 Douglas N. Martini1*
Douglas N. Martini1* Rosie Morris1Valerie E. Kelly2
Rosie Morris1Valerie E. Kelly2 Amie Hiller1,3Kathryn A. Chung1,3Shu-Ching Hu4,5
Amie Hiller1,3Kathryn A. Chung1,3Shu-Ching Hu4,5 Cyrus P. Zabetian4,5John Oakley4
Cyrus P. Zabetian4,5John Oakley4 Kathleen Poston6
Kathleen Poston6 Ignacio F. Mata4,5,7
Ignacio F. Mata4,5,7 Karen L. Edwards8Jodi A. Lapidus9Thomas J. Grabowski10Thomas J. Montine11
Karen L. Edwards8Jodi A. Lapidus9Thomas J. Grabowski10Thomas J. Montine11 Joseph F. Quinn1,3
Joseph F. Quinn1,3 Fay Horak1
Fay Horak1- 1Department of Neurology, Oregon Health and Science University, Portland, OR, United States
- 2Department of Rehabilitation Medicine, University of Washington School of Medicine, Seattle, WA, United States
- 3Portland Veterans Affairs Health Care System, Portland, OR, United States
- 4Department of Neurology, University of Washington School of Medicine, Seattle, WA, United States
- 5Veterans Affairs Puget Sound Health Care System, Seattle, WA, United States
- 6Department of Neurology and Neurological Sciences, Stanford School of Medicine, Palo Alto, CA, United States
- 7Lerner Research Institute, Genomic Medicine, Cleveland Clinic Foundation, Cleveland, OH, United States
- 8Department of Epidemiology, University of California, Irvine, Irvine, CA, United States
- 9Biostatistics & Design Program, Oregon Health and Science University, Portland, OR, United States
- 10Department of Radiology, University of Washington School of Medicine, Seattle, WA, United States
- 11Department of Pathology, Stanford University School of Medicine, Palo Alto, CA, United States
Background: Mobility and sensorimotor inhibition impairments are heterogeneous in Parkinson's disease (PD). Genetics may contribute to this heterogeneity since the apolipoprotein (APOE) ε4 allele and glucocerebrosidase (GBA) gene variants have been related to mobility impairments in otherwise healthy older adult (OA) and PD cohorts. The purpose of this study is to determine if APOE or GBA genetic status affects sensorimotor inhibition and whether the relationship between sensorimotor inhibition and mobility differs in genetic sub-groups of PD.
Methods: Ninety-three participants with idiopathic PD (53 non-carriers; 23 ε4 carriers; 17 GBA variants) and 72 OA (45 non-carriers; 27 ε4 carriers) had sensorimotor inhibition characterized by short-latency afferent inhibition. Mobility was assessed in four gait domains (pace/turning, rhythm, trunk, variability) and two postural sway domains (area/jerkiness and velocity) using inertial sensors.
Results: Sensorimotor inhibition was worse in the PD than OA group, with no effect of genetic status. Gait pace/turning was slower and variability was higher (p < 0.01) in PD compared to OA. Postural sway area/jerkiness (p < 0.01) and velocity (p < 0.01) were also worse in the PD than OA group. Genetic status was not significantly related to any gait or postural sway domain. Sensorimotor inhibition was significantly correlated with gait variability (r = 0.27; p = 0.02) and trunk movement (r = 0.23; p = 0.045) in the PD group. In PD non-carriers, sensorimotor inhibition related to variability (r = 0.35; p = 0.010) and trunk movement (r = 0.31; p = 0.025). In the PD ε4 group, sensorimotor inhibition only related to rhythm (r = 0.47; p = 0.024), while sensorimotor inhibition related to pace/turning (r = −0.49; p = 0.046) and rhythm (r = 0.59; p = 0.013) in the PD GBA group. Sensorimotor inhibition was significantly correlated with gait pace/turning (r = −0.27; p = 0.04) in the OA group. There was no relationship between sensorimotor inhibition and postural sway.
Conclusion: ε4 and GBA genetic status did not affect sensorimotor inhibition or mobility impairments in this PD cohort. However, worse sensorimotor inhibition was associated with gait variability in PD non-carriers, but with gait rhythm in PD ε4 carriers and with gait rhythm and pace in PD with GBA variants. Impaired sensorimotor inhibition had a larger effect on mobility in people with PD than OA and affected different domains of mobility depending on genetic status.
Introduction
Mobility impairment is intrinsically linked to Parkinson's disease (PD). The severity of mobility impairment in PD is heterogeneous, suggesting that underlying factors that play a role in mobility impairment. In addition to basal ganglia dysfunction, people with PD exhibit reduced cortical sensorimotor inhibition as assessed by short-latency afferent inhibition (SAI) with transcranial magnetic stimulation (TMS) (1–3). However, these studies reveal inconsistencies regarding the impact of disrupted sensorimotor inhibition on mobility disability in people with PD.
Sensorimotor inhibition is associated with decreased gait speed in people with PD (1), increased dual-task cost on stride length in PD fallers (2), and increased gait variability in people with PD (Martini et al., under review at J Gerontol: Med Sci), though not with freezing of gait (4). Within genetic subgroups of people with PD, neither a leucine-rich repeat kinase 2 mutation nor a Parkin gene mutation was associated with worse SAI (5–7). Importantly, none of these investigations assessed relationships among genetic status, SAI, and mobility. A potential reason for heterogeneity of mobility disability and of sensorimotor inhibition in PD could be the presence of the apolipoprotein (APOE) ε4 allele or glucocerebrosidase (GBA) gene variants (8, 9). The disparity in which specific domains are affected could be tied to genetic differences within PD to which specific gait or postural sway variables are characterized. Assessing relatively independent domains of gait or postural sway constructed by combinations of individual gait measures could help provide a more complete picture of gait performance (10).
The APOE ε4 allele has been associated with accelerated decline in gait speed in healthy, older men (11). Since PD is twice as likely to occur in men (12), it is pertinent to look at the effect of the ε4 allele status on gait and postural sway in people with PD, and men with PD, in particular. People with Alzheimer's disease, a disease inherently linked to the ε4 allele and known to be associated with cholinergic insufficiency, have worse SAI than people without Alzheimer's and cholinergic medication improves SAI, suggesting that acetylcholine loss may contribute to poor SAI (13, 14). To date, no attempt has been made to assess the link between ε4 allele carrier status and gait dysfunction and their relationships to sensorimotor inhibition in people with PD. Similarly, variants of the GBA gene are also associated with accelerated mobility decline in the elderly and with decline in the clinical postural instability and gait disorder phenotype in people with PD, regardless of sex (15–17). However, no investigation has approached sensorimotor inhibition in GBA carriers with PD and related SAI heterogeneity to their mobility performance.
