 Jianfeng Liang1Wanni Zhao2Changyu Lu1Danni Liu3Ping Li4Xun Ye1Yuanli Zhao1,5Jing Zhang3
Jianfeng Liang1Wanni Zhao2Changyu Lu1Danni Liu3Ping Li4Xun Ye1Yuanli Zhao1,5Jing Zhang3 Dong Yang6,7*
Dong Yang6,7*- 1Department of Neurosurgery, Peking University International Hospital, Beijing, China
- 2Department of General Surgery, Beijing Hospital, National Center of Gerontology, Beijing, China
- 3HaploX Biotechnology, Shenzhen, China
- 4Department of Hematology, Tongji Hospital of Tongji University, Shanghai, China
- 5Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 6Department of Neurosurgery, China-Japan Friendship Hospital, Beijing, China
- 7The 2nd People's Hospital of Tibet Autonomous Region, Lhasa, China
Background and aims: The next-generation sequencing technologies and their related assessments of circulating tumor DNA in both glioma and metastatic brain tumors remain largely limited.
Methods: Based largely on a protocol approved by the institutional review board at Peking University International Hospital, the current retrospective, single-center study was conducted. Genomic DNA was extracted from blood samples or tumor tissues. With the application of NextSeq 500 instrument (Illumina), Sequencing was performed with an average coverage of 550-fold. Paired-end sequencing was employed utilized with an attempt to achieve improved sensitivity of duplicate detection and therefore to increase the detection reliability of possible fusions.
Results: A total of 28 patients (21 men and 7 women) with brain tumors in the present study were involved in the study. The patients enrolled were assigned into two groups, including glioma group (n = 21) and metastatic brain tumor group (n = 7). The mean age of metastatic brain tumor group (59.86 ± 8.85 y), (43.65 ± 13.05 y) reported significantly higher results in comparison to that of glioma group (45.3 ± 12.3 years) (P < 0.05). The mutant genes in metastatic brain tumor group included ALK, MDM2, ATM, BRCA1, FGFR1, MDM4 and KRAS; however, there were no glioma-related mutant genes including MGMT, IDH1, IDH2, 1p/19q, and BRAF et al. Interesteringly, only two patient (28.3%) was detected blood ctDNA in metastatic brain tumor group; In contrast, blood ctDNA was found in ten glioma patients (47.6%) including 1p/19q, MDM2, ERBB2, IDH1, CDKN2A, CDK4, PDGFRA, CCNE1, MET. The characterizations of IDH mutations in the glioma included IDH1 mutation (p.R132H) and IDH2 mutation (p.R172K). The mutation rate of IDH in tumor tissues was 37.06 ± 8.32%, which was significantly higher than blood samples (P < 0.05).
Conclusion: The present study demonstrated that the mutant genes among glioma and metastatic brain tumors are shown to be different. Moreover, the ctDNAs in the metastatic brain tumors included ALK and MDM2, and glioma-related ctDNAs included 1p/19q and MDM2 followed by frequencies of ERBB2, IDH1, CDKN2A, CDK4, PDGFRA, CCNE1, MET. These ctDNAs might be biomarkers and therapeutic responders in brain tumor.
Background
Brain tumors are a highly heterogeneous disease with significant morbidity and mortality, which contains a collection of neoplasms arising largely from within the brain (glioma). On the other hand, brain tumors can also occur because of systemic tumors that have metastasized to the brain (metastatic brain tumors) (1). As for adults, primary brain tumors are predicted to represent 1.4% of all new cancer diagnoses and account for 2.6% of all cancer deaths (2). The overall incidence of glioma throughout the globe is estimated to be 6.4 per 100,000 persons annually, and the disease has been reported with an overall 5-year survival rate of 33.4%.
In addition, age between 55 and 64 years is considered as peak prevalence, and glioma is the most common primary brain tumor in adults (3). Metastatic brain tumors are estimated to occur as much as 10 times more frequently in comparison to glioma, which is ~53.7 per 100,000 persons (4). Either glioma or metastatic brain tumors are associated with poor prognosis (median overall survival of only 4–15 months), progressive neurological deterioration, and reduced quality of life (5, 6). Therefore, the early diagnosis, accurate differentiation, and dynamic monitoring progression of primary and metastatic brain tumors are of great importance. However, traditional methods, such as clinical examination, magnetic resonance imaging, and histopathological biopsy, are often limited to meet the requirement for clinical practice (7).
Non-invasive or minimal invasive technology to detect circulating tumor DNA (ctDNA) derived from blood (liquid biopsy) has several advantages. First, the technology is able to reduce invasive damages and avoid spatial heterogeneity and difficulties of harvesting brain tumor tissues. Second, it is more feasible and accessible, allowing for repeat blood sampling and providing dynamic insight of brain tumors progression, which becomes a promising and convincing tool to analyze the genomic characterization of brain tumors (8, 9). Recently, according to the revised fourth edition of the World Health Organization (WHO) classification of central nervous system (CNS) tumors, the integration of histology and genetic analysis for the diagnosis of specific neoplastic entities are recommended, such as isocitrate dehydrogenase 1 and 2 (IDH1/2) mutations, 1p/19q chromosomal codeletion, point mutations in tumor protein 53 (TP53), and O6-methylguanine methyltransferase (MGMT) promoter methylation for adults diffuse glioma (10). Other genetic alterations are meaningful for the molecular characterization of different types of brain tumors, including mutation in the promoter of telomerase reverse transcriptase (TERT) for oligodendroglioma, the v-RAF murine sarcoma viral oncogene homolog B1 (BRAF) V600E mutation for non-diffuse glioma, and v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA) fusion for supratentorial ependymomas (11, 12). Recently, the next-generation sequencing (NGS) technologies have drawn increasing attention as a result of several advantages, such as globally interrogating the genetic composition of biological samples, significantly reduced sequencing cost, improved accuracy of detection, and real-time monitoring progression of tumors, with high sensitivity for detecting extremely low levels of mutation frequency; therefore, the technology allows early screening and diagnosis of brain tumors (13). However, limited reports exist considering the NGS-related assessments in both glioma and metastatic brain tumors.
