 Xiaoli Yu
Xiaoli Yu Michael Graner1
Michael Graner1 Peter G. E. Kennedy
Peter G. E. Kennedy- 1Department of Neurosurgery, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow, United Kingdom
- 3Department of Neurology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
The presence of persistent intrathecal oligoclonal immunoglobulin G (IgG) bands (OCBs) and lesional IgG deposition are seminal features of multiple sclerosis (MS) disease pathology. Despite extensive investigations, the role of antibodies, the products of mature CD19+ B cells, in disease development is still controversial and under significant debate. Recent success of B cell depletion therapies has revealed that CD20+ B cells contribute to MS pathogenesis via both antigen-presentation and T-cell-regulation. However, the limited efficacy of CD20+ B cell depletion therapies for the treatment of progressive MS indicates that additional mechanisms are involved. In this review, we present findings suggesting a potential pathological role for increased intrathecal IgGs, the relation of circulating antibodies to intrathecal IgGs, and the selective elevation of IgG1 and IgG3 subclasses in MS. We propose a working hypothesis that circulating B cells and antibodies contribute significantly to intrathecal IgGs, thereby exerting primary and pathogenic effects in MS development. Increased levels of IgG1 and IgG3 antibodies induce potent antibody-mediated cytotoxicity to central nervous system (CNS) cells and/or reduce the threshold required for antigen-driven antibody clustering leading to optimal activation of immune responses. Direct proof of the pathogenic roles of antibodies in MS may provide opportunities for novel blood biomarker identification as well as strategies for the development of effective therapeutic interventions.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease that affects the central nervous system (CNS), especially the brain, spinal cord, and optic nerves. About 1 million people in the US and 2.5 million worldwide live with a diagnosis of MS (1). After its first description in 1868 by Jean-Martin Charcot, MS has been classified into different types such as clinically isolated syndrome (CIS, a clinical syndrome highly suggestive of a first manifestation of MS), relapsing remitting (RR), secondary progressive (SP), and primary progressive (PP) (2). Extensive pathological studies have classified MS lesions as active, chronic active, inactive, and pre-active stages (3). Despite the heterogeneous features of MS lesions, a consensus has emerged that the pathogenic mechanisms of the disease are contributed by CNS inflammation and infiltration of peripheral immune cells, resulting in neuronal and glial cell injury and subsequent loss of myelin sheath around nerves, interruption of axonal communication, and neurologic deficits (4). However, the exact cause of MS is unknown, and currently there is no cure for the disease.
The presence of persistent CNS oligoclonal immunoglobulin G (IgG) bands (OCBs) and lesional IgG deposition are hallmarks of MS. OCBs consist of clonally restricted immunoglobulins detected by isoelectric focusing (IEF) and are a key feature of ongoing inflammatory events in CNS in a number of neuro-inflammatory conditions and viral infections (5). Although the pathological effects of OCBs have been implicated since their discovery (6), the role of antibodies in the pathogenesis of MS is controversial. In this review, we discuss pathological and immunological studies regarding the role of antibodies in MS. We propose a novel framework regarding the pathogenic mechanism of disease, which could be mediated by increased levels of serum IgG1 and IgG3 antibodies.
Increased Intrathecal Synthesis of OCB is the Most Characteristic Feature of MS
Early biochemical studies of MS autopsy brain plaques with active lesions have demonstrated the presence of excessive amounts of IgG antibodies in both free/soluble and tissue-bound/particulate forms (7–9). The IgGs extracted from corresponding soluble and particulate samples displayed OCBs (9). Extensive pathological characterization of heterogeneous MS autopsy brain samples has demonstrated the co-localization of IgG antibodies, complement, and Fc gamma receptors (FcγR) in the active lesions, suggesting a role for these antibodies in the early stages of the disease (10, 11). Furthermore, complement activation is found in PPMS cortical gray matter lesions (12), indicating that antibodies may contribute to the worsening pathology that underlies the irreversible progression of MS. These lines of evidence suggest that the excessive presence of IgG antibodies in MS lesions may induce complement-mediated and immune-cell-mediated cytotoxicity, resulting in lesion formation.