The purpose of this study was to determine if ε4 carrier or GBA variant status is associated with reduced sensorimotor inhibition in people with PD and whether worse sensorimotor inhibition is associated with worse gait and postural sway performance. We hypothesized that sensorimotor inhibition and mobility will be worse in ε4 carriers, especially those with PD, and also in those with PD and GBA variants. Further, we hypothesize that decreased gait pace and increased gait variability will be related to worse sensorimotor inhibition.
Materials and Methods
This study was approved by Institutional Review Boards at both University of Washington and Oregon Health & Science University. All participants were provided and signed written informed consent prior to participation.
Subjects and Clinical Assessments
Participants were recruited from an ongoing Pacific Udall Center project at two sites: the University of Washington/Veterans Affairs Puget Sound Health Care System and Oregon Health Sciences University/Veterans Affairs Portland Health Care System. Each participant was screened for TMS eligibility before enrollment. Inclusion criteria included diagnosis of idiopathic PD using the United Kingdom Parkinson's Disease Society Brain Bank (UKBB) criteria (18) and ability to stand unsupported for 30 s. Exclusion criteria were inability to walk for 2 min without an assistive device, any TMS contraindication, any musculoskeletal injury/abnormality that would affect mobility, or any neurodegenerative disorder aside from PD. All participants with PD were tested “ON” dopaminergic medications due to the interaction with SAI (19), and no participants on cholinergic medication was recruited to the TMS portion of the Pacific Udall Center project.
The Movement Disorders Society Unified Parkinson's Disease Rating Scale (MDS-UPDRS) part III, the modified Hoehn & Yahr (H&Y) score, and the Montreal Cognitive Assessment (MoCA) were used to assess motor severity and global cognition, respectively. A consensus committee comprised of movement disorder neurologists and a neuropsychologist reviewed data from each participant to determine if UKBB criteria were satisfied and to assign a cognitive diagnosis (normal, mild cognitive impairment, or dementia) (20).
Genotyping
Genomic DNA was extracted from peripheral blood or saliva samples using standard procedures. The entire GBA coding region was screened using Sanger sequencing to capture all known pathogenic mutations (defined as those reported in patients with Gaucher disease) and the E326K polymorphism (rs2230288). APOE rs429358 and rs7412, which define the ε2, ε3, and ε4 alleles, were genotyped using TaqMan assays. All sequencing and genotyping was performed at a single laboratory in Seattle implementing methods previously described (8, 21).
Transcranial Magnetic Stimulation
TMS of the motor cortex was performed with a Magstim 200 (Magstim Co.). A figure-of-eight coil (external loop diameter of 9 or 7 cm, site specific) was positioned over the hemisphere associated with the most affected side in PD participants and the dominant side in control participants. Motor-evoked potentials (MEPs) were recorded from the first dorsal interosseous muscle through disposable, Ag/AgCl surface electrodes. Samples were amplified (gain: 2000) and bandpass filtered (100 Hz−5 kHz) using BIOPAC MP150 system (BIOPAC Systems, Inc) or amplified (CED 1902 isolated preamplifier, Cambridge Electronics), converted from analog to digital (Sampling rate 40 KHz, PowerLab, ADInstruments), and recorded for offline analysis (LabChart, ADInstruments). Resting motor threshold was determined as the percentage of the minimum stimulator output to elicit an MEP of 50 μV in five out of 10 trials.
Short Latency Afferent Inhibition (SAI)
SAI was performed using a modified version of a protocol previously described (22). A peripheral, electric conditioning stimulus was applied over the median nerve followed by the central test stimulus, TMS. The intensity of the conditioning stimulus was set at the amplitude required to elicit a visible twitch of the thenar muscles. The N20 latency was not individualized by somatosensory evoked potentials, instead, the time of 20 ms for the N20 was used across all participants. The interstimulus intervals (ISI) from N20 + 0 ms to N20 + 5 ms were applied in a randomized block format. Five unconditioned trials were collected within each block of conditioned trials, in a pseudo-random order, for a total of 30 unconditioned trials. A total of 10 trials were collected for each condition and the conditioned peak-to-peak MEP magnitudes averaged for each ISI. There was a minimum of seven seconds between each trial. A grand mean of the ISIs is expressed as the percentage of the unconditioned MEP magnitudes. Participants were instructed to remain at rest, while sitting as still as possible, and refrain from keeping their eyes closed.
Gait and Postural Sway
Inertial sensors (Opals, APDM Inc.) were placed on each wrist and foot, around the waist, and over the sternum to record mobility measures (23). Gait was characterized over a 2-min walk, over a 7-meter path, requiring 180° turns at the ends of the marked path. Comprehensive measures of gait and balance were combined into four domains based on our previous principle component analysis using a larger cohort, which included people with PD in this study (10). The four domains of gait were pace/turning, rhythm, variability, and trunk movement. Postural sway was characterized while participants stood quietly for 1 min looking straight ahead with feet width standardized by a template (24). Similar to the gait variables, sway variables of interest were divided into two domains, sway area/jerkiness and sway velocity.
Statistical Analyses
Data were inspected for normality using histograms and the Kolmogorov-Smirnov test of normality. All non-normally distributed data were log-base 10 transformed. Domain scores were calculated by averaging the Z-scores for each gait and sway variable. Z-scores were multiplied by −1 to reverse scaling if needed for consistent sign in domain score calculations. A one-way ANOVA compared PD vs. control group differences for demographic information and SAI, with post hoc analyses Tukey corrected for multiple comparisons. A Chi-Squared test assessed the sex differences among the groups. Two-by-two general linear models (GLM) were used to assess the effects of PD status and APOE ε4 carrier status on SAI, gait domains, and sway domains, controlling for age, sex, and collection site. The GBA group was excluded from this GLM model. To determine if GBA variants affected SAI and gait/postural sway within the PD cohort, we used a separate GLM, controlling for age, sex, and collection site. Neither of the OA groups were included in this GLM. Participants who carried a pathogenic mutation or the E326K polymorphism where combined into a single GBA group. Alpha was set a priori to p < 0.05. IBM SPSS version 25 was used for statistical analyses.