In the present study, the genetic characterization of both glioma and metastatic brain tumors was comprehensively analyzed by using tumor tissue or blood samples based on the NGS technology, including mutant gene, microsatellite instability (MSI), mismatch repair, tumor mutational burden (TMB), and PD-L1 expression. Our research was conducted to help provide insight for the genetic alterations in both primary and metastatic brain tumors.
Methods
Participants
Based on a protocol approved by the institutional review board at Peking University International Hospital, the current retrospective, single-center study was performed according to the Good Clinical Practice guidelines, as well as the principles of the Declaration of Helsinki. The medical records of adult patients harboring brain tumors were reviewed. The patients enrolled undergoing the whole treatments in this hospital from August 2018 to June 2019 (N = 213). In order to be included in the current study, the following inclusion criteria should be met: (1) patients with age ranging from 18 to 75 years old; (2) patients were pathologically confirmed with primary or metastatic brain tumors, and with 5-year cancer-free history (excluding melanoma); (3) patients with normal functions of multiple vital organs (including heart, liver, lung, kidney, and bone marrow) without severe or vital illness; (4) patients scored from 0 to 1 based on the Eastern Cooperative Oncology Group (ECOG); (5) patients were in agreement with complete ctDNA tests for tissue or blood samples; (6) informed consent form was signed voluntarily by participants. The exclusion criteria included history of other malignant tumors or CNS benign tumors within 5 years (excluding melanoma), participating in other clinical trials within 3 months, organ transplant or blood transfusion recipients within 3 months, pregnant or lactating women, hepatitis B virus/hepatitis C virus/human immunodeficiency virus positive, autoimmune diseases, severe or vital illness, ECOG scoring from 2 to 5, incomplete clinical evaluations, incomplete ctDNA tests, unsigned informed consent form, and other unsuitable circumstances.
Sampling and Sequencing
Genomic DNA was extracted from blood samples or tumor tissues. As for blood sampling, at least 10 mL of peripheral blood (anticoagulated with EDTA) was drawn from participants and separated through centrifugation (1,600 × g, 10 min) at room temperature. Circulating tumor DNA of blood samples was extracted with the use of QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). As for tumor tissues, ctDNA from 10-μm formalin-fixed paraffin-embedded (FFPE) tissue was extracted through the use of QIAamp DNA FFPE Tissue Kit (Qiagen) following manufacturer's instructions. After DNA quantification, take more than 20 ng of DNA from the instructions of the kit for DNA library construction (Kapa HTP library preparation kit). The steps include ctDNA large fragment separation, small fragment recovery, DNA end repair and a-connector connection, adding special connector of Illumina sequencing kit (California, USA) at both ends of DNA, magnetic bead screening according to the required DNA fragment size, polymerase chain reaction (PCR) amplification library for probe hybridization capture, and sequencing experiment. In the panel, the target region is designed according to the reference genome sequence of Hg 19 to detect point mutation, insertion, fusion, and deletion. With the application of a NextSeq 500 instrument (Illumina), sequencing was performed with an average coverage of 550-fold. When choosing the respective adapters, the sample in the panel could be generally detected on other sequencing devices. Flow cells were selected on the basis of desired read length (150 bp), number of samples, and required target coverage for the Illumina reagent selection algorithm. The sequencing data are first processed by base calling to extract base information, and then data quality control is carried out, including removing low-quality data, tailoring data, removing poly X and other error information; data comparison, deduplication, and error correction are processed by RWA, PICARD algorithms; GATK and VarScan2 are used for variation information, genotype information, SNP, indel, et al. (14–16) are obtained. Finally, annotate the variation information. The specific methods are as follows: RAW sequencing reads were preprocessed by fastp v0.18.0 and then aligned to the reference genome (hg19/GRch37) using BWA-MEM v0.7.15 with default settings. Gencore v0.12.0 was used to remove duplicated reads. Pileup files for properly paired reads with mapping quality ≥60 were generated using Samtools v0.1.19. Somatic variants were called by VarScan2 v2.3.8 and GATK 4.0. The called somatic variants were filtered with following criteria: read depth >20 ×; variant allele frequency (VAF) ≥2% for tumor tissue DNA and ≥0.05% for cfDNA from blood samples; somatic P ≤ 0.01; strand filter ≥1. Allele frequencies were calculated with all bases of quality >Q30. CNVkit v0.9.3 was applied for copy number variation detection, and GeneFuse v0.6.1 was used to detect actionable gene fusions. Paired-end sequencing was employed and applied in order to improve the sensitivity of duplicate detection as well as increase the detection reliability of possible fusions (17).
Statistics Analysis
The present study applied Statistical Product and Service Solutions software (SPSS 15.0, Inc., Chicago, IL, USA) for statistical analysis. The aggregated results were expressed as mean ± standard deviation (SD). We also utilized one-way analysis of variance (ANOVA) and two-way ANOVA and Student t-test for continuous data, and χ2 test was used for categorical data. In addition, Kruskal–Wallis test and Wilcoxon two-sample tests were used for non-normal distribution samples. P < 0.05 represented significant statistical difference.