The single most consistent laboratory abnormality in MS is the presence of OCBs in the cerebrospinal fluid (CSF) of up to 95% of patients (13, 14). Once present, the pattern of OCB is characteristic for each individual and does not change within patients over years, despite therapeutic interventions (5, 13, 15, 16). Besides OCBs, the intrathecal IgG can also be visualized with Reiber diagram, which uses CSF/serum quotient diagrams with hyperbolic discrimination lines for IgG (17). Plasma cells are found in the chronically inflamed MS CNS (18). Long-lived plasma cells were demonstrated in chronically inflamed CNS and chemokine CXCL12 is involved in plasma cell persistence (19). For detailed review regarding the complex nature of resident plasma cells and mechanism driving the persistence in CNS, please see the review by Pryce and Baker (5). Using a phage-displayed random peptide library approach, we demonstrated that CSF IgGs obtained longitudinally from MS patients recognized identical epitopes over time, supporting the notion of a temporal stability of CSF IgG specificity (20).
Accumulating evidence supports the pathological role of CSF immunoglobulins. CSF OCBs were found to be associated with increased levels of disease activity and disability, with the conversion from a CIS to early RRMS, with greater brain atrophy, and with increased levels of disease activity (21–28). Further, CSF of MS patients induced inflammatory demyelination and axonal damage in mice (29, 30). We demonstrated that a subset of myelin-specific recombinant antibodies constructed from clonal expanded plasma cells in MS CSF caused robust complement-dependent cytotoxicity in oligodendrocytes and induced rapid demyelination in mouse organotypic cerebellar slices (31, 32). These studies support the pathogenic effects of CSF IgGs in MS. Despite the significance of OCB in MS, no statistically significant differences of both number of OCBs and IgG index were found among subtypes of MS (CIS, RRMS, PPMS, and SPMS) (33). New technologies such as recombinant antibodies generated from clonally expanded single B cells/plasma cells and directly from IgG sequences of OCBs provided promises for determining the specificities of OCB, but have so far failed to reveal a common targets of MS (34–37) (https://www.jni-journal.com/article/S0165-5728(20)30298-8/pdf).
Except for myelin, convincing CNS target antigens for OCBs that are specific to MS are not known. Recently, it was shown that some OCBs targeted ubiquitous self-proteins and intracellular antigens (37–39), suggesting that CSF antibodies may develop as a passive response to CNS injury, rather than mediating primary pathogenic effects. Besides MS, CSF OCBs have been reported in a number of neuro-inflammatory conditions and viral infections (40). It has been argued that this intrathecal, poly-specific, and oligoclonal immune response possibly indicates that it is not a specific antigen that drives the development of OCBs in MS, but rather a non-specific activation of CSF-localized B cells (41).
The Sources of Increased Intrathecal IgG, a Controversy In MS
Correlation of Serum Antibodies With Intrathecal IgGs
OCBs are thought to be produced by intrathecal parenchymal B lymphocytes, as the CSF Ig proteome and transcriptome of CSF-located B cells matched each other. In addition, intrathecal B cells show signs of somatic hyper-mutation and clonal expansion, pointing toward a germinal center-like reaction with antigen-driven affinity maturation within the CNS (42, 43). However, there is new evidence that terminally differentiated B cells in MS CSF are not solely derived from intrathecal maturation, but can emerge from the CNS compartment and interact with the peripheral immune system (44–46). Recent deep-immune repertoire studies revealed that MS CSF OCBs were not merely produced by CNS B cells, and some OCB specificities were related only to peripheral B cells, which indicate that disease-relevant B cells circulate between the CNS and peripheral compartments (47). We recently demonstrated that serum IgG in MS was significantly elevated and there was a strong correlation between CSF IgG and CSF albumin, and also between CSF IgG and serum IgG (48). Since CSF albumin is exclusively derived from the blood in MS, this correlation suggests that most of the CSF IgG is derived from the blood. It has to be noted that about 50% of MS sera did not show OCBs and patients with OCBs in sera had patterns either partially similar or completely different from those seen in matching CSF (49). Because serum IgG is about 200 times more concentrated than CSF IgG (48), it is possible that serum OCBs are masked by massive amounts of polyclonal IgGs.