Results
Ninety-three participants with idiopathic PD (53 non-carriers; 23 ε4 carriers; 17 GBA variants) and 72 healthy older adults (OA; 45 non-carriers; 27 ε4 carriers) participated (Table 1). The consensus committee assigned 50 participants with MCI and nine participants with dementia in the PD, while 23 participants in the OA group were assigned with MCI and one with dementia. 13/50 PD MCI and 2/9 PD dementia were ε4 carriers, while 9/50 PD MCI and 3/9 PD dementia were GBA variants. 10/23 OA MCI and the one OA with dementia were ε4 carriers. Age was a significant factor [F(1, 166) = 5.75; p = 0.02], and post hoc analyses showed the PD GBA group was significantly younger than the rest of the groups (all p-values ≤ 0.03). Only the MDS-UPDRS III was significantly different between the PD [24.2 (12.7)] and OA [1.9 (2.9)] groups [F(1, 160) = 211.98; p < 0.001]. Post hoc analyses reveled the OA genetic groups were not different from each other, but both OA groups had lower MDS-UPDRS III than all the PD groups (all p-values < 0.001).
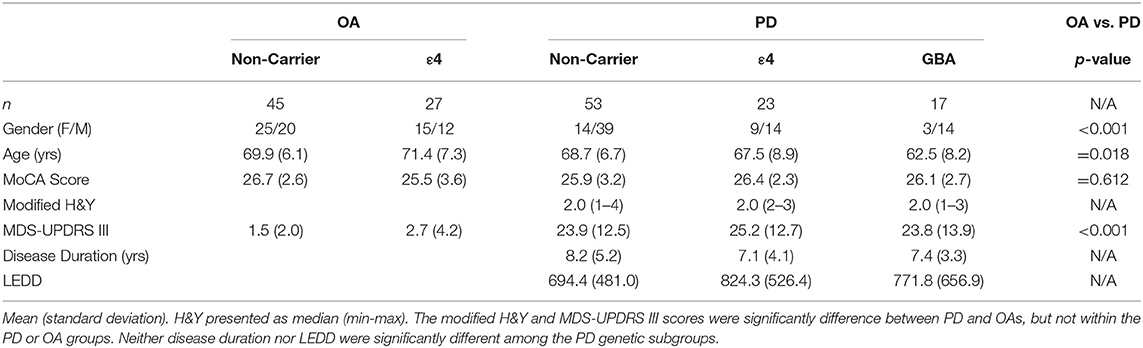
Table 1. Demographic and clinical information.
Short-Latency Afferent Inhibition (SAI)
The PD group exhibited worse sensorimotor inhibition than the control group [PD: 77.6 (18.2)% inhibition; OA: 69.4 (20.8)% inhibition; F(1, 157) = 8.31]. There was no significant main effect of ε4 carrier status or interaction between PD status and genetics on SAI. Within the PD group, there was no significant main effect of genetics for SAI. SAI for each genetic subgroup of OA and PD are shown in Figure 1.
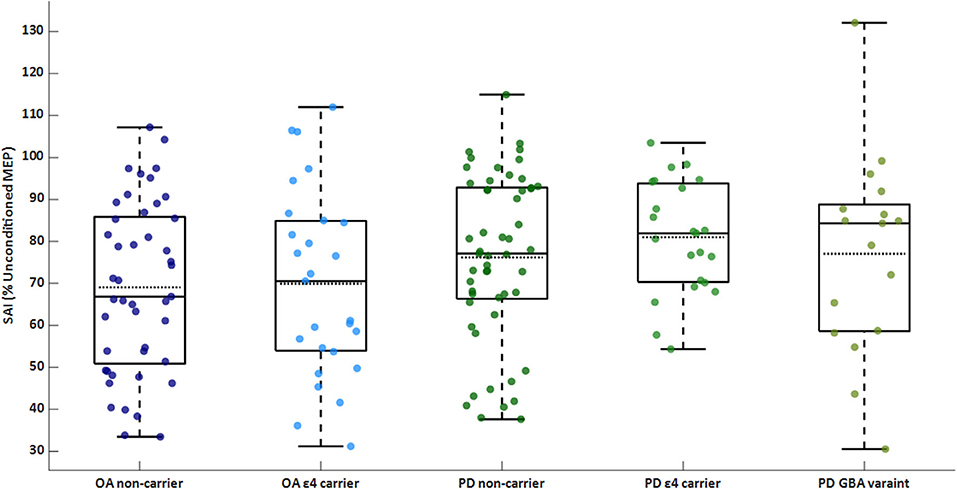
Figure 1. Box/scatter plot of sensorimotor inhibition (SAI) for each group. A higher percentage is associated with less (i.e., worse) inhibition. Dashed lines represent group means. SAI is significantly different between the PD and OA groups (p = 0.004). OA, older adults; PD, Parkinson's disease.
Gait Domains
The main effect of PD status was significant [F(4, 138) = 7.49; p < 0.001] for the pace/turning domain [F(1, 141) = 22.61; p < 0.001] and variability domain [F(1, 141) = 10.05; p = 0.002]. Specifically, gait was slower and more variable in the PD than the control group. The main effect for ε4 carrier status on gait domains was not significant. Further, there was no main effect for genetic status within the PD group. The gait domain scores are presented in Table 2. Individual gait characteristics that comprise each gait domain are provided in Table 3 to provide greater detail about gait performance.
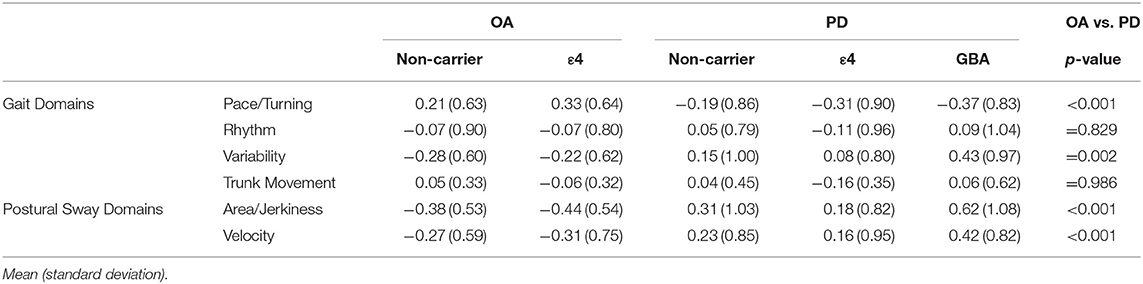
Table 2. Gait and postural sway domain scores.
