementarity, base-pair recognition, and binding, with the 3′-untranslated region (3′-UTR) of their target mRNA 3′-UTRs and in doing so decrease the capability of that specific mRNA to be expressed (15, 37–40). It appears that multiple regions of single-stranded mRNAs, such as the leader sequence or the 5′-untranslated region, or introns, are capable of interacting with miRNAs, but the 3′-UTRs of mature mRNAs are preferred miRNA targets and multiple miRNAs may interact with this 3′-untranslated region, sometimes even at overlapping mRNA binding sites (15, 20, 21, 40–42). Under homeostatic neurophysiological conditions, free energies of association (EA) of about −20 kcal/mol or less between any miRNA and its mRNA 3'-UTR target are highly favorable, and miRNA-146a has been shown to strongly interact with multiple AD- and/or PrD-relevant mRNA targets including those mRNAs encoding the interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 (14) or complement factor H (CFH) (41, 42) (Figure 1). In addition, miRNA-146a targets both human and murine tumor necrosis factor receptor-associated factor 6 (TRAF6), a key downstream signaling protein that interacts with both IRAK1 and IRAK2 in the mediation of inflammatory signaling, although one study showed no significant changes in TRAF6 abundance in AD (14). Other notable attributes of miRNA-146a are as follows: (i) miRNA-146a is abundant in both mouse and human brain so that experimentation with miRNA-146a in both species, as well as in transgenic murine models carrying human genes, is directly comparable (42); (ii) despite about ~96 million years of evolutionary divergence between Rodentia and Homo sapiens, both mouse and human miRNA-146a have identical ribonucleotide sequences (43); (iii) miRNA-146a is particularly abundant in human brain neurons and immunological cells such as in microglial and in monocyte cells; in fact, human miRNA-146a was originally described about 14 years ago as a critical mediator of inflammatory signaling in human monocytes in the laboratory of David Baltimore (16, 17); and (iv) this miRNA has been specifically associated with an upregulation of inflammatory degenerative neuropathology in both neocortical and hippocampal regions in AD, in transgenic murine models of AD and in PrD. As such, miRNA-146a was the first “pro-inflammatory microRNA” characterized because its induction and upregulation are significantly linked to the onset of a dysfunctional innate-immune response and increased inflammatory signaling in multiple neurological disease states (2, 17, 18, 26).
miRNA-146a and Inflammatory Neurodegeneration
Many if not all human diseases and microbial infections, including viral and bacterial, and prion-induced disorders appear to be initiated and/or propagated via a number of critical inflammatory components and the “immunologically privileged” brain and CNS are no exception (37–40). Inflammation is the body's initial response to dyshomeostasis and/or infection and is a key factor in the participation of host defenses against infectious agents, traumatic injury, and the excess of reactive oxidative stress and reactive oxygen species (ROS) that they trigger. However, disordered inflammatory signaling can also contribute to and drive the pathophysiology of chronic neurodegenerative diseases with an age-related, progressive, and inflammatory component such as those encountered in AD and PrD (29, 44). Complex intra- and extracellular interactions of the innate- and adaptive-immune systems and inflammatory mediators including microglial activation, pro-inflammatory cytokines, and chemokines coordinate critical aspects of acute and chronic inflammation. These in turn orchestrate a series of common interactive effector mechanisms of tissue inflammation that contribute interactively to angiogenesis, amyloidogenesis, remodeling of the extracellular matrix, tissue injury moderated in part by reactive oxidative species (ROS), and the recruitment of blood leukocytes that (i) in many tissues characterize the initiation of inflammation, and (ii) influence critical temporal aspects of disease initiation and progression (45–51).
AD like PrD possesses a progressive neuropathology, in part based on the aggregation of abnormal lipoprotein assemblies that result in their recognition by microglia, microglial activation, and the induction of pro-inflammatory signaling cascades that support irreversible neurodegenerative pathways. It appears that the neuroinflammation associated with AD and PrD, and other human neurodegenerative maladies may represent classical examples of an important pathological process involving cyclic, self-reinforcing, and relentless propagation of neuroglial cell activation, release of pro-inflammatory cytokines and related pathogenic factors and neuronal damage, atrophy, dysfunction, and degeneration (47–53). It has recently been suggested that the provision of highly pathogenic prokaryotic signals from the GI tract microbiome involving secreted pro-inflammatory glycolipids such as Bacteroides fragilis LPS and enterotoxins such as the hydrolase fragilysin (EC 3.4.24.74) often found in abundance in the systemic circulation and brain parenchyma of AD and/or PrD patients may well contribute to the initiation and/or propagation of these progressive and irreversible age-related neurodegenerative disease processes (20, 29, 44–46).
