 Erica Parrotta
Erica Parrotta Ilya Kister
Ilya Kister- 1Saint Peter's Health Partners, Saint Peter's MS & Headache Center, Albany, NY, United States
- 2New York University Langone Medical Center, Multiple Sclerosis Comprehensive Care Center, New York, NY, United States
Introduction
The ability of MOG antibody (MOG-Ab) to induce autoimmune disease in animals has been known for decades (1), but it is only recently since the cell-based assay for MOG-Ab IgG1 has been developed and commercialized, that it became possible to characterize clinical syndromes associated with MOG-Ab in humans. Early reports of MOG Associated Disease (MOGAD) emphasized its similarity to Neuromyeliits Optica Spectrum Disorder (NMOSD) (2–4). Indeed, a minority of patients with Aquaporin-4 antibody (AQ4-ab)-seronegative NMOSD−42% in one series–test positive for MOG-Ab (5). However, because the spectrum of MOGAD encompasses many NMOSD-atypical presentations, and because of differences in pathophysiology–AQ4-ab-positive NMOSD being an astrocytopathy and MOGAD being an oligodendrocytopathy—there is an increasing tendency to recognize AQ4-Ab-positive NMOSD and MOGAD as distinct entities (6–10).
In this review, we organize the clinical presentations of MOGAD by neuroanatomic compartments, while emphasizing the wide range of reported presentations. While this organization is useful for didactic purposes, it should be borne in mind that MOGAD may involve multiple regions of the CNS simultaneously– much more often than other CNS inflammatory diseases, and that half of MOGAD patients have active lesions in more than one location at the time of initial presentation (11–14).
While no phenotype is restricted to any specific age group, some generalizations about clinical presentations of MOGAD in children and adults are possible. In children under the age of 11, ADEM-like phenotypes (encephalopathy, multifocal neurologic deficits and “fluffy” supratentorial cerebral lesions in a bilateral distribution) predominate, while in adolescents and adults, focal syndromes of optic neuritis or longitudinally extensive myelitis are more common (11, 15, 16). Unlike Multiple Sclerosis (MS), where relapse rates are higher in children and decline with older age, in MOGAD the majority of children are not prone to frequent relapses, with 80% of having a monophasic course (17). However, the high rate of monophasic disease may be an overestimate due short follow up (right censoring) as recent case reports documented disease reemergence years and even decades after the initial episode in childhood (18, 19). Given the important differences in pediatric and adult MOGAD, we will qualify discussion of specific syndromes with reference to the respective age group (with the caveat that the clinical distinctions across age groups are only generalizations).
Optic Neuritis and Other Visual Pathway Presentations
Optic neuritis (ON) is the most common initial presentation of MOGAD in adolescence and adulthood, and a frequent presentation in pediatric patients (11, 16, 20). It is associated with a higher risk of subsequent relapse compared to other clinical presentations (11–13, 18). At the onset, vision loss is often severe and up to 80% of patients have bilateral optic nerve involvement, which is highly unusual in MS (12, 14, 21–24). Despite the severity of vision loss in the acute phase, recovery is usually good, especially in children: 89–98% of children had visual acuity to 20/25 or better at 6 months (14, 25). In adults, 6–14% of patients had permanent loss of vision (≤ 20/200) in the affected eye (11, 13, 24).
Optic disc edema is rare in MS or NMOSD but is present in up to 86% of patients with MOGAD-ON (13, 21, 22, 24, 26, 27). Rarely, bilateral ON with disc edema can be mistaken for idiopathic intracranial hypertension especially if the patient also complains of headache and has elevated opening pressure on lumbar puncture; however lymphocytic pleocytosis in CSF and enhancement of optic nerve on orbital MRI point toward an inflammatory etiology and should prompt testing for MOG-Ab (28). Fulminant disc edema with peripapillary hemorrhages and “macular star” have been described in MOGAD-ON (29–31). Both of these findings are considered highly atypical for other inflammatory-demyelinating diseases and are more often associated with infectious and ischemic etiologies (29, 30).
Up to 50% of adults with MOG-ON have a recurrence of optic neuritis (11–13, 18), which may be the only manifestations of MOGAD. Two rare previously described phenotypes, chronic relapsing inflammatory optic neuropathy (CRION)– a rare condition characterized by relapsing, steroid-dependent optic neuritis (32), and relapsing isolated optic neuritis (RION), have been associated with MOG-Ab in some cases (33, 34).