Results
Demographic Characteristics
The current retrospective study involved a total number of 28 patients (21 men and 7 women) harboring brain tumors. The patients enrolled in the present study were divided into two groups including primary brain tumor group (n = 21) and metastatic brain tumor group (n = 7). The average age of all included 28 patients was 47.5 ± 13.8 years (range, 22–75 years). The mean age of patients in metastatic brain tumor group (61.2 ± 9.4 years) was calculated to be significantly higher when comparing that in primary brain tumor group (Age: glioma 43.65 ± 13.05, Metastatic brain tumor: 59.86 ± 8.85) (P < 0.05). As laid out in Table 1, the pathological type of all glioma was diffuse glioma, and the pathological types of metastatic brain tumors included lung adenocarcinoma, lung squamous carcinoma, renal cell carcinoma, intestinal adenocarcinoma, and endometrial cancer.
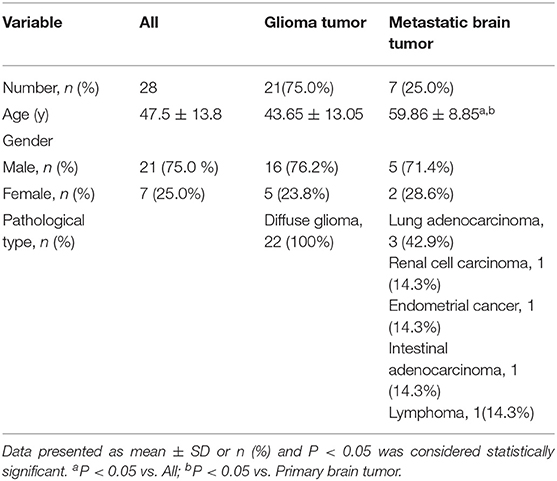
Table 1. Demographic characteristics of patients with brain tumors.
Data Processing Results
The results of data processing are shown in Tables 2, 3. The raw data and mapped data of cfDNA in patients' peripheral blood are shown in Table 2. All patients had more than 99.90% mapped rate and 92.00% unique mapped rate, among which patient 11 had the lowest unique mapped rate (92.40%). Patient gDNA data information is shown in Table 3. All patients had mapped rate >99.8% and unique mapped rate >98.00%.
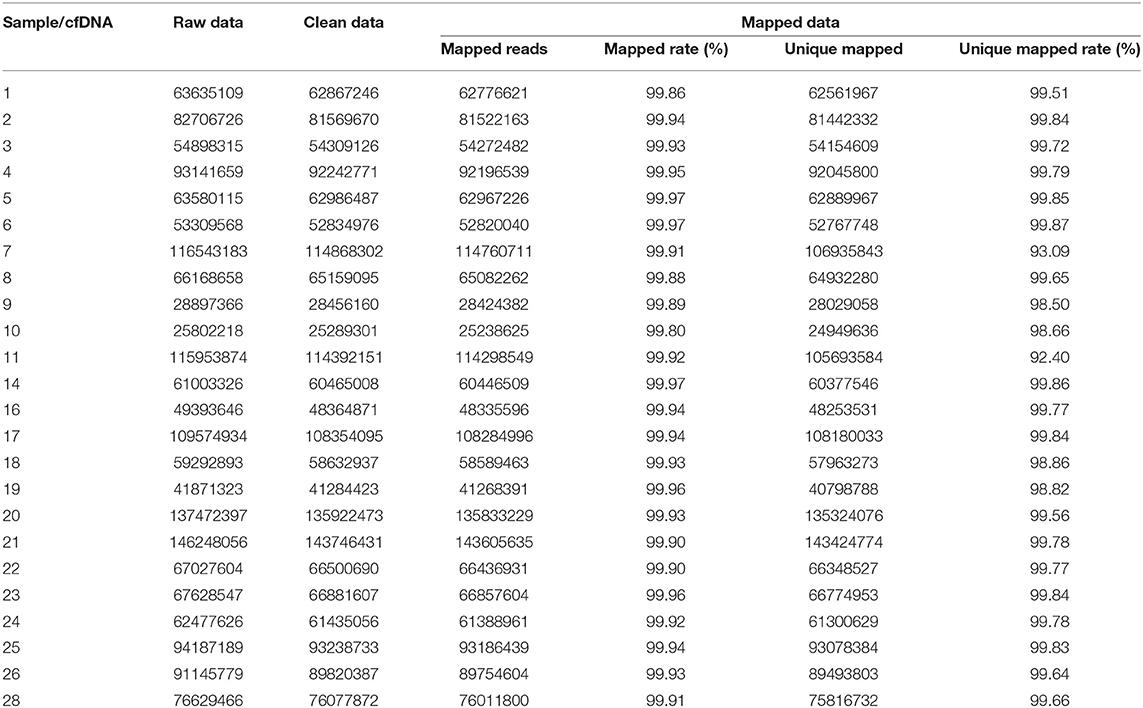
Table 2. Patient cfDNA raw data and mapped data.