miRNA-146a in AD and PrD
It may not be too surprising that the single-copy human miRNA-146a gene precursor encoded from a well-characterized chr 5q33.3 locus, with three tandem NF-kB binding sites in the immediate promoter (upstream from exon 1 of the miRNA-146a coding region), is not only extremely sensitive to NF-kB upregulation but might be an important and critical initial NF-kB-sensor for the onset and initiation of pro-inflammatory signaling in these above-described disease processes (Figure 1). The activation of NF-kB and miRNA-146a is indeed very tightly coupled and is representative of a very rapid neurogenetic signaling mechanism. Kinetically, increases in NF-kB are closely followed by increases in miRNA-146a as measured by Western and gel-shift assay, microfluidic gene array analysis, and RNA sequencing in multiple neurological cell systems (2, 14, 16, 54, 55). These experimental test systems include cytokine and Aβ42-peptide stressed human neuronal–glial (HNG) cells in primary co-culture, transgenic murine models for AD (TgAD; including the amyloid over-expressing 5xFAD TgAD model carrying 5 familial AD mutations), and in AD and PrD themselves (2, 11, 12, 14, 24–26, 31, 34). Interestingly, both the cytoplasmic and nuclear abundance of the NF-kB (p50/p65) transcription complex and miRNA-146a entity are tandemly induced by a remarkable array of multiple classes of potentially pathogenic agents such as (i) pro-inflammatory cytokines and chemokines (including IL-1β, TNFα, and other inflammatory mediators such as prostaglandins); (ii) at least three different broad classes of single- or double-stranded DNA or RNA neurotrophic human CNS- and microbiome-resident viruses ranging from herpes simplex 1 (HSV-1; Herpesviridae; dsDNA genome) to Hantavirus [HTV; Bunyaviridae; (–) ssRNA genome] to human immunodeficiency virus [HIV; Retroviridae; (+) ssRNA genome] (56); (iii) secretory products of GI tract abundant Gram-negative bacteria such as enterotoxins and LPS; (iv) neurotoxic end-stage pathogenic peptides such as the AD abundant Aβ42 peptides and related senile plaque extracts; (v) neurotrophic environmental metallotoxins that include ROS-generating aluminum and mercury sulfates; (vi) multiple biophysical agents that include traumatic shock, heat stress, and hypoxia; or (vii) any combinations of these internally, externally, or environmentally derived factors (15, 20, 31–40). These findings help define and delineate a role for miRNA-146a as an important modulator of the innate-immune response and inflammatory signaling in specific immunological and brain cell types and suggests a broad role for miRNA-146a in the brain's innate-immune signaling response, anti-neurotoxin and inflammatory response, and anti-viral immunity. Besides AD and PrD, miRNA-146a is found to be significantly increased in Aβ42 peptide- and neurotoxic metal-induced, oxidatively stressed HNG cells in primary culture and in other neurodegeneration-associated diseases such as status epilepticus (SE) (56, 57). Importantly, in diseases that have been well characterized at the molecular–genetic and epigenetic level such as AD and PrD, miRNA-146a levels are found to progressively increase with disease severity and co-localize to brain anatomical compartments enriched in inflammatory neuropathology and end-stage amyloid and proteolipid deposition (29, 31, 32, 44). This further supports the concept that an upregulated miRNA-146a and its potential for pathogenic signaling may be integral to innate immune or inflammatory brain cell responses in AD- and PrD-mediated infections and to progressive and irreversible neurodegeneration in both the murine and human brain and CNS.