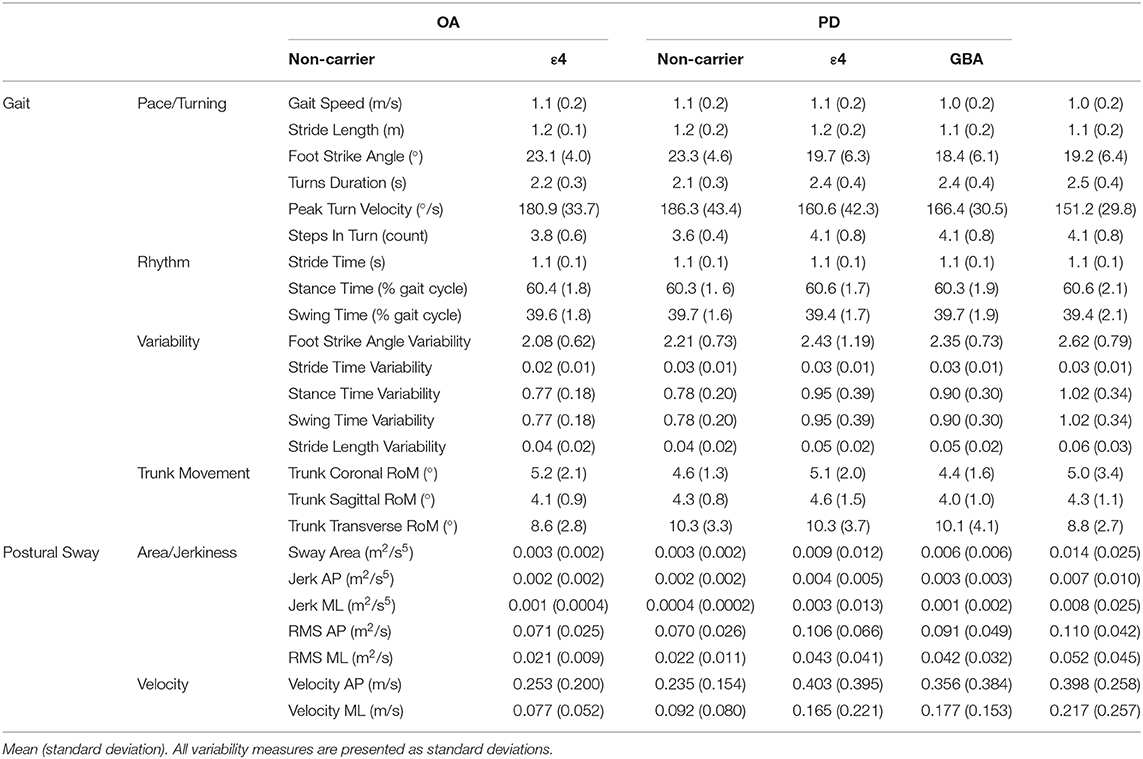
Table 3. Individual gait characteristics.
Sway Domains
Similar to the gait results, the main effect for PD status was significant [F(2, 140) = 12.18; p < 0.001]. The PD group showed larger sway area/jerkiness [F(1, 141) = 24.22; p < 0.001] and larger sway velocity [F(1, 141) = 13.08; p < 0.001] compared to the OA group. There was no main effect for ε4 carrier status on the sway domains. The sway domain scores are presented in Table 2. No significant main effect for genetic status within the PD group was found for any of the sway domains. Individual postural sway characteristics that comprise each gait domain are provided in Table 3 to provide greater detail about postural sway performance.
Relationship Between SAI and Mobility Domains
SAI significantly correlated with gait variability (Pearson's r = 0.27; p = 0.017) and trunk movement (Pearson's r = 0.23; p = 0.045) in the PD group. Though mobility performance was not different genetic groups in The PD cohort, the relationships between SAI and variability (Pearson's r = 0.35; p = 0.010) and between SAI and trunk movement (Pearson's r = 0.31; p = 0.025) only remained for the PD non-carriers. SAI only related to rhythm (Pearson's r = 0.47; p = 0.024) for the PD ε4 group, while SAI related to pace/turning (Pearson's r = −0.49; p = 0.046) and rhythm (Pearson's r = 0.59; p = 0.013) in the PD GBA group. There was no relationship between SAI and postural sway domains in the PD group. Scatter plots for significant relationships within the PD are in Figure 2.
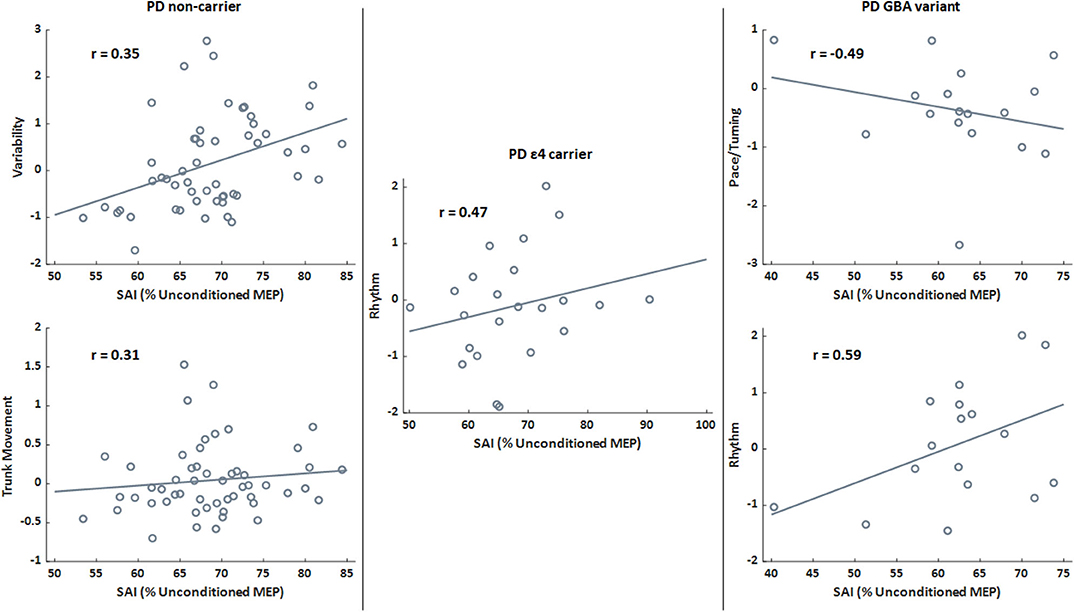
Figure 2. Scatter plots presenting the relationships between sensorimotor inhibition (SAI) and gait domains for the PD genetic subgroups only. Lines indicate the best fit line. A higher percentage (SAI) is associated with less (i.e., worse) inhibition. Higher domain scores indicate faster pace/turning, increased rhythm (increased times for stride, stance, and swing phases), and greater variability. r is the Pearson's correlation coefficient for the relationship between SAI and relevant gait domain score. All correlations presented are significant (p < 0.05).