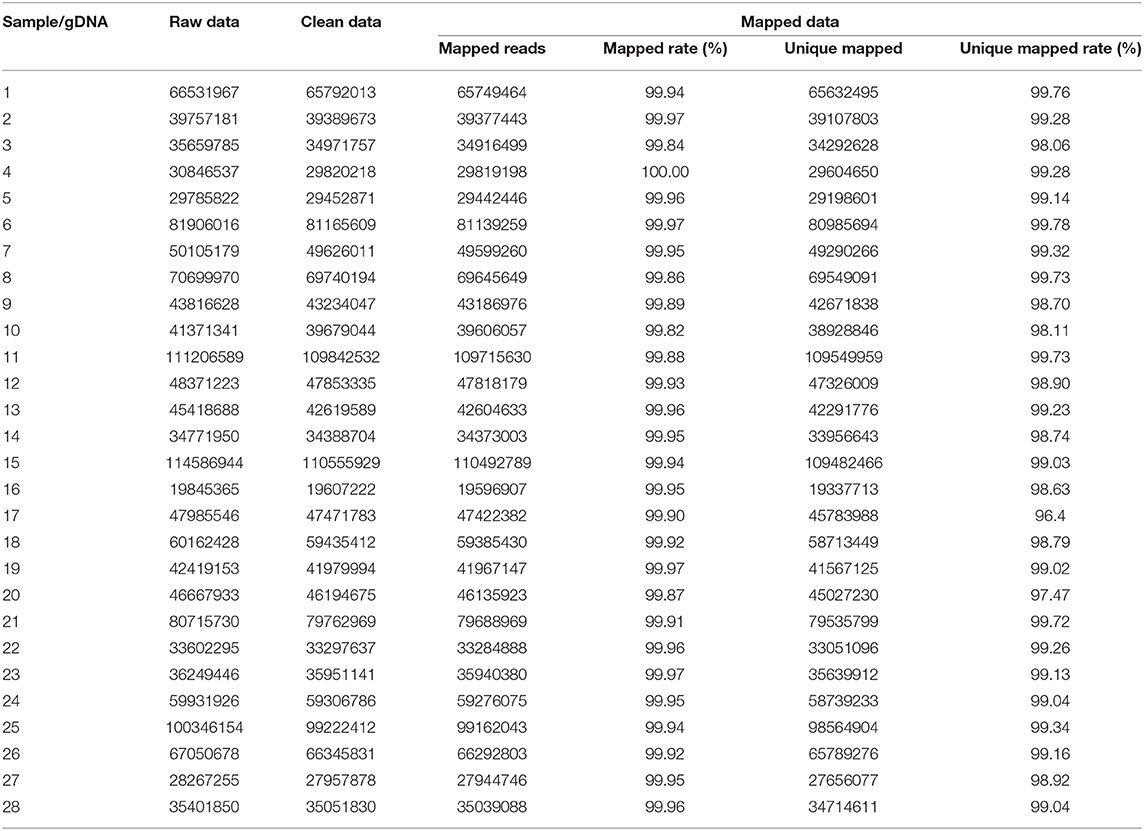
Table 3. Patient gDNA raw data and mapped data.
Genetic Alterations
As seen in Figure 1, the most frequent genetic alterations identified were MGMT (46.7%), followed by IDH1 (26.7%), TP53 (26.7%), CDKN2A (16.7%), H3F3A (13.3%), MDM2 (13.3%), 1p/19q (10%), ATM (10%), EGFR (10%), ALK (6.7%), BRAF (6.7%), CDK4 (6.7%), ERBB2 (6.7%), MDM4 (6.7%), MET (6.7%), NF1 (6.7%), PDGFRA (6.7%), PTEN (6.7%), ARID1A (3.3%), BRCA1 (3.3%), CCNE1 (3.3%), FGFR1 (3.3%), IDH2 (3.3%), KIT (3.3%), KRAS (3.3%), and PIK3CA (3.3%). Different somatic mutations occur in all genes, including amplification and fusion, chromosomal structural variation, insertion and deletion, and point mutation; among them, germline mutations occur in genes ATM, BRCA1, IDH1, PTEN, EGFR, IDH2, and TP53; ctDNA mutation rate is lower than tissue, among which TP53, ATM, BRAF, and PTEN do not occur in ctDNA.
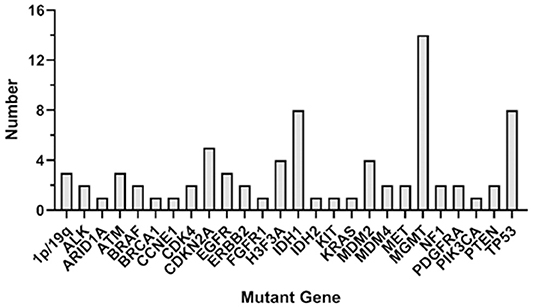
Figure 1. Genetic alterations in the whole participants. The figure shows the overall gene mutation statistics of 28 patients, among which MGMT has the greatest mutation probability, with a total of 14 patients; IDH1 and TP53 have the second mutation in eight patients each; CDK4 gene mutation in five patients; H3F3A and MDM2 have mutations in four patients each; 1p/19q, ALM, EGFR have mutations in three patients each, and the number of mutations in other genes is small.
The genetic alterations in metastatic brain tumors are shown in Table 4. The mutant genes in this group included ALK, MDM2, ATM, BRCA1, FGFR1, and KRAS. Among them, ALK mutation is the fusion of EML4-exon6 and ALK-exon 20 genes, MDM2 and FGFR1 mutation is copy number variation; ATM and BRCA1 mutations are germline heterozygous variants. The results of peripheral blood and tissue were basically similar. MDM2 did not detect variation in tissue, but the copy number in peripheral blood was 4. FGFR1 in the same patient did not detect variation in peripheral blood, but the copy number in tissue was 3.8. However, there were no glioma-related mutant genes. Remarkably, the MSI type of endometrial cancer metastatic brain tumor is MSI-H; the other MSI type of metastatic brain tumor is MSS.
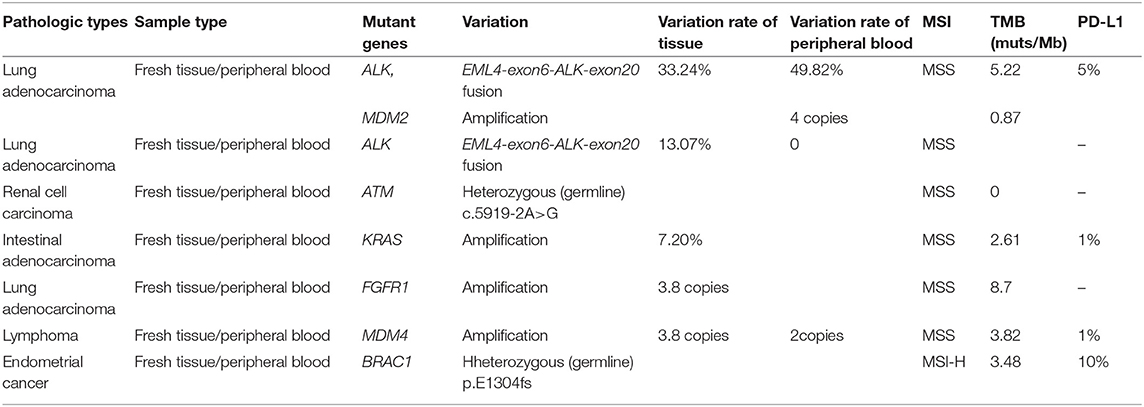
Table 4. Genetic alternations in metastatic brain tumors.