Unanswered Questions
Multiple unanswered questions remain in the study of the epigenetics and molecular neurobiology of NF-kB–miRNA-146a signaling and its pathogenic contribution to progressive and ultimately lethal age-related neurodegeneration of the mouse and human CNS. While miRNA-146a upregulation is clearly associated with the onset and/or propagation of inflammatory neurodegeneration, it is not entirely clear if this is “the critical pathological basis” for the support of neuroinflammation or in some cases an actual “atypical” neuroprotective response of target brain cells. Like NF-kB, miRNA-146a can have both pro- and anti-inflammatory effects and downstream consequences (54, 55). In HNG cells in primary culture, any externally applied stimulus, whether it be in the form of pro-inflammatory cytokines, highly neurotoxic ROS-generating aluminum or mercury sulfates, pro-inflammatory Aβ42 peptides, prion amyloids, or a viral challenge by HSV-1, or other stress factors such as hypoxia all elicit a robust pathology-inducing response. This is reflected in the rapid upregulation of NF-kB–miRNA-146a signaling over a time scale of minutes, although more chronic low-level NF-kB–miRNA-146a signaling over long time periods has also been observed in some experimental situations (2, 14, 20, 21, 41, 42). It has been further suggested that a small family of other upregulated NF-kB-sensitive miRNAs and the mRNAs of other genes whose transcription is under NF-kB-mediated transcriptional control may contribute to these pathological signaling pathways (37,52,53; unpublished observations). The fact that many of these upregulated responses involving the pathological stimulation of NF-kB (p50/p65) and miRNA-146a-5p in primary HNG brain cells, followed by highly pathogenic, chronic, and fatal consequences, are sensitive to and inhibited (i) by custom-designed metal chelators (in the case of divalent or trivalent neurotoxic metal-induced ROS and NF-kB upregulation); (ii) by anti-viral compounds such as acyclovir, zanamivir, and/or remdesivir (59; unpublished observations), (iii) by anti-NF-kB agents (22, 23, 58, 59); and/or (iv) by anti-miRNA-146a pharmacological treatment strategies that underscore the singular importance of the NF-κB (p50/p65)-miRNA-146a-mediated link in the initiation and/or propagation of pro-inflammatory signaling that supports an irreversible neurodegeneration (22, 22, 23, 59). These downstream events include the NF-kB–miRNA-146a-mediated downregulation of the IRAK-signaling system, deficiencies in CFH, ensuing dysfunction in complement signaling, and the loss of the cell's innate-immunological control (2, 13, 14, 22, 23). Indeed, well-directed ROS-quenching antioxidants and neurotoxic metal chelation, anti-viral, anti-NF-kB, anti-miRNA-146a (antagomir) pharmacotherapeutic approaches, or any combinations of these may be of use in the clinical management of AD, PrD, and other related progressive and terminal neurodegenerative disease processes with an inflammatory component (15, 22, 22, 23, 54, 55, 57–62). Very recently, intranasal administration of a miRNA-146a antagomir was shown to diminish pathological processes and improve cognitive impairment in an APP/PS1 AD transgenic mouse model (13).
Concluding Remarks
Recent and emerging data collectively support the observation that an inducible NF-kB (p50/p65)-miRNA-146a signaling system is upregulated in neurological disorders associated with progressive, age-related inflammatory neurodegeneration including AD. This pro-inflammatory transcription factor–miRNA pairing also displays increased activity in a wide range of murine and human viral and PrD-based infections. These same data suggest that a significant pathological upregulation of NF-kB (p50/p65)–miRNA-146a signaling coupled to downregulation of pathogenic mRNA targets such as IRAK and CFH may directly contribute to altered innate-immune or inflammatory responses in both viral- and prion-mediated infections, features that also accompany progressive, irreversible inflammatory neurodegeneration in a surprisingly wide range of human brain diseases including AD and PrD and in murine transgenic models for these diseases. Currently, there is (i) considerable research effort involving miRNA-based pre-symptomatic predictive and/or diagnostic biomarkers in the cerebrospinal fluid (CSF) and blood serum of the systemic circulation; and (ii) related therapeutic opportunities that address a wide range of these insidious, progressive, and lethal neurological disorders in which signaling along the NF-kB–miRNA-146a axis appears to be playing a critical role in brain cell fate (8, 9, 24, 56). Although much of the data obtained to date are relatively preliminary and need to be further confirmed in suitable brain cell-culture models, transgenic animal model studies, and controlled clinical trials, they draw considerable and thought-provoking attention to a rapidly evolving field of neurobiology and neurogenetics that should acquire increasing relevance in the future as to the nature of the pathogenic signaling mechanisms involved in these insidious and lethal neuropathological states.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher, and are also in the process of being compiled and organized into manuscript form and will available in an upcoming publication “manuscript in preparation” (Autumn, 2020).