Another line of evidence supporting serum and intrathecal IgG exchange comes from the discrepancy between number of CNS B cells and the quantities of intrathecal IgG. A careful examination of MS plaques concluded that there were far too few cells in the plaques to contribute to IgG (50). As calculated by Tourtellotte's formula, the normal values of the CNS IgG synthesis rate were lower than 3.3 mg/day and the median value in MS patients was 29 mg/day (51). It would take 3.2 billion lymphocytes in MS to generate such large amounts of CNS IgG (30 mg in 500 ml CSF) (52). We and others have demonstrated that CSF leukocyte counts in most MS patients were <50 cells/μl (about 2.5 million cells in 500 ml CSF), of which 5% were B cells (48, 53, 54). Therefore, CSF lymphocytes could only account for <0.1% of the IgG in the MS CSF per day. The low number of lymphocytes in MS CSF and the high level of intrathecal IgG raise the question as to whether CNS B cells in MS can be responsible for the massive amounts of elevated intrathecal IgG. This apparent knowledge gap suggests that most of the intrathecal IgG in MS may in fact be derived from the blood.
MRI Detection of the Central Vein Sign in MS Lesions Supports a Peripheral Blood Contribution to Disease Activity
Early histopathological studies detected a unique character of MS lesions described as “centrifugally-spreading” that an MS plaque did not spread or grow at its edges. Plaques began as collars of demyelination around small veins and enlarged thereafter (50). This perivenous distribution of MS plaques was confirmed by ultra-high-field magnetic resonance imaging (MRI) (55–57). The MRI-detectable central vein sign inside white matter lesions can distinguish MS from other CNS inflammatory disorders and has been proposed as a biomarker for inflammatory demyelination (58). Further, the serum protein fibrinogen was found frequently and extensively to be present diffusely in both extracellular and intracellular spaces of MS motor cortex and in close proximity to blood vessels, and was related to the extent of neurodegeneration in progressive forms of MS (59). This line of evidence supports the notion of peripheral blood contribution of B cells and antibodies to intrathecal IgGs and their potential role in disease development.
We propose that MS intrathecal IgGs are derived from B cells in both the CNS and peripheral blood and may thus be contributed by serum antibodies (Figure 1). Inside the CNS compartments, clonally expanded antigen-experienced plasma cells produce antibodies that may target cell surface antigens and exert a pathogenic effect by activating complement-dependent or immune-cell dependent cytotoxicity. A subpopulation of these antibodies may direct against intracellular autoantigens released during tissue destruction. On the other hand, serum B cells and antibodies migrate across the blood barriers either by active transportation or by barrier breakdown. Some of these serum antibodies in MS are clearly pathogenic as reviewed below.
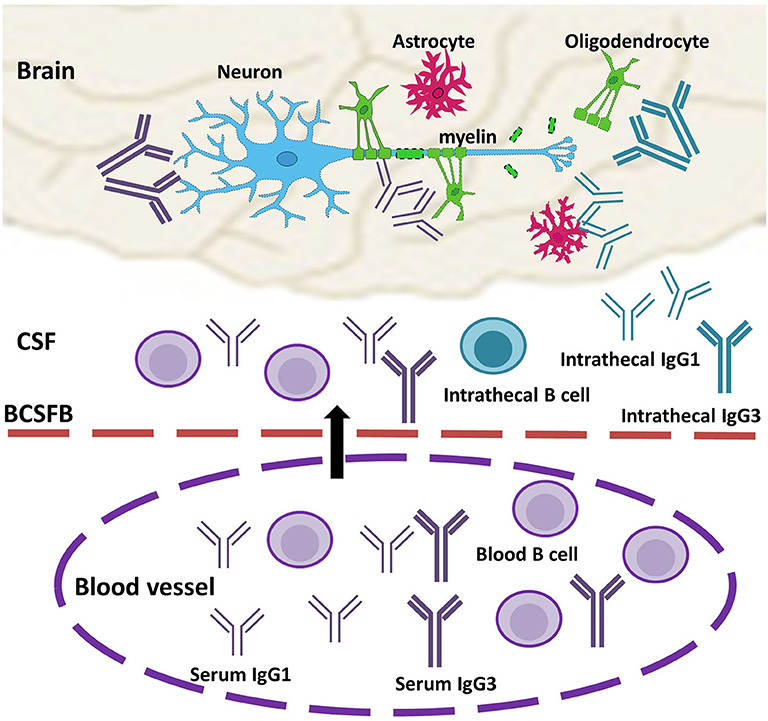
Figure 1. Model of the role of serum antibodies in MS disease pathogenesis. Circulating serum antibodies (IgG1 and IgG3, purple) and antibody-producing B cells migrate across the impaired blood-barrier (arrow), and they are present in CSF OCBs and CNS lesion together with intrathecal IgGs (IgG1 and IgG3, turquoise). In the brain, IgGs recognize antigens on the cell surfaces of neurons or/and glial cells and form immune complexes with complement factors and/or immune cells. Elevated levels of IgG1 and IgG3 induce enhanced cytotoxicity or reduced threshold to trigger injury response to CNS cells, which, in turn, result in loss of myelin sheath outside of axons. BCSFB, blood-CSF barrier.