MRI of the orbits during acute MOG-ON typically shows longitudinally extensive optic nerve enhancement with a predilection for the anterior portion of optic nerves; the chiasm and optic tracts are less frequently affected (21, 31). “Optic perineuritis,” characterized by inflammation of the optic nerve sheath and surrounding structures on MRI (35), is seen in up to 50% of cases of MOGAD-ON (Figure 1A) (13, 21, 25, 36, 37). Perineural enhancement is a feature that can help differentiate MOGAD from NMOSD or MS (13, 21, 25, 36, 37). Isolated cases of MOGAD perineuritis, involving the nerve sheath and surrounding structures but not the optic nerve, have also been reported (38, 39). Rarely, uveitis and keratitis can occur simultaneously or subsequently to MOG-ON (38).
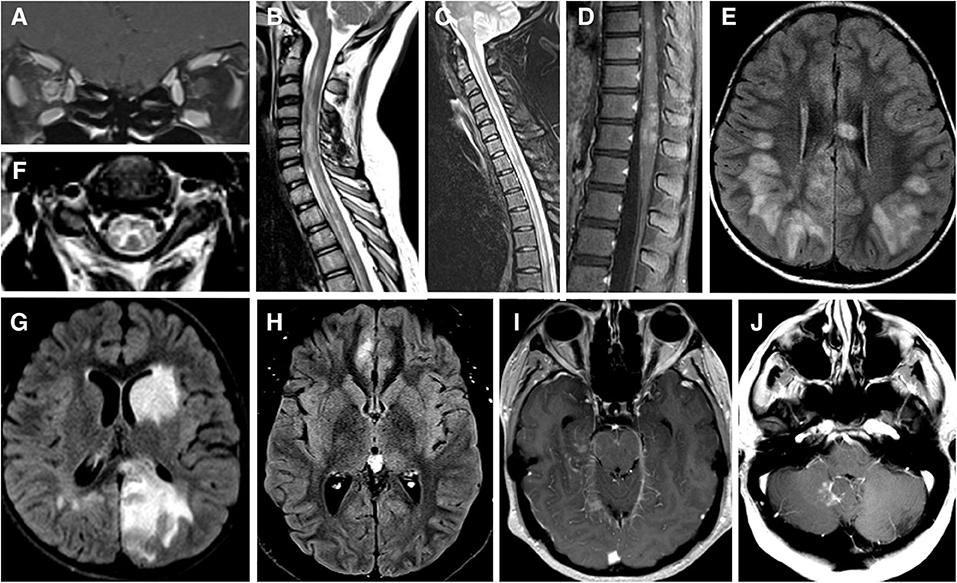
Figure 1. (A) MRI brain T1 coronal post gadolinium contrast showing contrast enhancement of bilateral optic nerves and right optic nerve sheath consistent with perioptic neuritis. (B) MRI spine sagittal STIR showing longitudinal extensive patchy lesion spaning from cervical to thoracic cord. (C) MRI spine sagittal T2 showing hyperintense longitudinally extensive “pseudo-dilation” of central canal. (D) MRI spine sagittal T1 post gadolinium contrast showing patchy enhancement of the conus medullaris. (E) MRI brain axial FLAIR showing large subcortical and septal white matter lesions in a pediatric patient presenting with ADEM. (F) MRI brain axial T2 with hyperintense “H” sign outlining the central gray matter of the upper cervical cord in a teenager with myelitis. (G) MRI brain axial T2 with “fluffy” hyperintense lesion of gray and white matter of the left caudate and left occipital parietal regions in a pediatric patient who presenting with ADEM. (H) MRI brain axial T2 showing unilateral FLAIR hyperintensity and edema of right mesial frontal cortex in a patient with FLAMES syndrome. (I) MRI brain axial T1 post gadolinium contrast showing leptomeningeal enhancement of the midrain and right mesial temporal lobe. (J) MRI brain axial T1 post gadolinium contrast showing a lesion adjacent to the cerebellar vermis and dorsal medulla in a patient with brainstem syndrome and no other lesions.