As seen in Figure 2, glioma-related mutant genes included MGMT (n = 14), IDH1 (n = 8), IDH2 (n = 1), 1p/19q (n = 3), BRAF (n = 2), and TP53(n = 6) in the glioma group. Among them, MGMT methylation, IDH mutation, 1p/19q deletion, BRAF mutation, TP53 mutation or splicing mutation, and all patients with IDH mutation showed MGMT methylation positive. All genes have different somatic mutations; among them, the genes causing germline variation are IDH1, PTEN, EGFR, BRAF, IDH2, and TP53. In the detection of glioma gene mutation, it was found that there was a great difference between the tissue mutation rate and the peripheral blood mutation rate. In general, the tissue mutation rate was higher than the peripheral blood mutation rate (Table 5). And TP53 gene was found to be highly variable in tissue in patients with TP53 mutation detected. Interestingly, 47.6% of glioma patients were detected ctDNA, but only two metastatic patients were found with somatic mutations in ctDNA. Glioma-related ctDNAs included 1p/19q, MDM2, ERBB2, IDH1, CDKN2A, CDK4, PDGFRA, CCNE1, MET. Among ctDNA positive glioma patients, 30% of them were detected 1p/19q codeletion and MDM2 amplification in both tissue and blood.
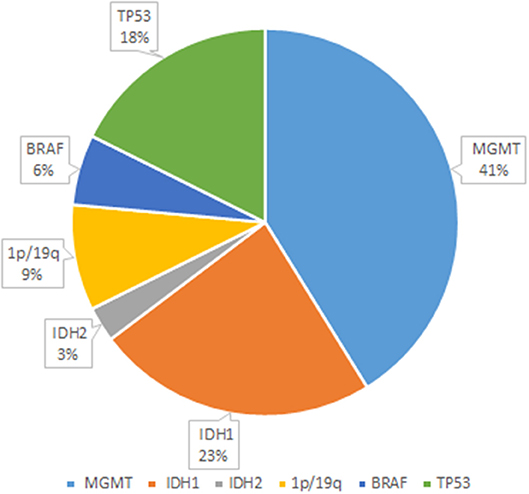
Figure 2. Glioma-related mutant genes. The figure shows several genes with higher probability of mutation and their respective probability of occurrence in 22 glioma patients. Among them, the genes prone to mutation were MGMT, IDH1, IDH2, 1p/19q, BRAF, and TP53, and their mutation changes were 41, 23, 3, 9, 6, and 18%, respectively.
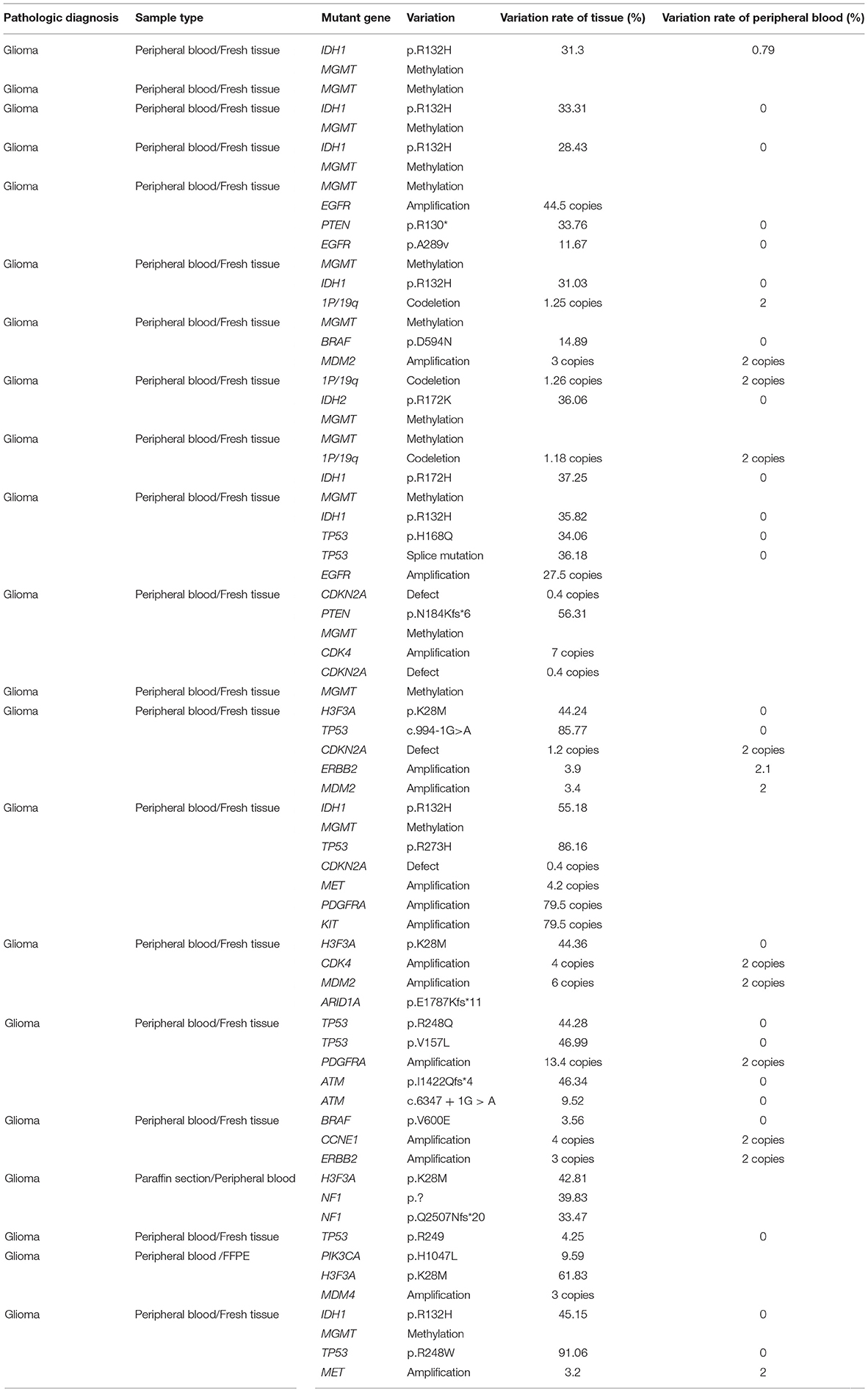
Table 5. Gene mutations in tissues and peripheral blood of patients with glioma.