Circulating Antibodies Contribute to MS Disease Pathogenesis
Pathogenic Effect of Serum Antibodies
The benefit shown in therapeutic plasma exchange and immune-adsorption therapy in some MS patients (41, 60, 61) suggests that serum antibodies in MS are pathogenic. Patients who had lesions with prominent Ig deposition and complement activation profited most from plasma exchange (60, 62). However, direct proof of the pathogenic role of serum antibody in MS is complicated by the marked heterogeneity of the disease and the variability of experimental procedures.
Early in vitro demyelination studies have provided evidence supporting a pathogenic role of serum antibodies (50) and that there is strong correlation between disease activity and demyelinative activity of MS serum. Lumsden (50) investigated sera from 450 MS patients and controls over 7 years with 1,300 tests. He found that over 80% of MS sera with natural complement produced demyelination in live cultures of newborn rat cerebellum. Further, he found patients' immunoglobulins and complement were fixed anti-mortem to CNS components, indicating that circulating antibody in MS binds to myelin and causes demyelination. Lumsden's data indicate that serum antibodies in their natural state are pathogenic when they penetrate the CNS parenchyma. Using ex vivo assays, a number of laboratories have reported that some MS patients have serum factors that demyelinate myelinating explants (63–66). Later, complement-dependent demyelinating IgG response was detected with purified serum IgGs in ~30% of 37 MS patients (67). However, the demyelinating effect of MS serum IgGs has been controversial, which is due in part to variable tissue culture and myelin imaging methods. Additionally, we postulate that the different results may reflect the sources of antibodies used. The antibody purification procedures may result in loss of the natural state of antibodies and may fail to efficiently recover specific IgG subclasses, resulting in a substantial reduction or a complete loss of demyelinating effect. Lumsden's work was carried out using unpurified native serum antibodies (50).
The potential pathogenic role of serum antibodies may also extend to enhancing inflammatory responses across the BBB in MS. For example, significantly higher levels of anti-endothelial cell antibodies and immune complexes were found in MS sera (68), and serum antibodies from MS patients were detected in micro-vessels in brain tissues and bound to endothelia cells (69, 70). Further, sera from RRMS and SPMS disrupt the BBB (71). In summary, over 50 years of extensive scientific investigations have provided accumulating evidence that serum antibodies in their natural state exert primary antibody-dependent cytotoxicity to glial cells, which leads to demyelinating effects that could contribute to MS disease pathogenesis.
Insights From B Cell Depletion Therapies
B cell depletion therapies using monoclonal antibodies against CD20; namely, Rituximab, Ocrelizumab, and Ofatumumab have shown profound success in controlling MS relapses (41). CD20 is a four-transmembrane protein expressed on the surface of B cells from the late pro-B cells through the memory cell stages, but not on antibody-producing plasma cells. Thus, the efficacy of this B cell depletion therapy has been considered to be mediated by B cell function independent of antibody production, such as antigen-presentation for the activation of T cells and pro-inflammatory cytokine secretion (72). Indeed, serum antibody level and CSF OCB often persist despite CD20-antibody depleting B cells (5, 73). In some instances, certain serum antibodies were reduced in a proportion of patients after intravenous Rituximab treatment (74). CSF plasma cell depletion was observed following repeated intrathecal Rituximab injection (75, 76). These reductions are thought to result from secondary effects such as depletion of plasma cell precursors, depletion of survival factors, or possibly destruction of B cell niches rather than a direct influence on plasma cells. Interestingly, Laquinimod, a T cell targeting oral disease-modifying therapy, has been shown to modulate myelin antigen-specific B cell immune response and inhibit development of MOG-specific IgG antibodies (77). And treatment of Cladribine (another T cell targeted drug) in MS is associated with depletion of memory B cells (78).