Transverse Myelitis
MOG-Ab associated acute transverse myelitis is a relatively common presentation of MOGAD in adults, and can be seen in children as well (11). In some cases of MOG-TM, there is an antecedent history of infection or vaccination, but in most patients, no such history can be elicited (11, 18, 40). While MOG-TM is typically steroid-responsive with favorable long-term recovery, around 9% of patients have poor recovery (11). Recurrent myelitis, without any other syndromes of MOGAD, is reported in up to 5% of patients (41).
MOG-TM can affect any segments of the spinal cord but has a greater predilection for conus medullaris–reported in 11–41% patients–than other CNS inflammatory-demyelinating diseases (11, 18, 40, 42). The involvement of the conus (Figure 1D) may explain the high incidence of neurogenic bowel and bladder symptoms (83%), and erectile dysfunction (54%) during acute phase (40), as well as in the long-term (11). There are also reports of a steroid-dependent myeloradiculitis in MOGAD with a longitudinally extensive transverse lesion from T12 to the conus with sacral nerve root enhancement (43).
Radiographically, MOG-TM is usually associated with a longitudinally extensive lesion spanning 3-4 vertebral segments (Figure 1B) (2, 18, 40, 44). In this respect, MOG-TM is similar to NMO-TM, but there are several radiographic differences between the two diseases. First, cord lesion of MOG-TM during the acute phase are much less likely to demonstrate gadolinium enhancement than in NMOSD: only 26% of MOG patients show enhancement vs. 78% of AQ4-ab-seropositive NMOSD (40). Secondly, spinal cord lesions in MOGAD can be multifocal: 62% of patients had ≥2 non-contiguous spinal cord lesions (40). The radiographic multifocality is in line with the notion that MOGAD has a tendency to affect multiple areas of CNS simultaneously.
MOG-TM affects both gray and white matter of the cord. The involvement of gray matter can manifest as linear hyperintensity of the central spinal canal (“pseudo-dilation,” Figure 1C) (44), or as H-shaped T2-hyperintensity that outlines the anterior and posterior horns (“H-sign,” Figure 1F) (2, 18, 40). The “H-sign” is suggestive, but not specific for MOGAD, reported in 29% of patients with MOG-TM and 8% of patients with NMO-TM (40). The predilection for the gray matter may explain why MOG-TM sometimes presents as acute flaccid paralysis (AFM) (45): in one series 10 out of 47 MOGAD patients (21%) met clinical criteria for AFM (40).
Acute Disseminated Encephalomyelitis (ADEM) and Other Cerebral Presentations
In young children, MOGAD frequently presents as ADEM or an ADEM-like syndrome (ADEM with optic neuritis, multiphasic disseminated encephalomyelitis) (16, 46–49). MRI of the brain typically shows large, ill-defined bilateral lesions frequently involving cortical and deep gray matter structures (Figure 1G) (50). Lesions may also involve subcortical white matter and corpus callosum as seen in Figure 1E. Optic nerves and spinal cord may be involved concurrently with brain (51). Recurrent ADEM or ADEM associated with recurrent optic neuritis (52, 53) are especially suggestive of MOGAD. Importantly, in children with clinical syndrome of encephalitis, MOGAD diagnosis is possible even when MRI findings are not compatible with ADEM—for example, exclusive cortical or symmetric thalamic/basal ganglia involvement, or even normal MRI (54).
Cerebral involvement in adults is both less common and more restricted than in children, though there are exceptions (55). Syndrome of encephalitis with steroid-responsive seizures, also termed FLAMES (FLAIR-hyperintense Lesions and Anti-MOG-associated Encephalitis with Seizures), appears to be specific to MOGAD (20, 56–58). FLAMES patients present with focal-onset, tonic-clonic seizures, and have unilateral FLAIR hyperintensities with edema on MRI (Figure 1H). A review by Budhram et al. found 20 cases of FLAMES in the literature. The most common symptoms were seizures (85%), headache (70%), and fever (55%). CSF pleocytosis and cortical leptomeningeal enhancement (Figure 1I) were present in a minority of patients (57). All patients with FLAMES responded to high dose steroids with resolution of FLAIR changes. Of note, a number of patients developed ON either before or after seizures (56, 58, 59). Thus, the emergence of seizures in the context of ON or focal brain inflammatory lesions should prompt testing for MOG-Ab (52).