The genetic alterations in glioma were laid out in Table 6. The mutant genes in this group included MGMT, IDH1, IDH2, 1p/19q, BRAF, TP53, CDKN2A, H3F3A, MDM2, ATM, EGFR, ALK, CDK4, ERBB2, MDM4, MET, NF1, PDGFRA, PTEN, ARID1A, BRCA1, CCNE1, FGFR1, KIT, KRAS, and PIK3CA. Based on Table 7, the characterizations of IDH mutations in the glioma included IDH1 mutation (p.R132H) and IDH2 mutation (p.R172K). The mutation abundance of IDH in tumor tissues was 37.06 ± 8.32%, which was significantly higher in comparison to that in blood samples (P < 0.05).
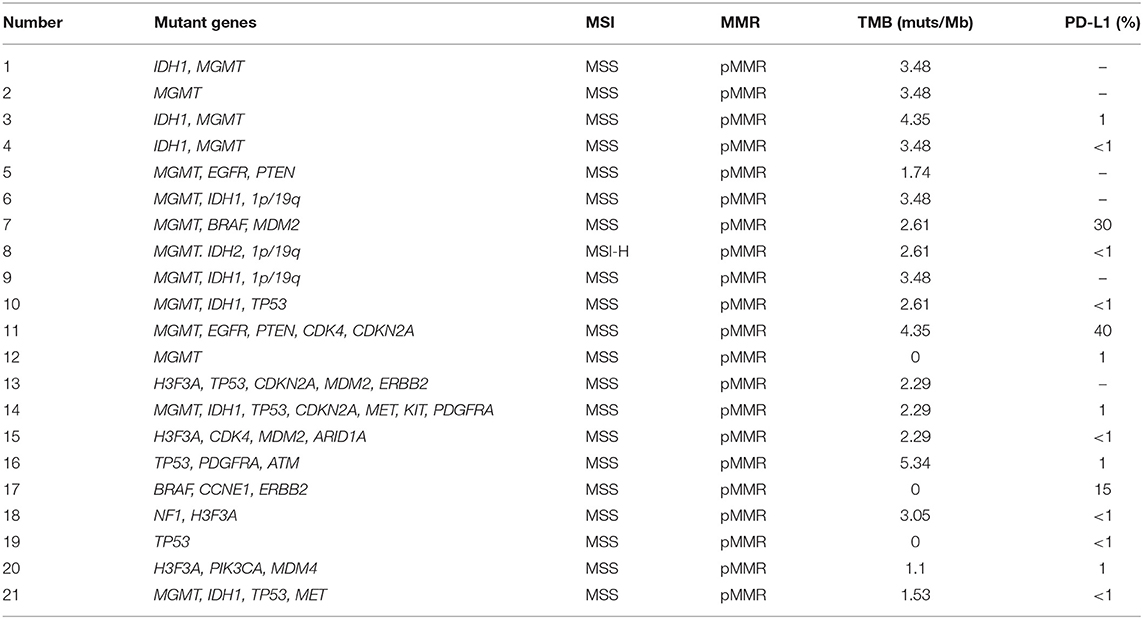
Table 6. Genetic alternations in primary brain tumors.
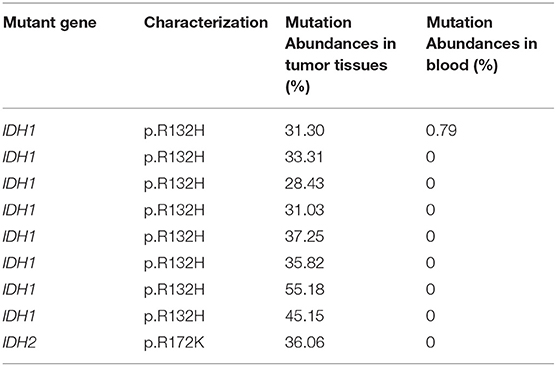
Table 7. IDH mutations in the glioma.