SAI was significantly correlated with the gait pace/turning domain (Pearson's r = −0.24; p = 0.046) in the OA group. Genetic subgroup analyses revealed no relationships for the OA group. There was no relationship between SAI and postural sway domains for the OA group.
Discussion
This study investigated how genetic factors influence sensorimotor inhibition and mobility in OAs and people with PD. Although people with PD had worse SAI, gait, and balance than OAs, our results suggest that carriers of APOE ε4 allele or GBA variants do not show worse SAI, nor worse gait and postural sway than non-carriers. However, worse sensorimotor inhibition was associated with worse gait (but not standing postural sway) in all genetic subgroups of PD, but not OA groups. In fact, heterogeneity of specific domains of gait most affected in people with PD (decreased pace/turning, increased variability, and increased rhythm) depended upon the genetic status of the PD group. Thus, cortical sensorimotor inhibition appears more critical for gait than for standing balance and more critical for people with PD than for older adults without PD.
We previously reported (Martini et al., under review at J Gerontol: Med Sci) that people with PD had significant mobility dysfunction compared to healthy OAs, including slower gait speed/turn velocity and greater gait variability, postural sway area/jerkiness, and sway velocity, consistent with previous investigations (25–27). Our previous report included 81 of the 93 people with PD and 69 of the 72 OAs in this report (Martini et al., under review at J Gerontol: Med Sci). The current study used mobility domain scores, based on principal component analysis (10), rather than individual metrics of postural sway and gait, which can lead to inflated, type-one errors.
Genetic status was not a significant factor for gait or sway performance in people with idiopathic PD, contrary to our hypothesis. GBA is a strong risk factor for PD and particularly PD with more severe cognitive and gait and balance decline than PD without GBA (8, 9, 17). Since ACh is known to be important for cognition (28) and cognition is known to be related to gait speed in both older adults and people with PD (1, 29), we expected people with PD who had GBA variants to show worse SAI as well as worse balance and gait than non-carriers A larger cohort of people with PD and GBA variants (n = 58) from our PUC investigation reported that people with PD and GBA a variant had a quicker progression through motor and cognitive decline than non—GBA variant PD (17). Our subset from that sample of people with GBA variants is smaller, with milder parkinsonian symptoms based on the MDS-UPDRS III (17).
Like GBA, APOE ε4 allele status was not associated with worse mobility. The APOE ε4 allele is not a risk factor for PD, but it is a risk factor for dementia. Since decline in cognition is associated with decline in balance control with aging and with PD (30, 31), we hypothesized that people with PD who are carriers of APOE ε4 allele would have more loss of ACh than non-carriers and therefore show worse SAI and worse balance and gait than non-carriers. In fact, APOE ε4 allele carriers with PD have been shown to have worse executive function than non-carriers and executive function has been associated with slower gait speed (32) in otherwise healthy OAs and with increased variability (33) and Alzheimer's disease. However, we found no evidence that APOE ε4 status in people with PD was associated with worse SAI nor worse balance or gait. Though, the MoCA scores for the genetic subsets were not statistically different and were above the cognitive impairment cut off score of 23, which was reported to have higher sensitivity and specificity than higher cut off scores (34). Longitudinal studies, which incorporate a comprehensive cognitive batteries, are needed to determine if PD APOE ε4 carriers would show faster progression of their balance and gait disorders over time, similar to older men without PD (11).
Genetic status was not a significant factor for gait or sway performance in healthy OAs. This contradicts previous reports that observed smaller stride length in male OA ε4 allele carriers than non-carriers (35). Longitudinal analyses reported similar observations, with an accelerated decline in gait speed and increase in gait variability in ε4 allele carriers over 1 year (11, 36). The accelerated decline in gait speed was only noted for OA males (11), which suggests that there may be a sex influence on ε4 allele carrier status and increased risk of gait dysfunction over time. Despite the ε4 allele being linked to an increased risk for dementia (37), a separate study found no change in MoCA scores over a year (36), suggesting an increased motor decline not directly linked to cognitive decline. Thus, assessing changes in gait and balance performance across time may be a better indicator of mobility dysfunction in male ε4 allele carriers than a cross-sectional approach.
Consistent with previous investigations, we show that people with PD do not inhibit the motor cortex as much with a sensory afferent signal compared to OAs (1, 2, 38) (Martini et al., under review at J Gerontol: Med Sci). GBA and ε4 allele status did not affect SAI, similar to leucine-rich repeat kinase 2 mutation nor a Parkin gene mutation (5–7). Sensorimotor inhibition, as measured with SAI, is thought to reflect levels of cortical ACh neurotransmitter activity (3), The basis for the notion that SAI is mediated by cholinergic activity is that SAI is impaired in Alzheimer's disease, a disorder known to result in loss of ACh, and because medications that increase ACh also improve the SAI (13, 39–41). Recent theories of the neural pathways responsible for SAI suggests that the subthalamic nucleus or paramedian thalamic nuclei are responsible for inhibition of the primary motor cortex either directly, or indirectly, via the primary sensory cortex (38, 42). The theoretical pathway that incorporates the subthalamic nucleus was posited as a result of the modulatory effects of deep brain stimulation on the subthalamic nucleus in human and animal studies (38). The theoretical pathway, involving the paramedian thalamic nuclei is based off of the connections between SAI and cholinergic pathways as observed in drug and disease studies (13, 41, 43). Importantly, dopamine replacement therapy is linked to worse SAI in people with PD (19), suggesting that cholinergic activity is not the only influence on the inhibitory response recorded in SAI. A reciprocal circuit among the basal ganglia (e.g., subthalamic nucleus), thalamus, and cortex could be the primary neural pathway responsible for SAI, as a measure of sensorimotor inhibition (38, 42, 44). Although the specific neural mechanisms underlying the SAI are a debated, a recent review of the topic details the complex neural pathways between the basal ganglia and motor cortices, moderated by thalamus that may be involved (44).
The effect of PD status on SAI suggests that the pathway between the substantia nigra pars compacta, ventral thalamus, and motor regions are responsible for the inhibitory response observed. The ventral lateral thalamic nuclei receive input from the basal ganglia, the substantia nigra in particular, as well as proprioceptive input (44). The substantia nigra degeneration with concomitant loss of dopamine neurotransmitter is responsible for Parkinsonism but dopamine replacement therapy does not alleviate all gait and balance impairments in PD, and may even worsen some domains of mobility (45–47). In addition, proprioception is a critical sensory input for control of gait and balance and is known to be impaired in PD (48).