Isolated seizures may rarely be an index event in MOGAD. In one case, an adult patient presented with aphasic status epilepticus with initial MRI showing no abnormalities. Six months later the patient developed a tumefactive demyelinating lesion, with MOG-Ab testing positive several months later (60). A similar presentation has been described in four pediatric patients who presented with isolated seizures and normal brain MRI and developed MRI brain lesions months, and in one case years, later (61).
Several studies document an association between MOGAD and autoimmune encephalitis with NMDA-antibody (62–64). In a retrospective case review by Titulaer et al., 12 of 691 with NMDAR encephalitis patients (1.6%) tested positive for MOG-Ab. Some patients presented with MOGAD syndrome followed by encephalitis, others with encephalitis followed by MOGAD, and in some NMDA encephalitis and MOGAD were diagnosed concurrently. Three patients with NMDAR encephalitis and no clinical or MRI features to suggest MOGAD also tested positive for MOG-Ab (62).
Finally, mention should be made of rare cases when MOG-Ab was found in patients with pathologically-proven CNS vasculitis (65, 66). Two patients presented with fever, headache, confusion, and focal neurologic deficits (66), and the third had 9 months of progressive cognitive and behavioral decline (65). MRI showed multifocal lesions in both the gray and white matter in two cases, one of whom also had open-ring contrast-enhancing lesions. The third case had findings of focal cortical encephalitis with gyriform FLAIR hyperintensities with edema, similar to findings seen in FLAMES. All three cases underwent brain biopsy, which showed small vessel perivascular inflammation, consistent with CNS vasculitis. However, fibrinoid necrosis, a pathologic requirement for small vessel CNS vasculitis, was absent in two of the cases (66, 67). Whether vasculitis should be regarded as a primary or secondary manifestation of MOGAD, or MOG-Ab is unrelated to vasculitis diagnosis, is difficult to determine given rarity of the association.
Brainstem and Cerebellar Presentations
Brainstem involvement is seen in 30% of MOGAD patients, and is a risk factor for a higher disability at long-term follow-up and more active disease (68). In one large series brainstem inflammation occurred concomitantly with inflammation in optic nerves in 40% of cases, spinal cord in 89% cases and cerebrum in 66% of cases (68). However, there are reports of isolated brainstem inflammation as well (Figure 1J) (68). Any part of the brainstem can be affected, medulla being the most common (11, 68). Brainstem lesions are usually associated with disabling symptoms—weakness, cranial nerve deficits, ataxia, hypoventilation syndrome, impaired consciousness and, and, exceptionally, a fatal outcome (68). Area postrema syndrome (APS), one of the core syndromes of NMOSD, has also been described in MOGAD (11, 68–70).
MOGAD can mimic infective rhomboencephalitis when a patient presents with fever, CSF leukocytosis, brainstem enhancing lesions and leptomeningeal enhancement (44, 68), or Chronic Lymphocytic Inflammation with Pontine Perivascular Enhancement Responsive to Steroids (CLIPPERS), when MRI shows punctate, curvilinear enhancement in the pons (71–73). Whether CLIPPERS is a form of MOGAD or elicits an immune response to MOG-Ab is uncertain (73).
Conclusion
Since the first reports of MOG-Ab associated neurologic diseases appeared just a few years ago (4), the floodgates of case reporting have been opened and our understanding of MOGAD has grown exponentially. We now recognize certain clinical and radiologic features that help to differentiate MOG-ON and MOG-TM from NMOSD syndromes; that pediatric ADEM is frequently associated with MOG-Ab, especially if followed by episodes of ADEM or ON; that in adults, MOG can be associated with seizures and focal cerebral edema (“FLAMES syndrome,” which appears to be unique to MOGAD); that brainstem inflammation is seen in a significant minority of MOGAD patients and may be an isolated finding; that MOG Ab is a common mimicker of infectious encephalitis (54) that MOG antibody is exceptionally rare in MS or AQ4 Ab positive NMOSD, but may co-exist with NMDA and other autoimmune encephalidites (64, 74). But many important questions remain. We need to determine sensitivity, specificity, positive and negative predictive value of MOG-Ab in the various neurologic syndromes; whether MOG-Ab shoud be tested in CSF, if it is negative in serum (75); whether various ultrarare presentions, such as isolated seizures without brain lesions, CLIPPERS, and a MOG-Ab-associated CNS vasculitis-type syndrome should be subsumed under MOGAD rubric. Most importantly, we need to better stratify risk of disease recurrence after the first or second episode and determine best treatments to prevent recurrence. With the rapid pace of progress, we can expect to answer these and other questions, and, no doubt, find new surprises along the way.