Discussion
The accurate differentiation of primary and metastatic brain tumors is considered pivotal, considering that the intervention and therapy approaches for patients with these two types of tumors are remarkably different for clinical practice (18, 19). The cancers with the highest propensity in terms of metastasizing to the brain are lung (50%), followed by breast (15%) and melanoma (5–10%), accounting for ~80% of all brain metastases (20). In this study, we found that the mutant genes in the metastatic brain tumors included ALK, MDM2, ATM, BRCA1, FGFR1, and KRAS, and there were no glioma-related mutant genes (MGMT, IDH1, IDH2, 1p/19q, BRAF, and TERT). According to the aggregated result, NGS-based genetic analysis might become a promising tool to differentiate primary and metastatic brain tumors. Based on a report supported by Bettegowda et al. (21), the sensitivity of ctDNA was 87.2% for detection of clinically relevant KRAS gene mutations, with specificity of 99.2% in the detection of metastatic cancers. Wang et al. (22) suggested that liquid biopsy such as ctDNA could be regarded a feasible alternative approach in terms of identifying sensitizing genomic alterations, and ALK translocation could be identified in the diffuse brain metastases. However, in this study ALK, MEM2 and MDM4 were detected in ctDNA of only two brain metastatic patients. It might be due to sample size, and we plan to expand the sample size in further study. Moreover, the results showed that glioma-related mutant genes included MGMT (n = 14), IDH1 (n = 8), IDH2 (n = 1), 1p/19q (n = 3), and BRAF (n = 2). As a DNA repair protein, MGMT is able to remove the alkylation of the O6 position of guanine which is also the most cytotoxic lesion induced by alkylating agent chemotherapy (23). Hypermethylation of the promoter of MGMT is considered to have predictive value to respond to the alkylating agent temozolomide among patients harboring glioblastoma (24). Piccioni et al. (25) reported that 50% of patients with glioma had ≥1 somatic alteration detected. Additionally, 61 genes were found with single-nucleotide variants, and amplifications were detected in EGFR MET, ERBB2, and others, indicating that plasma cfDNA genomic analysis might be used as a viable approach for clinical practice to identify genomically driven therapy options. According to the study of Schwaederle et al. (26), the most frequent alterations among diverse cancers were reported to be TP53 (29.8%), followed, respectively, by EGFR (17.5%), MET (10.5%), PIK3CA (7%), and NOTCH1 (5.8%). In addition, detectable ctDNA aberrations existed among 65% of diverse cancers (as well as 27% of glioblastomas), with the majority theoretically actionable by an approved agent. In this study, 47.6% of glioma patients were detected ctDNA including 1p/19q, MDM2, ERBB2, IDH1, CDKN2A, CDK4, PDGFRA, CCNE1, MET. These ctDNAs might be biomarkers and therapeutic responders in glioma and be worthy of further investigation.
Furthermore, we found that the characterizations of IDH mutations in the glioma included IDH1 mutation (p.R132H) and IDH2 mutation (p.R172K). The mutation abundance of IDH in tumor tissues was 37.06 ± 8.32%, which reported evidently higher results in comparison to that in blood samples (P < 0.05). IDH-mutated results were found in at least 80% of WHO grades II and III infiltrating astrocytomas and secondary GBMs, whereas all oligodendrogliomas were IDH-mutated and 1p/19q co-deleted (27). Similar results were observed in the study of Hartmann et al. (28) that codon 132 of the IDH1 gene, known as the R132H variant, was reported to account for 92.7% of IDH mutation, followed by R132C (4.1%), R132S (1.5%), R132G (1.4%), and R132L (0.2%). Moreover, residue R172 in exon 4 of the IDH2 gene was homologous to R132 in the IDH1 gene, and the most common IDH2 mutations included R172W (16%), R172M (19%), and R172K (65%). In current study, three glioma patients were oligodendrogliomas, and 1p/19q codeletion was detected in both blood and tissue of these patients. Interestingly, copy number of 1p/19q is higher in blood than tissue. Thus these findings provide new insights into verification 1p/19q codeleciton to glioma patient via a noninvasive approach. Lots of focus has been paid to the predictive value of TMB as biomarker in terms of the response to immune checkpoint blockade therapy among many clinical trials (29). High TMB was consistently selected for beneficial outcome with immune checkpoint blockade therapy, and we found that the mean TMB of glioma was 2.55 mutations per Mb. According to the study supported by Johnson et al. (30), 43 to 575 mutations per Mb existed in hypermutated gliomas characterized by TMBs.
Conclusion
The present study demonstrated that the mutant genes among glioma and metastatic brain tumors include are different. Moreover, the ctDNAs in the metastatic brain tumors included ALK and MDM2, and glioma-related ctDNAs included 1p/19q and MDM2 followed by frequencies of ERBB2, IDH1, CDKN2A, CDK4, PDGFRA, CCNE1, MET. These ctDNAs might be noninvasive biomarkers and therapeutic responders in brain tumor.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://bigd.big.ac.cn/search?dbId=&q=HRA000141, with accession no: HRA000141.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board at Peking University International Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JL, WZ, CL, and DL carried out the experiments. DL, PL, and JZ participated in data analysis. CL, XY, and YZ participated in the clinical investigation of the patient. JL, PL, and DY drafted the manuscript. JL, JZ, and DY offered opinions for discussions and reviewed the manuscript. YZ and DY critically reviewed the overall manuscript as well as supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by The National Natural Science Foundation of China (Nos. 81301092, 31301118, 81900431), The National Natural Science Foundation of Tibet Autonomous Region (No. XZ2017ZR-ZY002) and Peking University International Hospital Research Grant (No. YN2017ZD02).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Butowski NA. Epidemiology and diagnosis of brain tumors. Continuum. (2015) 21:301–13. doi: 10.1212/01.CON.0000464171.50638.fa
2. McNeill KA. Epidemiology of brain tumors. Neurol Clin. (2016) 34:981–8. doi: 10.1016/j.ncl.2016.06.014
3. Perkins A, Liu G. Primary brain tumors in adults: diagnosis and treatment. Am Fam Phys. (2016) 93:211–7.
4. Ostrom QT, Wright CH, Barnholtz-Sloan JS. Brain metastases: epidemiology. Handb Clin Neurol. (2018) 149:27–42. doi: 10.1016/B978-0-12-811161-1.00002-5
5. Liang J, Lv X, Lu C, Ye X, Chen X, Fu J, et al. Prognostic factors of patients with Gliomas - an analysis on 335 patients with Glioblastoma and other forms of Gliomas. BMC Cancer. (2020) 20:35. doi: 10.1186/s12885-019-6511-6.