In this same cohort of people with PD, we previously reported that sensorimotor inhibition was specifically related to stride length variability, foot strike angle variability, and jerkiness of sway (Martini et al., under review at J Gerontol: Med Sci). The relationship between SAI and gait domains, but not with balance domains, observed here suggests that sensorimotor inhibition is more critical for control of dynamic balance, than static, standing balance, which may be more controlled by thalamic and brainstem mechanisms (49). The relationship between worse sensorimotor increased gait variability could be an indicator of decreased gait automaticity, with increased reliance on frontal cortical areas (50, 51). Indeed, increased frontal cortex activity and gait variability is found in people with PD and freezing of gait (52). Ultimately, loss of gait automaticity is related to an increased risk of falls across gait disorders (53).
SAI was differently related to gait domains across the genetic subgroups in the PD group. Worse SAI was related to increased gait variability and trunk movement in the PD non-carrier group, which may reflect impaired dynamic balance as postural stepping responses to correct lateral body center of mass displacements interrupt steady state rhythmic gait (47, 54, 55). SAI was significantly related to increased rhythm in the PD ε4 group. Increased rhythm, which is derived from stride time, stance time, and swing time, suggests that the PD ε4 group has longer stride and stance times when sensorimotor inhibition is worse. Lastly, SAI related to decreased pace/turning and increased rhythm in the PD GBA variant group. This relationship may permit SAI to be a marker of slowed gait performance due to bradykinesia or an early adoption of a conservative gait strategy, in an attempt to prevent falls.
SAI was associated with the gait pace/turning domain in older adults, but there were no effects of the ε4 allele on gait or the relationship between sensorimotor inhibition and gait in the OA group. Previous investigations found no difference in sensorimotor inhibition between otherwise healthy young and OA groups (56, 57). However, both sensorimotor inhibition and gait speed were reduced in OA fallers compared to OA non-fallers (2). Adopting a slower gait pace may be a compensatory mechanism for poorer sensorimotor inhibition as a means for preventing falls. These results suggest that OAs with low sensorimotor inhibition may need an intervention to help reduce potential falls. SAI may be a quick, non-invasive assessment for identifying OAs who could benefit from an intervention. Though, the appropriate type of intervention would depend on the neurological mechanism responsible for SAI.
There are potential limitations to the interpretation of the results of this investigation. TMS was collected at two different academic centers. However, we controlled for this statistically and the SAI is calculated as a percentage of each participant's own MEPs, mitigating device or administrator influence. Further, we created a site variable and used it as a covariate in analyses. The effects of sex on SAI are not fully understood, so we adjusted for sex statistically. The sample sizes for the ε4 allele carriers and the GBA carriers were small. We attempted to mitigate this by analyzing the ε4 allele carriers and non-carriers across PD status, and ε4 allele and GBA variant status within the PD group only. GBA variants only exist in 8% of the idiopathic PD population, which makes this population difficult to recruit. The inclusion criteria requiring participants to stand for at least 30 s unassisted could have biased the sample away from more severely affected ε4 allele carriers and GBA carriers. Using the first dorsal interosseous muscle to measure SAI, opposed to a lower-limb muscle, when attempting to elucidate relationships between SAI and gait/sway characteristics is a limitation that may affect the generalizability of the results. However, previous groups implemented the same SAI methodology when relating SAI to gait performance (1, 2, 4).
Among people with PD, APOE and GBA genetic status did not affect either sensorimotor inhibition or measures of gait and postural sway. However, dependent on genetic status, worse sensorimotor inhibition was related to increased gait variability, impaired temporal rhythm and/or slower gait pace/turning in this PD group. This dichotomy of the relationships between sensorimotor inhibition and gait domains for people with PD with different genetic status could be related to the dependence of each group on cortical control of different aspects of gait. Longitudinal studies could elucidate more robust relationships between sensorimotor inhibition and mobility, while factoring in the role of cognition, in the ε4 and GBA variant subgroups of people with PD.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by This study was approved by Institutional Review Boards at both University of Washington and Oregon Health & Science University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
DM: research organization and execution, statistical design and execution, and writing first draft. RM: research organization and execution, and statistical design, and draft review and critique. VK, AH, KC, S-CH, and IM: research organization and execution, and draft review and critique. CZ: research conception, organization and execution and draft review and critique. JO: research organization and execution and draft review. KP: statistical critique and draft review and critique. KE: research conception, organization, statistical critique, and draft review and critique. JL: research conception, statistical critique, and draft review and critique. TG: research organization and draft review and critique. TM, JQ, and FH: research conception, organization and execution, statistical critique, and draft review and critique. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Neurological Disorders and Stroke (P50 NS062684) and the Department of Veterans Affairs (101 CX001702).
Conflict of Interest
FH has a significant financial interest in APDM, a company that may have a commercial interest in the results of this research and technology. This potential conflict has been reviewed and managed by OHSU.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rochester L, Yarnall AJ, Baker MR, David RV, Lord S, Galna B, et al. Cholinergic dysfunction contributes to gait disturbance in early Parkinson's disease. Brain. (2012) 135:2779–88. doi: 10.1093/brain/aws207