Author Contributions
EP and IK wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest
IK served on advisory boards for Biogen and Genentech and received consulting fees from Roche and research support for investigator-initiated grants from Sanofi Genzyme, Biogen, EMD Serono, National MS Society, and Guthy Jackson Charitable Foundation.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lebar R, Baudrimont M, Vincent C. Chronic experimental autoimmune encephalomyelitis in the guinea pig. presence of anti-M2 antibodies in central nervous system tissue and the possible role of M2 autoantigen in the induction of the disease. J Autoimmunity. (1989) 2:115–32. doi: 10.1016/0896-8411(89)90149-2
2. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. (2012) 79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e
3. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflamm. (2011) 8:184. doi: 10.1186/1742-2094-8-184
4. Rostasy K, Mader S, Hennes EM, Schanda K, Gredler V, Guenther A, et al. Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin-4 antibody negative pediatric neuromyelitis optica. Mult Sclero J. (2013) 19:1052–9. doi: 10.1177/1352458512470310
5. Hamid SH, Whittam D, Mutch K, Linaker S, Solomon T, Das K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. (2017) 264:2088–94. doi: 10.1007/s00415-017-8596-7
6. Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front Neurol. (2018) 9:217. doi: 10.3389/fneur.2018.00217
7. You Y, Zhu L, Zhang T, Shen T, Fontes A, Yiannikas C, et al. Evidence of müller glial dysfunction in patients with aquaporin-4 immunoglobulin g-positive neuromyelitis optica spectrum disorder. Ophthalmology. (2019) 126:801–10. doi: 10.1016/j.ophtha.2019.01.016
8. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. (2014) 24:83–97. doi: 10.1111/bpa.12099
9. Fujihara K, Misu T, Nakashima I, Takahashi T, Bradl M, Lassmann H, et al. Neuromyelitis optica should be classified as an astrocytopathic disease rather than a demyelinating disease. Clin Experimental Neuroimmunol. (2012) 3:58–73. doi: 10.1111/j.1759-1961.2012.00030.x
10. Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm. (2015) 2:e62. doi: 10.1212/NXI.0000000000000062
11. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
12. de Mol CL, Wong Y, van Pelt ED, Wokke B, Siepman T, Neuteboom RF, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler. (2020) 26:806–14. doi: 10.1177/1352458519845112
13. Chen JJ, Flanagan EP, Jitprapaikulsan J, López-Chiriboga AS, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody–positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. (2018) 195:8–15. doi: 10.1016/j.ajo.2018.07.020
14. Chen Q, Zhao G, Huang Y, Li Z, Sun X, Lu P, et al. Clinical characteristics of pediatric optic neuritis with myelin oligodendrocyte glycoprotein seropositive: a cohort study. Pediatric Neurol. (2018) 83:42–9. doi: 10.1016/j.pediatrneurol.2018.03.003
15. Cobo-Calvo Á, Ruiz A, D'Indy H, Poulat AL, Carneiro M, Philippe N, et al. MOG antibody-related disorders: common features and uncommon presentations. J Neurol. (2017) 264:1945–55. doi: 10.1007/s00415-017-8583-z
16. Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. (2017) 89:900–8. doi: 10.1212/WNL.0000000000004312
17. Waters P, Fadda G, Woodhall M, O'Mahony J, Brown RA, Castro DA, et al. Serial anti–myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. (2019) 77:82–93. doi: 10.1001/jamaneurol.2019.2940
18. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
19. Numa S, Kasai T, Kondo T, Kushimura Y, Kimura A, Takahashi H, et al. An adult case of anti-myelin oligodendrocyte glycoprotein (MOG) antibody-associated multiphasic acute disseminated encephalomyelitis at 33-year intervals. Internal Med. (2016) 55:699–702. doi: 10.2169/internalmedicine.55.5727
20. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. (2018) 90:e1858–e1869. doi: 10.1212/WNL.0000000000005560
21. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Sclero J. (2016) 22:470–82. doi: 10.1177/1352458515593406
22. Ramanathan S, Reddel SW, Henderson A, Parratt JD, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e40. doi: 10.1212/NXI.0000000000000040
23. Lock JH, Newman NJ, Biousse V, Peragallo JH. Update on Pediatric Optic Neuritis. Curr Opinion Ophthalmol. (2019) 30:418–25. doi: 10.1097/ICU.0000000000000607
24. Zhao Y, Tan S, Chan TC, Xu Q, Zhao J, Teng D, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. British J Ophthalmol. (2018) 102:1372–7. doi: 10.1136/bjophthalmol-2017-311177
25. Song H, Zhou H, Yang M, Tan S, Wang J, Xu Q, et al. Clinical characteristics and prognosis of myelin oligodendrocyte glycoprotein antibody-seropositive paediatric optic neuritis in China. British J Ophthalmol. (2019) 103:831–6. doi: 10.1136/bjophthalmol-2018-312399
26. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
27. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
28. Narayan RN, Wang C, Sguigna P, Husari K, Greenberg B. Atypical Anti-MOG syndrome with aseptic meningoencephalitis and pseudotumor cerebri-like presentations. Mult Sclero Relat Disorde. (2019) 27:30–3. doi: 10.1016/j.msard.2018.10.003
29. Chen JJ, Bhatti MT. Clinical phenotype, radiological features, and treatment of myelin oligodendrocyte glycoprotein-immunoglobulin G (MOG-IgG) optic neuritis. Curr Opin Neurol. (2020) 33:47–54. doi: 10.1097/WCO.0000000000000766
30. Moura FC, Sato DK, Rimkus CM, Apóstolos-Pereira SL, de Oliveira LM, Leite CC, et al. Anti-MOG (Myelin Oligodendrocyte Glycoprotein)–positive severe optic neuritis with optic disc ischaemia and macular star. Neuro-Ophthalmology. (2015) 39:285–8. doi: 10.3109/01658107.2015.1084332
31. Biotti D, Bonneville F, Tournaire E, Ayrignac X, Dallière CC, Mahieu L, et al. Optic neuritis in patients with anti-MOG antibodies spectrum disorder: MRI and clinical features from a large multicentric cohort in France. J Neurol. (2017) 264:2173–5. doi: 10.1007/s00415-017-8615-8
32. Kidd D. Chronic relapsing inflammatory optic neuropathy (CRION). Brain. (2003) 126:276–84. doi: 10.1093/brain/awg045
33. Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. J Neurol Neurosurg Psychiatry. (2019) 90:1021–6. doi: 10.1136/jnnp-2019-320493
34. Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. J Neuroinflamm. (2018) 15:302. doi: 10.1186/s12974-018-1335-x
35. Purvin V. Optic perineuritis: clinical and radiographic features. Arch Ophthalmol. (2001) 119:1299. doi: 10.1001/archopht.119.9.1299
36. Zhou L, Huang Y, Li H, Fan J, Zhangbao J, Yu H, et al. MOG-antibody associated demyelinating disease of the CNS: a clinical and pathological study in Chinese Han patients. J Neuroimmunol. 305: 19–28. doi: 10.1016/j.jneuroim.2017.01.007
37. Akaishi T, Sato DK, Nakashima I, Takeshita T, Takahashi T, Doi H, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: a comparative study. J Neurol Neurosurg Psychiatry. (2016) 87:446–8. doi: 10.1136/jnnp-2014-310206
38. Ramanathan S, Fraser C, Curnow SR, Ghaly M, Leventer RJ, Lechner-Scott J, et al. Uveitis and optic perineuritis in the context of myelin oligodendrocyte glycoprotein antibody seropositivity. Eur J Neurol. (2019) 26:1137. doi: 10.1111/ene.13932
39. Yanagidaira M, Hattori T, Emoto H, Kiyosawa M, Yokota T. Optic perineuritis with anti-myelin oligodendrocyte glycoprotein antibody. Mult Sclero Relat Disorde. (2020) 38:101444. doi: 10.1016/j.msard.2019.101444
40. Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301. doi: 10.1001/jamaneurol.2018.4053
41. Anis S, Regev K, Pittock SJ, Flanagan EP, Alcalay Y, Gadoth A. Isolated recurrent myelitis in a persistent MOG positive patient. Mult Sclero Relat Disorde. (2019) 30:163–4. doi: 10.1016/j.msard.2019.02.016