However, reports of MS patients' failure to respond to anti-CD20 therapies, or even disease exacerbation thereafter, have also been published. Anti-CD20 therapies have limited efficacy in inhibiting disease progression (41). It is possible that these therapies do not effectively target the antibody-producing B cells, or do not significantly reduce the antibody levels in serum and CSF. Another issue concerns the increased risk of infection that is likely to accumulate with continuous B cell depletion with time. In MS, only a fraction of B cells and antibodies are pathogenic, while other subsets of B cells and antibodies exert essential regulatory functions to limit chronic inflammation. For in-depth reviews regarding B cell therapies and B cell biology between subtypes of MS, please see review papers by Gelfand et al. (79), Fraussen et al. (80), and Myhr et al. (81). It will be very important to develop innovative strategies selectively abrogating pathogenic B cells and antibodies. Thus, identifying the specific features of pathogenic antibodies in MS is crucial for the development of successful therapeutic interventions.
Elevated Levels of IgG1 and IgG3 Antibodies in CSF And Serum, An MS Specific Feature
Selective Elevation of IgG1 and IgG3 in MS
The glycoprotein IgG can be separated into four subclasses: IgG1 (60–70% in plasma), IgG2 (20–30%), IgG3 (5–8%), and IgG4 (1–3%) (82). A selective elevation of the IgG1 in MS CSF was observed (83, 84). The elevation of IgG1 and IgG3 indices in MS were found more frequently than the elevation of the general IgG index (21). Patients with a relapse were significantly more frequently seropositive for anti-MOG and anti-MBP IgG3 than those in remission (85). Further, the IgG3 allotype G3m was MS-specific and present in active brain plaques (86). Subsequent studies demonstrated that the susceptibility to MS was associated with an IgG3 restriction fragment length polymorphism (87), and a GWAS study showed that intrathecal IgG synthesis in MS was significantly associated with the intronic region of the IgG3 heavy chain gene SNPs (88). The significance of IgG3 in MS was recently highlighted by a findings that higher serum IgG3 levels may predict the development of MS from CIS (89) and IgG3 + B cells are associated with the development of MS (90). These data suggest that the presence of higher levels of IgG1 and IgG3 antibodies may play a significant role in MS disease activity.
Increased IgG1 and IgG3 Enhances Effector Functions
IgG3 has an extended hinge region with highest flexibility compared to other antibody subclasses. This subclass can probe less exposed antigens. This feature could contribute to the higher potential of IgG3, followed by IgG1, to antibody oligomerization and activation of effector functions, including enhanced antibody-mediated cellular cytotoxicity (ADCC); opsonophagocytosis; complement activation; and neutralization. Additionally, IgG3 has superior affinity to FcγR and the first component of complement cascade, C1q (78). Thus, increased IgG1 and IgG3 in MS serum and CSF may enhance immune-mediated cytotoxicity to CNS cells or may reduce the thresholds for antigen-driven antibody clustering for optimal activation of immune responses.
Conclusions
The role of antibodies in MS disease mechanisms has been disputed over several decades due to the lack of direct and reproducible proof of pathogenic effects. The limited efficacy of CD20-B cell therapies in progressive MS patients and in controlling disease progression implicates that CD20-negative antibody-producing B cells, as well as antibodies, play an important role in disease pathogenesis. The combined accumulating data that there is a strong correlation between serum and CSF IgG, insufficient B cells to produce large quantities of intrathecal IgG, MRI detection of the central vein sign in MS, and presence of pathogenic serum antibodies provide evidence for the hypothesis that circulating antibodies contribute to increased intrathecal IgG synthesis in MS. We further propose that (1) serum antibodies exert primary and pathogenic effects in MS development; (2) increased IgG1 and IgG3 result in enhanced cytotoxicity to CNS cells and produce antibody-mediated injury in MS pathogenesis (Figure 1). This novel hypothesis may help to resolve the current controversy regarding the roles of antibodies in MS and may draw attention to the possibly pathogenic role of IgG3. It may also provide novel opportunities for blood biomarker identification and the development of effective therapeutic interventions for MS.