6. Maher EA, Mietz J, Arteaga CL, DePinho RA, Mohla S. Brain metastasis: opportunities in basic and translational research. Cancer Res. (2009) 69:6015–20. doi: 10.1158/0008-5472.CAN-08-4347
7. Baris MM, Celik AO, Gezer NS, Ada E. Role of mass effect, tumor volume and peritumoral edema volume in the differential diagnosis of primary brain tumor and metastasis. Clin Neurol Neurosurg. (2016) 148:67–71. doi: 10.1016/j.clineuro.2016.07.008
8. Gorgannezhad L, Umer M, Islam MN, Nguyen NT, Shiddiky MJA. Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip. (2018) 18:1174–96. doi: 10.1039/C8LC00100F
9. von Bubnoff N. Liquid biopsy: approaches to dynamic genotyping in cancer. Oncol Res Treat. (2017) 40:409–16. doi: 10.1159/000478864
10. Weller M, Stupp R, Hegi ME, van den Bent M, Tonn JC, Sanson M, et al. Personalized care in neuro-oncology coming of age: why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol. (2012) 14(Suppl. 4):100–8. doi: 10.1093/neuonc/nos206
11. Priesterbach-Ackley LP, Wesseling P, Snijders TJ, de Vos FYFL, de Leng WWJ. Molecular tools for the pathologic diagnosis of central nervous system tumors. Neurooncol Pract. (2019) 6:4–16. doi: 10.1093/nop/npy041
12. Capper D, Stichel D, Sahm F, Jones DT, Schrimpf D, Sill M, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. (2018) 136:181–210. doi: 10.1007/s00401-018-1879-y
13. Chen M, Zhao H. Next-generation sequencing in liquid biopsy: cancer screening and early detection. Hum Genomics. (2019) 13:34. doi: 10.1186/s40246-019-0220-8
14. Yu X, Sun S. Comparing a few SNP calling algorithms using low-coverage sequencing data. BMC Bioinformatics. (2013) 14:274. doi: 10.1186/1471-2105-14-274
15. Rasal KD, Chakrapani V, Pandey AK, Rasal AR, Sundaray JK, Ninawe A, et al. Status and future perspectives of single nucleotide polymorphisms (SNPs) markers in farmed fishes: way ahead using next generation sequencing. Gene Rep. (2017) 6:81–6. doi: 10.1016/j.genrep.2016.12.004
17. Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. (2016) 131:903–10. doi: 10.1007/s00401-015-1519-8
18. Mouthuy N, Cosnard G, Abarca-Quinones J, Michoux N. Multiparametric magnetic resonance imaging to differentiate high-grade gliomas and brain metastases. J Neuroradiol. (2012) 39:301–7. doi: 10.1016/j.neurad.2011.11.002
19. Liang J, Meng Q, Zhao W, Tong P, Li P, Zhao Y, et al. An expression based REST signature predicts patient survival and therapeutic response for glioblastoma multiforme. Sci Rep. (2016) 6:34556. doi: 10.1038/srep34556
20. Parrish KE, Sarkaria JN, Elmquist WF. Improving drug delivery to primary and metastatic brain tumors: strategies to overcome the blood-brain barrier. Clin Pharmacol Ther. (2015) 97:336–46. doi: 10.1002/cpt.71
21. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. (2014) 6:224ra24. doi: 10.1126/scitranslmed.3007094
22. Wang VE, Young L, Ali S, Miller VA, Urisman A, Wolfe J, et al. Case of Metastatic atypical neuroendocrine tumor with ALK translocation and diffuse brain metastases. Oncologist. (2017) 22:768–73. doi: 10.1634/theoncologist.2017-0054
23. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. (2000) 343:1350–4. doi: 10.1056/NEJM200011093431901
24. Horbinski C, Ligon KL, Brastianos P, Huse JT, Venere M, Chang S, et al. The medical necessity of advanced molecular testing in the diagnosis and treatment of brain tumor patients. Neuro Oncol. (2019) 21:1498–508. doi: 10.1093/neuonc/noz119
25. Piccioni DE, Achrol AS, Kiedrowski LA, Banks KC, Boucher N, Barkhoudarian G, et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. (2019) 8:CNS34. doi: 10.2217/cns-2018-0015
26. Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget. (2016) 7:9707–17. doi: 10.18632/oncotarget.7110
27. Velázquez Vega JE, Brat DJ. Incorporating advances in molecular pathology into brain tumor diagnostics. Adv Anat Pathol. (2018) 25:143–71. doi: 10.1097/PAP.0000000000000186
28. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. (2009) 118:469–74. doi: 10.1007/s00401-009-0561-9
29. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. evelopment of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. (2019) 30:44–56. doi: 10.1093/annonc/mdy495
Keywords: ctDNA, brain tumors, NGS, MGMT, IDH1/2
Citation: Liang J, Zhao W, Lu C, Liu D, Li P, Ye X, Zhao Y, Zhang J and Yang D (2020) Next-Generation Sequencing Analysis of ctDNA for the Detection of Glioma and Metastatic Brain Tumors in Adults. Front. Neurol. 11:544. doi: 10.3389/fneur.2020.00544
Received: 05 January 2020; Accepted: 14 May 2020;
Published: 21 August 2020.
Edited by:
Hailiang Tang, Huashan Hospital Affiliated to Fudan University, ChinaReviewed by:
Arif Ozgun Harmanci, University of Texas Health Science Center at Houston, United StatesMatthew Tate, Northwestern University, United States
Copyright © 2020 Liang, Zhao, Lu, Liu, Li, Ye, Zhao, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Yang, eWFuZ2RvbmcxOTc1QHNpbmEuY29t