42. Salama S, Khan M, Levy M, Izbudak I. Radiological characteristics of myelin oligodendrocyte glycoprotein antibody disease. Mult Sclero Relat Disorde. (2019) 29:15–22. doi: 10.1016/j.msard.2019.01.021
43. Sundaram S, Nair SS, Jaganmohan D, Unnikrishnan G, Nair M. Relapsing lumbosacral myeloradiculitis: an unusual presentation of mog antibody disease. Mult Sclero J. (2019) 26:509–11. doi: 10.1177/1352458519840747
44. Denève M, Biotti D, Patsoura S, Ferrier M, Meluchova Z, Mahieu L, et al. MRI features of demyelinating disease associated with anti-MOG antibodies in adults. J Neuroradiol. (2019) 46:312–8. doi: 10.1016/j.neurad.2019.06.001
45. Wang C, Narayan R, Greenberg B. Anti-myelin oligodendrocyte glycoprotein antibody associated with gray matter predominant transverse myelitis mimicking acute flaccid myelitis: a presentation of two cases. Pediatric Neurol. (2018) 86:42–5. doi: 10.1016/j.pediatrneurol.2018.06.003
46. Pröbstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology. (2011) 77:580–8. doi: 10.1212/WNL.0b013e318228c0b1
47. Di Pauli F, Mader S, Rostasy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. (2011) 138:247–54. doi: 10.1016/j.clim.2010.11.013
48. Baumann M, Sahin K, Lechner C, Hennes EM, Schanda K, Mader S, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry. (2015) 86:265–72. doi: 10.1136/jnnp-2014-308346
49. Hacohen Y, Mankad K, Chong WK, Barkhof F, Vincent A, Lim M, et al. Diagnostic algorithm for relapsing acquired demyelinating syndromes in children. Neurology. (2017) 89:269–78. doi: 10.1212/WNL.0000000000004117
50. Wegener-Panzer A, Cleaveland R, Wendel EM, Baumann M, Bertolini A, Häusler M, et al. Clinical and imaging features of children with autoimmune encephalitis and MOG antibodies. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e731. doi: 10.1212/NXI.0000000000000731
51. Baumann M, Hennes EM, Schanda K, Karenfort M, Kornek B, Seidl R, et al. Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): extending the spectrum of MOG antibody positive diseases. Mult Sclero J. (2016) 22:1821–9. doi: 10.1177/1352458516631038
52. Gutman JM, Kupersmith M, Galetta S, Kister I. Anti-myelin oligodendrocyte glycoprotein (MOG) antibodies in patients with optic neuritis and seizures. J Neurol Sci. (2018) 387:170–3. doi: 10.1016/j.jns.2018.01.042
53. Huppke P, Rostasy K, Karenfort M, Huppke B, Seidl R, Leiz S, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Sclero J. (2013) 19:941–6. doi: 10.1177/1352458512466317
54. Armangue T, Olivé-Cirera G, Martínez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Muñoz-Batista M, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol. (2020) 19:234–46. doi: 10.1016/S1474-4422(19)30488-0
55. Uchigami H, Arai N, Sekiguchi M, Ogawa A, Yasuda T, Seto A, et al. Anti-myelin oligodendrocyte glycoprotein antibody-positive acute disseminated encephalomyelitis mimicking limbic encephalitis: a case report. Mult Scler Relat Disord. (2020) 38:101500. doi: 10.1016/j.msard.2019.101500
56. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody–positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
57. Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Sharma M, Nicolle MW. Unilateral cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol. (2019) 266:2481–7. doi: 10.1007/s00415-019-09440-8
58. Ikeda T, Yamada K, Ogawa R, Takai Y, Kaneko K, Misu T, et al. The pathological features of MOG antibody-positive cerebral cortical encephalitis as a new spectrum associated with MOG antibodies: a case report. J Neurol Sci. (2018) 392:113–5. doi: 10.1016/j.jns.2018.06.028
59. Taraschenko O, Zabad R. Overlapping demyelinating syndrome and anti-n-methyl-d-aspartate receptor encephalitis with seizures. Epilepsy Behav Rep. (2019) 12:100338. doi: 10.1016/j.ebr.2019.100338
60. Katsuse K, Kurihara M, Sugiyama Y, Kodama S, Takahashi M, Momose T, et al. Aphasic status epilepticus preceding tumefactive left hemisphere lesion in anti-MOG antibody associated disease. Mult Sclero Relat Disorde. (2019) 27:91–4. doi: 10.1016/j.msard.2018.10.012