Author Contributions
XY and YL wrote and edited the manuscript. PK and MG critically reviewed and edited the manuscript. All authors contributed to the manuscript.
Funding
The work was supported by Department of Neurosurgery Research Funds, University of Colorado Anschutz Medical Campus.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology. (2019) 92:e102940. doi: 10.1212/WNL.0000000000007035
2. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. (2014) 83:278–86. doi: 10.1212/WNL.0000000000000560
3. van der Valk P, de Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. (2000) 26:2–10. doi: 10.1046/j.1365-2990.2000.00217.x
4. Liu Y. New ex vivo demyelination/remyelination models to defeat multiple sclerosis and neuromyelitis optica. Neural Regen Res. (2019) 14:1715–6. doi: 10.4103/1673-5374.257525
5. Pryce G, Baker D. Oligoclonal bands in multiple sclerosis; functional significance and therapeutic implications. Does the specificity matter? Mult Scler Relat Disord. (2018) 25:131–7. doi: 10.1016/j.msard.2018.07.030
6. Kabat EA, Moore DH, Landow H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J Clin Invest. (1942) 21:571–7. doi: 10.1172/JCI101335
7. Gilden D, Devlin M, Wroblewska Z. A technique for the elution of cell-surface antibody from human brain tissue. Ann Neurol. (1978) 3:403–5. doi: 10.1002/ana.410030507
8. Mehta PD, Frisch S, Thormar H, Tourtellotte WW, Wisniewski HM. Bound antibody in multiple sclerosis brains. J Neurol Sci. (1981) 49:91–8. doi: 10.1016/0022-510X(81)90191-X
9. Glynn P, Gilbert HM, Newcombe J, Cuzner ML. Analysis of immunoglobulin G in multiple sclerosis brain: quantitative and isoelectric focusing studies. Clin Exp Immunol. (1982) 48:102–10.
10. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
11. Breij EC, Brink BP, Veerhuis R, van den Berg C, Vloet R, Yan R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. (2008) 63:16–25. doi: 10.1002/ana.21311
12. Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, Rees MI, et al. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflammation. (2016) 13:161. doi: 10.1186/s12974-016-0611-x
13. Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol. (2006) 180:17–28. doi: 10.1016/j.jneuroim.2006.07.006
14. Dobson R, Ramagopalan S, Davis A, Giovannoni G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: a meta-analysis of prevalence, prognosis and effect of latitude. J Neurol Neurosurg Psychiatry. (2013) 84:909–14. doi: 10.1136/jnnp-2012-304695
15. Olsson JE, Link H. Immunoglobulin abnormalities in multiple sclerosis. Relation to clinical parameters: exacerbations and remissions. Arch Neurol. (1973) 28:392–9. doi: 10.1001/archneur.1973.00490240052009
16. Walsh MJ, Tourtellotte WW. Temporal invariance and clonal uniformity of brain and cerebrospinal IgG, IgA, and IgM in multiple sclerosis. J Exp Med. (1986) 163:41–53. doi: 10.1084/jem.163.1.41
17. Reiber H. External quality assessment in clinical neurochemistry: survey of analysis for cerebrospinal fluid (CSF) proteins based on CSF/serum quotients. Clin Chem. (1995) 41:256–63. doi: 10.1093/clinchem/41.2.256
18. Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest. (1978) 38:409–21.