Ethics Statement
All acquisition, handling, experimental and analytical procedures involving postmortem human brain tissues were carried out in an ethical manner in strict accordance with the ethics review board policies at brain and tissue donor institutions and at the Louisiana State University (LSU) Health Sciences Center. Informed consent from next of kin was obtained at brain and tissue donor institutions for all tissue samples prior to autopsy and donation; coded postmortem brain tissue samples (containing no personal identifying information of the donors) were obtained from brain tissue banks listed in the Acknowledgments section. The ethical use of postmortem human brain tissues and their analyses were also carried out in strict accordance with the Institutional Biosafety Committee and the Institutional Review Board Committee (IBC/IRBC) ethical guidelines IBC#18059 and IRBC#6774 at the Louisiana State University Health Sciences Center (LSUHSC), New Orleans, LA 70112, United States.
Author Contributions
WL compiled and distilled the results from all laboratory experiments and those of many collaborators both domestic and international, and performed literature searches of recent peer-reviewed publications in this microRNA area of neurobiological research and wrote the article.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research in this article was presented in part at the Vavilov Institute of General Genetics Autumn 2018 Seminar Series in Moscow, Russia, October 2018, at the Society for Neuroscience (SFN) Annual Meeting, Chicago, IL, USA, November 2019. Sincere thanks are extended to Drs. L Cong, F Culicchia, C Eicken, K Navel, AI Pogue, W Poon, T Bond, E Head, Y Zhao, and the late Drs. JM Hill and PN Alexandrov for helpful discussions in this research area, for short post-mortem interval (PMI) human brain and retinal tissues or extracts, and for initial bioinformatics and data interpretation, and to AI Pogue and D Guillot for expert technical assistance and medical artwork. We would like to further thank the following brain and tissue banks for access to high-quality post-mortem tissues and valuable analytical advice: the National Institute of Neurological Disorders and Stroke (NINDS), Bethesda MD USA; the Oregon Health Sciences University, Portland OR, USA; the Southern Eye Bank, Metairie LA, USA; the University of California (UCI) MIND Institute, Irvine CA, USA; and the many neuropathologists, physicians, and researchers in the US and Canada who have provided high-quality, short PMI human brain tissue fractions for scientific analysis. Research on microRNAs, ethnobiology, botanical neurotoxins, pro-inflammatory, and pathogenic signaling in the Lukiw laboratory involving the microbiome, the innate-immune response, amyloidogenesis, synaptogenesis, and neuro-inflammation in AD, prion, and other human neurological- and plant-viroid-based diseases was supported through an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN); and NIH grants NEI EY006311, NIA AG18031, and NIA AG038834 (WL). The content of this manuscript is solely the responsibility of the author and does not necessarily represent the official views of the National Institute on Aging, the National Center for Research Resources, or the National Institutes of Health.
References
1. Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. (2007) 18:297–300. doi: 10.1097/WNR.0b013e3280148e8b
2. Lukiw WJ, Zhao Y, Cui JG. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. (2008) 283:31315–22. doi: 10.1074/jbc.M805371200
3. Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer's disease temporal lobe neocortex. Neurosci Lett. (2009) 459:100–4. doi: 10.1016/j.neulet.2009.04.052