2. Pelosin E, Ogliastro C, Lagravinese G, Bonassi G, Mirelman A, Hausdorff JM, et al. Attentional control of gait and falls: is cholinergic dysfunction a common substrate in the elderly and parkinson's disease? Front Aging Neurosci. (2016) 8:104. doi: 10.3389/fnagi.2016.00104
3. Morris R, Martini DN, Madhyastha T, Kelly VE, Grabowski TJ, Nutt J, et al. Overview of the cholinergic contribution to gait, balance and falls in Parkinson's disease. Parkinsonism Relat Disord. (2019) 63:20–30. doi: 10.1016/j.parkreldis.2019.02.017
4. Picillo M, Dubbioso R, Iodice R, Iavarone A, Pisciotta C, Spina E, et al. Short-latency afferent inhibition in patients with Parkinson's disease and freezing of gait. J Neural Transm. (2015) 122:1533–40. doi: 10.1007/s00702-015-1428-y
5. Bäumer T, Pramstaller P, Siebner H, Schippling S, Hagenah J, Peller M, et al. Sensorimotor integration is abnormal in asymptomatic Parkin mutation carriers: a TMS study. Neurology. (2007) 69:1976–81. doi: 10.1212/01.wnl.0000278109.76607.0a
6. Dubbioso R, De Rosa A, Esposito M, Peluso S, Iodice R, De Michele G, et al. Does motor cortex plasticity depend on the type of mutation in the leucine-rich repeat kinase 2 gene? Mov Disord. (2017) 32:947–8. doi: 10.1002/mds.27012
7. Ponzo V, Di Lorenzo F, Brusa L, Schirinzi T, Battistini S, Ricci C, et al. Impaired intracortical transmission in G2019S leucine rich-repeat kinase Parkinson patients. Mov Disord. (2017) 32:750–6. doi: 10.1002/mds.26931
8. Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson's disease. Mov Disord. (2016) 31:95–102. doi: 10.1002/mds.26359
9. Mata IF, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Van Deerlin VM, et al. Large-scale exploratory genetic analysis of cognitive impairment in Parkinson's disease. Neurobiol Aging. (2017) 56:211.e211–7. doi: 10.1016/j.neurobiolaging.2017.04.009
10. Morris R, Martini DN, Smulders K, Kelly VE, Zabetian CP, Poston K, et al. Cognitive associations with comprehensive gait and static balance measures in Parkinson's disease. Parkin Relat Disord. (2019) 69:104–10. doi: 10.1016/j.parkreldis.2019.06.014
11. Verghese J, Holtzer R, Wang C, Katz MJ, Barzilai N, Lipton RB. Role of APOE genotype in gait decline and disability in aging. J Gerontol A Biol Sci Med Sci. (2013) 68:1395–401. doi: 10.1093/gerona/glt115
12. Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, et al. Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. (2003) 157:1015–22. doi: 10.1093/aje/kwg068
13. Di Lazzaro V, Pilato F, Dileone M, Saturno E, Oliviero A, Marra C, et al. In vivo cholinergic circuit evaluation in frontotemporal and Alzheimer dementias. Neurology. (2006) 66:1111–3. doi: 10.1212/01.wnl.0000204183.26231.23
14. Marra C, Quaranta D, Profice P, Pilato F, Capone F, Iodice F, et al. Central cholinergic dysfunction measured “in vivo” correlates with different behavioral disorders in Alzheimer's disease and dementia with Lewy body. Brain Stimul. (2012) 5:533–8. doi: 10.1016/j.brs.2011.08.009
15. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community-based incident cohort. Brain. (2013) 136:392–9. doi: 10.1093/brain/aws318
16. Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, et al. GBA-associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. (2015) 30:407–11. doi: 10.1002/mds.26071
17. Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson Disease. JAMA Neurol. (2016) 73:1217–24. doi: 10.1001/jamaneurol.2016.2245
18. Gibb W, Lees A. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. (1988) 51:745–52. doi: 10.1136/jnnp.51.6.745
19. Sailer A, Molnar GF, Paradiso G, Gunraj CA, Lang AE, Chen R. Short and long latency afferent inhibition in Parkinson's disease. Brain. (2003) 126:1883–94. doi: 10.1093/brain/awg183
20. Cholerton BA, Zabetian CP, Quinn JF, Chung KA, Peterson A, Espay AJ, et al. Pacific Northwest Udall Center of excellence clinical consortium: study design and baseline cohort characteristics. J Parkinsons Dis. (2013) 3:205–14. doi: 10.3233/JPD-130189
21. Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, Van Deerlin VM, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. (2014) 71:1405–12. doi: 10.1001/jamaneurol.2014.1455
22. Tokimura H, Di Lazzaro V, Tokimura Y, Oliviero A, Profice P, Insola A, et al. Short latency inhibition of human hand motor cortex by somatosensory input from the hand. J Physiol. (2000) 523:503–13. doi: 10.1111/j.1469-7793.2000.t01-1-00503.x
23. Mancini M, King L, Salarian A, Holmstrom L, McNames J, Horak FB. Mobility lab to assess balance and gait with synchronized body-worn sensors. J Bioeng Biomed Sci Suppl. (2011) 1:007. doi: 10.4172/2155-9538.S1-007
24. Maki BE, Holliday PJ, Fernie GR. Aging and postural control. A comparison of spontaneous- and induced-sway balance tests. J Am Geriatr Soc. (1990) 38:1–9. doi: 10.1111/j.1532-5415.1990.tb01588.x
25. Bloem BR, Hausdorff JM, Visser JE, Giladi N. Falls and freezing of gait in Parkinson's disease: a review of two interconnected, episodic phenomena. Mov Disord. (2004) 19:871–84. doi: 10.1002/mds.20115
26. Mancini M, Horak FB, Zampieri C, Carlson-Kuhta P, Nutt JG, Chiari L. Trunk accelerometry reveals postural instability in untreated Parkinson's disease. Parkinsonism Relat Disord. (2011) 17:557–62. doi: 10.1016/j.parkreldis.2011.05.010
27. Mancini M, Carlson-Kuhta P, Zampieri C, Nutt JG, Chiari L, Horak FB. Postural sway as a marker of progression in Parkinson's disease: a pilot longitudinal study. Gait Posture. (2012) 36:471–6. doi: 10.1016/j.gaitpost.2012.04.010
28. Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci. (2005) 6:48–56. doi: 10.1038/nrn1588
29. Buracchio T, Dodge HH, Howieson D, Wasserman D, Kaye J. The trajectory of gait speed preceding mild cognitive impairment. Arch Neurol. (2010) 67:980–6. doi: 10.1001/archneurol.2010.159
30. Seidler RD, Bernard JA, Burutolu TB, Fling BW, Gordon MT, Gwin JT, et al. Motor control and aging: links to age-related brain structural, functional, and biochemical effects. Neurosci Biobehav Rev. (2010) 34:721–33. doi: 10.1016/j.neubiorev.2009.10.005
31. Pantall A, Suresparan P, Kapa L, Morris R, Yarnall A, Del Din S, et al. Postural dynamics are associated with cognitive decline in Parkinson's Disease. Front Neurol. (2018) 9:1044. doi: 10.3389/fneur.2018.01044