19. Pollok K, Mothes R, Ulbricht C, Liebheit A, Gerken JD, Uhlmann S, et al. The chronically inflamed central nervous system provides niches for long-lived plasma cells. Acta Neuropathol Commun. (2017) 5:88. doi: 10.1186/s40478-017-0487-8
20. Yu X, Burgoon M, Green M, Barmina O, Dennison K, Pointon T, et al. Intrathecally synthesized IgG in multiple sclerosis cerebrospinal fluid recognizes identical epitopes over time. J Neuroimmunol. (2011) 240–1:129–36. doi: 10.1016/j.jneuroim.2011.10.009
21. Caroscio JT, Kochwa S, Sacks H, Makuku S, Cohen JA, Yahr MD. Quantitative cerebrospinal fluid IgG measurements as a marker of disease activity in multiple sclerosis. Arch Neurol. (1986) 43:1129–31. doi: 10.1001/archneur.1986.00520110029009
22. Joseph FG, Hirst CL, Pickersgill TP, Ben-Shlomo Y, Robertson NP, Scolding NJ. CSF oligoclonal band status informs prognosis in multiple sclerosis: a case control study of 100 patients. J Neurol Neurosurg Psychiatr. (2009) 80:292–6. doi: 10.1136/jnnp.2008.150896
23. Avasarala JR, Cross AH, Trotter JL. Oligoclonal band number as a marker for prognosis in multiple sclerosis. Arch Neurol. (2001) 58:2044–5. doi: 10.1001/archneur.58.12.2044
24. Calabrese M, Agosta F, Rinaldi F, Mattisi I, Grossi P, Favaretto A, et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch Neurol. (2009) 66:1144–50. doi: 10.1001/archneurol.2009.174
25. Ferreira D, Voevodskaya O, Imrell K, Stawiarz L, Spulber G, Wahlund LO, et al. Multiple sclerosis patients lacking oligoclonal bands in the cerebrospinal fluid have less global and regional brain atrophy. J Neuroimmunol. (2014) 274:149–54. doi: 10.1016/j.jneuroim.2014.06.010
26. Heussinger N, Kontopantelis E, Gburek-Augustat J, Jenke A, Vollrath G, Korinthenberg R, et al. Oligoclonal bands predict multiple sclerosis in children with optic neuritis. Ann Neurol. (2015) 77:1076–82. doi: 10.1002/ana.24409
27. Farina G, Magliozzi R, Pitteri M, Reynolds R, Rossi S, Gajofatto A, et al. Increased cortical lesion load and intrathecal inflammation is associated with oligoclonal bands in multiple sclerosis patients: a combined CSF and MRI study. J Neuroinflammation. (2017) 14:40. doi: 10.1186/s12974-017-0812-y
28. Seraji-Bozorgzad N, Khan O, Cree BAC, Bao F, Caon C, Zak I, et al. Cerebral gray matter atrophy is associated with the CSF IgG index in African American with multiple sclerosis. J Neuroimaging. (2017) 27:476–80. doi: 10.1111/jon.12435
29. Alcazar A, Regidor I, Masjuan J, Salinas M, Alvarez-Cermeno JC. Axonal damage induced by cerebrospinal fluid from patients with relapsing-remitting multiple sclerosis. J Neuroimmunol. (2000) 104:58–67. doi: 10.1016/S0165-5728(99)00225-8
30. Cristofanilli M, Rosenthal H, Cymring B, Gratch D, Pagano B, Xie B, et al. Progressive multiple sclerosis cerebrospinal fluid induces inflammatory demyelination, axonal loss, and astrogliosis in mice. Exp Neurol. (2014) 261:620–32. doi: 10.1016/j.expneurol.2014.07.020
31. Liu Y, Given KS, Harlow DE, Matschulat AM, Macklin WB, Bennett JL, et al. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun. (2017) 5:25. doi: 10.1186/s40478-017-0428-6
32. Liu Y, Given KS, Owens GP, Macklin WB, Bennett JL. Distinct patterns of glia repair and remyelination in antibody-mediated demyelination models of multiple sclerosis and neuromyelitis optica. Glia. (2018) 66:2575–88. doi: 10.1002/glia.23512
33. Sladkova V, Mares J, Hlustik P, Langova J, Kanovsky P. Intrathecal synthesis in particular types of multiple sclerosis. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2014) 158:124–6. doi: 10.5507/bp.2012.054
34. Yu X, Gilden DH, Ritchie AM, Burgoon MP, Keays KM, Owens GP. Specificity of recombinant antibodies generated from multiple sclerosis cerebrospinal fluid probed with a random peptide library. J Neuroimmunol. (2006) 172:121–31. doi: 10.1016/j.jneuroim.2005.11.010