4. Prodromidou K, Matsas R. Species-specific miRNAs in human brain development and disease. Front Cell Neurosci. (2019) 13:559. doi: 10.3389/fncel.2019.00559
5. Giorgi Silveira R, Perelló Ferrúa C, do Amaral CC, Fernandez Garcia T, de Souza KB, Nedel F. MicroRNAs expressed in neuronal differentiation and their associated pathways: Systematic review and bioinformatics analysis. Brain Res Bull. (2020) 157:140–8. doi: 10.1016/j.brainresbull.2020.01.009
6. Kinser HE, Pincus Z. MicroRNAs as modulators of longevity and the aging process. Hum Genet. (2020) 139:291–308. doi: 10.1007/s00439-019-02046-0
7. Silvestro S, Bramanti P, Mazzon E. Role of miRNAs in Alzheimer's disease and possible fields of application. Int J Mol Sci. (2019) 20:E3979. doi: 10.3390/ijms20163979
8. Zhao Y, Zhang Y, Zhang L, Dong Y, Ji H, Shen L. The potential markers of circulating microRNAs and long non-coding RNAs in Alzheimer's disease. Aging Dis. (2019) 10:1293–301. doi: 10.14336/AD.2018.1105
9. Swarbrick S, Wragg N, Ghosh S, Stolzing A. Systematic review of miRNA as biomarkers in Alzheimer's disease. Mol Neurobiol. (2019) 56:6156–67. doi: 10.1007/s12035-019-1500-y
10. Quinlan S, Kenny A, Medina M, Engel T, Jimenez-Mateos EM. microRNAs in neurodegenerative diseases. Int Rev Cell Mol Biol. (2017) 334:309–43. doi: 10.1016/bs.ircmb.2017.04.002
11. Lukiw WJ. NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp Neurol. (2012) 235:484–90. doi: 10.1016/j.expneurol.2011.11.022
12. Lukiw WJ. NF-κB-regulated, pro-inflammatory miRNAs in Alzheimer's disease. Alzheimers Res Ther. (2012) 4:47. doi: 10.1186/alzrt150
13. Mai H, Fan W, Wang Y, Cai Y, Li X, Chen F, et al. Intranasal administration of miR-146a agomir rescued the pathological process and cognitive impairment in an AD mouse model. Mol Ther Nucleic Acids. (2019) 18:681–95. doi: 10.1016/j.omtn.2019.10.002
14. Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kB in stressed human astroglial cells and in Alzheimer disease. J Biol Chem. (2010) 285:38951–60. doi: 10.1074/jbc.M110.178848
15. Hill JM, Clement C, Zhao Y, Lukiw WJ. Induction of the pro-inflammatory NF-kB-sensitive miRNA-146a by human neurotrophic viruses. Front Microbiol. (2015) 6:43. doi: 10.3389/fmicb.2015.00043
16. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
17. Li L, Chen XP, Li YJ. MicroRNA-146a and human disease. Scand J Immunol. (2010) 71:227–31. doi: 10.1111/j.1365-3083.2010.02383.x
18. Arena A, Iyer AM, Milenkovic I, Kovacs GG, Ferrer I, Perluigi M, et al. Developmental expression and dysregulation of miRNA-146a and miRNA-155 in Down's syndrome and mouse models of down's syndrome and Alzheimer's disease. Curr Alzheimer Res. (2017) 14:1305–17. doi: 10.2174/1567205014666170706112701
19. miRBASE. Cambridge. Available online at: http://www.mirbase.org/index.shtml; miRBase is a biological database that acts as an archive of microRNA sequences and annotations (Last accessed 5 March, 2020).
20. Alexandrov P, Zhao Y, Li W, Lukiw W. Lipopolysaccharide-stimulated, NF-kB-, miRNA-146a- and miRNA-155-mediated molecular-genetic communication between the human gastrointestinal tract microbiome and the brain. Folia Neuropathol. (2019) 57:211–9. doi: 10.5114/fn.2019.88449
21. Nagaraj S, Zoltowska KM, Laskowska-Kaszub K, Wojda U. microRNA diagnostic panel for Alzheimer's disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res Rev. (2019) 49:125–43. doi: 10.1016/j.arr.2018.10.008
22. Ju Hwang C, Choi DY, Park MH, Hong JT. NF-κB as a key mediator of brain inflammation in Alzheimer's Disease. CNS Neurol Disord Drug Targets. (2019) 18:3–10. doi: 10.2174/1871527316666170807130011
24. Zhao Y, Bhattacharjee S, Dua P, Alexandrov PN, Lukiw WJ. microRNA-based biomarkers and the diagnosis of Alzheimer's disease. Front Neurol. (2015) 6:162. doi: 10.3389/fneur.2015.00162