61. Ramanathan S, O'grady GL, Malone S, Spooner CG, Brown DA, Gill D, et al. Isolated seizures during the first episode of relapsing myelin oligodendrocyte glycoprotein antibody-associated demyelination in children. Dev Med Child Neurol. (2019) 61:610–4. doi: 10.1111/dmcn.14032
62. Titulaer MJ, Höftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, et al. Overlapping demyelinating syndromes and anti-N-Methyl-D-aspartate receptor encephalitis: anti-NMDAR encephalitis. Ann Neurol. (2014) 75:411–28. doi: 10.1002/ana.24117
63. Carroll E, Wallach A, Kurzweil A, Frucht S, Berk T, Boffa M, et al. Care report: seizure, fever, hallucinations & vision loss, a circuitous route to dual diagnoses. Pract Neurol. (2019). Available online at: https://practicalneurology.com/articles/2019-nov-dec/case-report-seizure-fever-hallucinations-vision-loss
64. Martinez-Hernandez E, Guasp M, García-Serra A, Maudes E, Ariño H, Sepulveda M, et al. Clinical significance of anti-NMDAR concurrent with glial or neuronal surface antibodies. Neurology. (2020) 94:e2302–e2310. doi: 10.1212/WNL.0000000000009239
65. Baba T, Shinoda K, Watanabe M, Sadashima S, Matsuse D, Isobe N, et al. MOG antibody disease manifesting as progressive cognitive deterioration and behavioral changes with primary central nervous system vasculitis. Mult Sclero Relat Disorde. (2019) 30:48–50. doi: 10.1016/j.msard.2019.01.053
66. Patterson K, Iglesias E, Nasrallah M, González-Álvarez V, Suñol M, Anton J, et al. Anti-MOG encephalitis mimicking small vessel CNS vasculitis. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e538. doi: 10.1212/NXI.0000000000000538
67. Giannini C, Salvarani C, Hunder G, Brown RD. Primary central nervous system vasculitis: pathology and mechanisms. Acta Neuropathol. (2012) 123:759–72. doi: 10.1007/s00401-012-0973-9
68. Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: brainstem involvement - frequency, presentation and outcome. J Neuroinflamm. (2016) 13:281. doi: 10.1186/s12974-016-0719-z
69. Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. (2017) 140:617–27. doi: 10.1093/brain/aww350
70. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
71. Berzero G, Taieb G, Marignier R, Younan N, Savatovsky J, Leclercq D, et al. CLIPPERS mimickers: relapsing brainstem encephalitis associated with anti-MOG antibodies. Eur J Neurol. (2018) 25:e16–e17. doi: 10.1111/ene.13483
72. Symmonds M, Waters PJ, Küker W, Leite MI, Schulz UG. Anti-MOG antibodies with longitudinally extensive transverse myelitis preceded by CLIPPERS. Neurology. (2015) 84:1177–9. doi: 10.1212/WNL.0000000000001370
73. Taieb G, Pierre L. ANti-MOG antibodies with longitudinally extensive transverse myelitis preceded by CLIPPERS. Neurology. (2015) 85:2. doi: 10.1212/01.wnl.0000472753.44905.98
74. Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ. Coexistence of myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies in adult and pediatric patients. JAMA Neurol. (2019) 77:257–9. doi: 10.1001/jamaneurol.2019.3656
Keywords: MOG (myelin oligodendrocyte glyco protein), MOG antibody disease, ADEM, myelitis, optic neuritis, CRION, brainstem encephalitis
Citation: Parrotta E and Kister I (2020) The Expanding Clinical Spectrum of Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Associated Disease in Children and Adults. Front. Neurol. 11:960. doi: 10.3389/fneur.2020.00960
Received: 12 December 2019; Accepted: 24 July 2020;
Published: 09 September 2020.
Edited by:
Yael Hacohen, University College London, United KingdomReviewed by:
Rinze Neuteboom, Erasmus Medical Center, NetherlandsKevin Rostasy, Vestische Kinder- und Jugendklinik Datteln, Germany
Copyright © 2020 Parrotta and Kister. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erica Parrotta, ZXJpY2EuaC5wYXJyb3R0YUBnbWFpbC5jb20=