32. Watson NL, Rosano C, Boudreau RM, Simonsick EM, Ferrucci L, Sutton-Tyrrell K, et al. Executive function, memory, and gait speed decline in well-functioning older adults. J Gerontol A Biol Sci Med Sci. (2010) 65:1093–100. doi: 10.1093/gerona/glq111
33. Sheridan PL, Solomont J, Kowall N, Hausdorff JM. Influence of executive function on locomotor function: divided attention increases gait variability in Alzheimer's disease. J Am Geriatr Soc. (2003) 51:1633–7. doi: 10.1046/j.1532-5415.2003.51516.x
34. Luis CA, Keegan AP, Mullan M. Cross validation of the Montreal Cognitive Assessment in community dwelling older adults residing in the Southeastern US. Int J Geriatr Psychiatry. (2009) 24:197–201. doi: 10.1002/gps.2101
35. MacAulay RK, Allaire T, Brouillette R, Foil H, Bruce-Keller AJ, Keller JN. Apolipoprotein E genotype linked to spatial gait characteristics: predictors of cognitive dual task gait change. PLoS ONE. (2016) 11:e0156732. doi: 10.1371/journal.pone.0156732
36. Sakurai R, Montero-Odasso M. Apolipoprotein E4 allele and gait performance in mild cognitive impairment: results from the gait and brain study. J Gerontol A Biol Sci Med Sci. (2017) 72:1676–82. doi: 10.1093/gerona/glx075
37. Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM, Van Broeckhoven C, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol. (1998) 55:964–8. doi: 10.1001/archneur.55.7.964
38. Dubbioso R, Manganelli F, Siebner HR, Di Lazzaro V. Fast intracortical sensory-motor integration: a window into the pathophysiology of Parkinson's disease. Front Hum Neurosci. (2019) 13:111. doi: 10.3389/fnhum.2019.00111
39. Di Lazzaro V, Oliviero A, Profice P, Pennisi M, Di Giovanni S, Zito G, et al. Muscarinic receptor blockade has differential effects on the excitability of intracortical circuits in the human motor cortex. Exp Brain Res. (2000) 135:455–61. doi: 10.1007/s002210000543
40. Di Lazzaro V, Oliviero A, Tonali PA, Marra C, Daniele A, Profice P, et al. Noninvasive in vivo assessment of cholinergic cortical circuits in AD using transcranial magnetic stimulation. Neurology. (2002) 59:392–7. doi: 10.1212/WNL.59.3.392
41. Di Lazzaro V, Oliviero A, Saturno E, Dileone M, Pilato F, Nardone R, et al. Effects of lorazepam on short latency afferent inhibition and short latency intracortical inhibition in humans. J Physiol. (2005) 564:661–8. doi: 10.1113/jphysiol.2004.061747
42. Turco CV, El-Sayes J, Savoie MJ, Fassett HJ, Locke MB, Nelson AJ. Short- and long-latency afferent inhibition; uses, mechanisms and influencing factors. Brain Stimul. (2018) 11:59–74. doi: 10.1016/j.brs.2017.09.009
43. Di Lazzaro V, Pilato F, Dileone M, Ranieri F, Ricci V, Profice P, et al. GABAA receptor subtype specific enhancement of inhibition in human motor cortex. J Physiol. (2006) 575:721–6. doi: 10.1113/jphysiol.2006.114694
44. Haber SN, Calzavara R. The cortico-basal ganglia integrative network: the role of the thalamus. Brain Res Bull. (2009) 78:69–74. doi: 10.1016/j.brainresbull.2008.09.013
45. Horak FB, Frank J, Nutt J. Effects of dopamine on postural control in parkinsonian subjects: scaling, set, and tone. J Neurophysiol. (1996) 75:2380–96. doi: 10.1152/jn.1996.75.6.2380
46. Peterson DS, Horak FB. Neural control of walking in people with Parkinsonism. Physiology. (2016) 31:95–107. doi: 10.1152/physiol.00034.2015
47. Peterson DS, Horak FB. The effect of levodopa on improvements in protective stepping in people with Parkinson's Disease. Neurorehabil Neural Repair. (2016) 30:931–40. doi: 10.1177/1545968316648669
48. Konczak J, Corcos DM, Horak F, Poizner H, Shapiro M, Tuite P, et al. Proprioception and motor control in Parkinson's disease. J Mot Behav. (2009) 41:543–52. doi: 10.3200/35-09-002
49. Boisgontier MP, Cheval B, Chalavi S, van Ruitenbeek P, Leunissen I, Levin O, et al. Individual differences in brainstem and basal ganglia structure predict postural control and balance loss in young and older adults. Neurobiol Aging. (2017) 50:47–59. doi: 10.1016/j.neurobiolaging.2016.10.024
50. Vandenbossche J, Deroost N, Soetens E, Coomans D, Spildooren J, Vercruysse S, et al. Freezing of gait in Parkinson's disease: disturbances in automaticity and control. Front Hum Neurosci. (2012) 6:356. doi: 10.3389/fnhum.2012.00356
51. Gilat M, Bell PT, Ehgoetz Martens KA, Georgiades MJ, Hall JM, Walton CC, et al. Dopamine depletion impairs gait automaticity by altering cortico-striatal and cerebellar processing in Parkinson's disease. Neuroimage. (2017) 152:207–20. doi: 10.1016/j.neuroimage.2017.02.073
52. Maidan I, Bernad-Elazari H, Gazit E, Giladi N, Hausdorff JM, Mirelman A. Changes in oxygenated hemoglobin link freezing of gait to frontal activation in patients with Parkinson disease: an fNIRS study of transient motor-cognitive failures. J Neurol. (2015) 262:899–908. doi: 10.1007/s00415-015-7650-6
53. Gschwind YJ, Bridenbaugh SA, Kressig RW. Gait disorders and falls. GeroPsych. (2010) 112:265–74. doi: 10.1024/1662-9647/a000004
54. Horak FB, Dimitrova D, Nutt JG. Direction-specific postural instability in subjects with Parkinson's disease. Exp Neurol. (2005) 193:504–21. doi: 10.1016/j.expneurol.2004.12.008
55. King LA, Horak FB. Lateral stepping for postural correction in Parkinson's disease. Arch Phys Med Rehabil. (2008) 89:492–9. doi: 10.1016/j.apmr.2007.11.017
56. Oliviero A, Profice P, Tonali PA, Pilato F, Saturno E, Dileone M, et al. Effects of aging on motor cortex excitability. Neurosci Res. (2006) 55:74–7. doi: 10.1016/j.neures.2006.02.002
Keywords: short-latency afferent inhibition, SAI, postural sway, balance, gait, GBA, APOE
Citation: Martini DN, Morris R, Kelly VE, Hiller A, Chung KA, Hu S-C, Zabetian CP, Oakley J, Poston K, Mata IF, Edwards KL, Lapidus JA, Grabowski TJ, Montine TJ, Quinn JF and Horak F (2020) Sensorimotor Inhibition and Mobility in Genetic Subgroups of Parkinson's Disease. Front. Neurol. 11:893. doi: 10.3389/fneur.2020.00893
Received: 22 May 2020; Accepted: 13 July 2020;
Published: 04 September 2020.
Edited by:
Steven Frucht, Mount Sinai Hospital, United StatesReviewed by:
Raffaele Dubbioso, Federico II University Hospital, ItalyChien Tai Hong, Taipei Medical University, Taiwan
Copyright © 2020 Martini, Morris, Kelly, Hiller, Chung, Hu, Zabetian, Oakley, Poston, Mata, Edwards, Lapidus, Grabowski, Montine, Quinn and Horak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas N. Martini, ZG1hcnRpbmlAdW1hc3MuZWR1