25. Kanata E, Thüne K, Xanthopoulos K, Ferrer I, Dafou D, Zerr I, et al. MicroRNA alterations in the brain and body fluids of humans and animal prion disease models: current status and perspectives. Front Aging Neurosci. (2018) 10:220. doi: 10.3389/fnagi.2018.00220
26. Lukiw WJ, Dua P, Pogue AI, Eicken C, Hill JM. Upregulation of micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt-Jakob disease (sCJD) and Gerstmann-Straussler-Scheinker (GSS) syndrome. J Toxicol Environ Health A. (2011) 74:1460–8. doi: 10.1080/15287394.2011.618973
27. Llorens F, Lopez-Gonzalez I, Thune K, Carmona M, Zafar S, Andeoletti O. Subtype and regional-specific neuroinflammation in sporadic Creutzfeldt-Jakob disease. Front Aging Neurosci. (2014) 6:198. doi: 10.3389/fnagi.2014.00198
28. Shah SZA, Zhao D, Hussain T, Sabir N, Yang L. Regulation of microRNAs-mediated autophagic flux: a new regulatory avenue for neurodegenerative diseases with focus on prion diseases. Front Aging Neurosci. (2018) 10:139. doi: 10.3389/fnagi.2018.00139
29. Carroll JA, Chesebro B. Neuroinflammation, microglia, and cell-association during prion disease. Viruses. (2019) 11:E65. doi: 10.3390/v11010065
30. Slota JA, Booth SA. MicroRNAs in neuroinflammation: implications in disease pathogenesis, biomarker discovery and therapeutic applications. Noncoding RNA. (2019) 5:E35. doi: 10.3390/ncrna5020035
31. Saba R, Gushue S, Huzarewich RLCH, Manguiat K, Medina S, Robertson C, et al. MicroRNA 146a (miR-146a) is over-expressed during prion disease and modulates the innate immune response and the microglial activation state. PLoS ONE. (2012) 7:e30832. doi: 10.1371/journal.pone.0030832
32. Moreno JA, Telling GC. Molecular mechanisms of chronic wasting disease prion propagation. Cold Spring Harb Perspect Med. (2018) 8:a024448. doi: 10.1101/cshperspect.a024448
33. Juzwik CA, S Drake S, Zhang Y, Paradis-Isler N, Sylvester A, Amar-Zifkin A, et al. microRNA dysregulation in neurodegenerative diseases: a systematic review. Prog Neurobiol. (2019) 182:101664. doi: 10.1016/j.pneurobio.2019.101664
34. Alexandrov PN, Dua P, Lukiw WJ. Up-Regulation of miRNA-146a in progressive, age-related inflammatory neurodegenerative disorders of the human CNS. Front Neurol. (2014) 5:181. doi: 10.3389/fneur.2014.00181
35. Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer's disease. J Neuroinflammation. (2018) 15:276. doi: 10.1186/s12974-018-1313-3
36. Sochocka M, Zwolinska K, Leszek J. The infectious etiology of Alzheimer's disease. Curr Neuropharmacol. (2017) 15:996–1009. doi: 10.2174/1570159X15666170313122937
37. Barbato C, Ruberti F, Cogoni C. Searching for MIND: microRNAs in neurodegenerative diseases. J Biomed Biotechnol. (2009) 2009:871313. doi: 10.1155/2009/871313
38. Li YY, Cui JG, Hill JM, Bhattacharjee S, Zhao Y, Lukiw WJ. Increased expression of miRNA-146a in Alzheimer's disease transgenic mouse models. Neurosci. Lett. (2011) 487:94–8. doi: 10.1016/j.neulet.2010.09.079
39. Roshan R, Ghosh T, Scaria V, Pillai B. microRNAs: novel therapeutic targets in neurodegenerative diseases. Drug Discov Today. (2009) 14:1123–9. doi: 10.1016/j.drudis.2009.09.009
40. Saugstad JA. microRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. J Cereb Blood Flow Metab. (2010) 30:1564–76. doi: 10.1038/jcbfm.2010.101
41. He F, Liu B, Meng Q, Sun Y, Wang W, Wang C. Modulation of miR-146a/complement factor H-mediated inflammatory responses in a rat model of temporal lobe epilepsy. Biosci Rep. (2016) 36:e00433. doi: 10.1042/BSR20160290
42. Lukiw WJ, Surjyadipta B, Dua P, Alexandrov PN. Common micro RNAs (miRNAs) target complement factor H (CFH) regulation in Alzheimer's disease (AD) and in age-related macular degeneration (AMD). Int J Biochem Mol Biol. (2012) 3:105–16.
43. Nei M, Xu P, Glazko G. Estimation of divergence times from multi-protein sequences for a few mammalian species and several distantly related organisms. Proc Natl Acad Sci. (2001) 98:2497–502. doi: 10.1073/pnas.051611498
44. D'Argenio V, Sarnataro D. Microbiome influence in the pathogenesis of prion and Alzheimer's diseases. Int J Mol Sci. (2019) 20:E4704. doi: 10.3390/ijms20194704
45. Lukiw WJ. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer's disease. Front Microbiol. (2016) 7:1544. doi: 10.3389/fmicb.2016.01544
46. Lukiw WJ. The microbiome, microbial-generated pro-inflammatory neurotoxins, and Alzheimer's disease. J Sport Health Sci. (2016) 5:393–6. doi: 10.1016/j.jshs.2016.08.008
47. Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. (2007) 12(Pt 2):S140–6. doi: 10.1301/nr.2007.dec.S140-S146
48. Novellino F, Saccà V, Donato A, Zaffino P, Spadea MF, Vismara M, et al. Innate-Immunity: A common denominator between neurodegenerative and neuropsychiatric diseases. Int J Mol Sci. (2020) 21:E1115. doi: 10.3390/ijms21031115
49. Dansokho C, Heneka MT. Neuroinflammatory responses in Alzheimer's disease. J Neural Transm (Vienna). (2018) 125:771–79. doi: 10.1007/s00702-017-1831-7
50. Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer's disease. Immunol Cell Biol. (2020) 98:28–41. doi: 10.1111/imcb.12301
51. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: Current evidence and future directions. Alzheimers Dement. (2016) 12:719–32. doi: 10.1016/j.jalz.2016.02.010
52. Regen F, Hellmann-Regen J, Costantini E, Reale M. Neuroinflammation and Alzheimer's disease: implications for microglial activation. Curr Alzheimer Res. (2017) 14:1140–48. doi: 10.2174/1567205014666170203141717
53. Ahmad MH, Fatima M, Mondal AC. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer's disease: Rational insights for the therapeutic approaches. J Clin Neurosci. (2019) 59:6–11. doi: 10.1016/j.jocn.2018.10.034
54. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
55. Marwarha G, Ghribi O. Nuclear factor kappa-light-chain-enhancer of activated B Cells (NF-κB) - a friend, a foe, or a bystander - in the neurodegenerative cascade and pathogenesis of Alzheimer's disease. CNS Neurol Disord Drug Targets. (2017) 16:1050–65. doi: 10.2174/1871527316666170725114652
56. Brindley E, Hill TDM, Henshall DC. MicroRNAs as biomarkers and treatment targets in status epilepticus. Epilepsy Behav. (2019) 101:106272. doi: 10.1016/j.yebeh.2019.04.025
57. Zhang H, Qu Y, Wang A. Antagonist targeting microRNA-146a protects against lithium-pilocarpine-induced status epilepticus in rats by nuclear factor-κB pathway. Mol Med Rep. (2018) 17:5356–61. doi: 10.3892/mmr.2018.8465
58. Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. (2006) 25:6887–6899. doi: 10.1038/sj.onc.1209982
59. Dehghani R, Rahmani F, Rezaei N. MicroRNA in Alzheimer's disease revisited:implications for major neuropathological mechanisms. Rev Neurosci. (2018) 29:161–82. doi: 10.1515/revneuro-2017-0042
60. Lukiw WJ, Pogue AI. Induction of specific micro RNA (miRNA) species by ROS-generating metal sulfates in primary human brain cells. J Inorg Biochem. (2007) 101:1265–9. doi: 10.1016/j.jinorgbio.2007.06.004
61. Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TP, Percy ME, et al. Characterization of an NF-kappaB-regulated, miRNA-146a-mediated down-regulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J Inorg Biochem. (2009) 103:1591–5. doi: 10.1016/j.jinorgbio.2009.05.012
Keywords: Alzheimer's disease (AD), herpes simplex virus, microRNA (miRNA), miRNA-146a-5p, NF-kB (p50/p65), prion disease (PrD), microbiome
Citation: Lukiw WJ (2020) microRNA-146a Signaling in Alzheimer's Disease (AD) and Prion Disease (PrD). Front. Neurol. 11:462. doi: 10.3389/fneur.2020.00462
Received: 06 March 2020; Accepted: 29 April 2020;
Published: 25 June 2020.
Edited by:
Y-h. Taguchi, Chuo University, JapanReviewed by:
Jongkyun Kang, Harvard Medical School, United StatesJereme Grant Spiers, La Trobe University, Australia
Copyright © 2020 Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter J. Lukiw, d2x1a2l3QGxzdWhzYy5lZHU=