35. Owens GP, Bennett JL, Lassmann H, O'Connor KC, Ritchie AM, Shearer A, et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann Neurol. (2009) 65:639–49. doi: 10.1002/ana.21641
36. Willis SN, Stathopoulos P, Chastre A, Compton SD, Hafler DA, O'Connor KC. Investigating the antigen specificity of multiple sclerosis central nervous system-derived immunoglobulins. Front Immunol. (2015) 6:600. doi: 10.3389/fimmu.2015.00600
37. Brandle SM, Obermeier B, Senel M, Bruder J, Mentele R, Khademi M, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc Natl Acad Sci USA. (2016) 113:7864–9. doi: 10.1073/pnas.1522730113
38. Gastaldi M, Zardini E, Franciotta D. An update on the use of cerebrospinal fluid analysis as a diagnostic tool in multiple sclerosis. Expert Rev Mol Diagn. (2017) 17:31–46. doi: 10.1080/14737159.2017.1262260
39. Bashford-Rogers RJM, Smith KGC, Thomas DC. Antibody repertoire analysis in polygenic autoimmune diseases. Immunology. (2018) 155:3–17. doi: 10.1111/imm.12927
40. Gilden DH. Infectious causes of multiple sclerosis. Lancet Neurol. (2005) 4:195–202. doi: 10.1016/S1474-4422(05)70023-5
41. Lehmann-Horn K, Kinzel S, Weber MS. Deciphering the role of B cells in multiple sclerosis-towards specific targeting of pathogenic function. Int J Mol Sci. (2017) 18. PubMed PMID:28946620. doi: 10.3390/ijms18102048
42. Obermeier B, Mentele R, Malotka J, Kellermann J, Kumpfel T, Wekerle H, et al. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med. (2008) 14:688–93. doi: 10.1038/nm1714
43. Obermeier B, Lovato L, Mentele R, Bruck W, Forne I, Imhof A, et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J Neuroimmunol. (2011) 233:245–8. doi: 10.1016/j.jneuroim.2011.01.010
44. von Budingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest. (2012) 122:4533–43. doi: 10.1172/JCI63842
45. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med. (2014) 6:248ra106. doi: 10.1126/scitranslmed.3008930
46. Stern JN, Yaari G, vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. (2014) 6:248ra107. doi: 10.1126/scitranslmed.3008879
47. Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol. (2014) 75:266–76. doi: 10.1002/ana.24088
48. Beseler C, Vollmer T, Graner M, Yu X. The complex relationship between oligoclonal bands, lymphocytes in the cerebrospinal fluid, and immunoglobulin G antibodies in multiple sclerosis: indication of serum contribution. PLoS ONE. (2017) 12:e0186842. doi: 10.1371/journal.pone.0186842
49. Mehta PD, Miller JA, Tourtellotte WW. Oligoclonal IgG bands in plaques from multiple sclerosis brains. Neurology. (1982) 32:372–6. doi: 10.1212/WNL.32.4.372
50. Lumsden CE. The immunogenesis of the multiple sclerosis plaque. Brain Res. (1971) 28:365–90. doi: 10.1016/0006-8993(71)90052-7
51. Tourtellotte WW, Potvin AR, Fleming JO, Murthy KN, Levy J, Syndulko K, et al. Multiple sclerosis: measurement and validation of central nervous system IgG synthesis rate. Neurology. (1980) 30:240–4. doi: 10.1212/WNL.30.3.240
52. Tourtellotte WW, Tavolato B, Parker JA, Comiso P. Cerebrospinal fluid electroimmunodiffusion. An easy, rapid, sensitive, reliable, and valid method for the simultaneous determination of immunoglobulin-G and albumin. Arch Neurol. (1971) 25:345–50. doi: 10.1001/archneur.1971.00490040071008
53. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. (2005) 128(Pt 7):1667–76. doi: 10.1093/brain/awh486
54. Andersson M, Alvarez-Cermeno J, Bernardi G, Cogato I, Fredman P, Frederiksen J, et al. Cerebrospinal fluid in the diagnosis of multiple sclerosis: a consensus report. J Neurol Neurosurg Psychiatry. (1994) 57:897–902. doi: 10.1136/jnnp.57.8.897
55. Tallantyre EC, Brookes MJ, Dixon JE, Morgan PS, Evangelou N, Morris PG. Demonstrating the perivascular distribution of MS lesions in vivo with 7-Tesla MRI. Neurology. (2008) 70:2076–8. doi: 10.1212/01.wnl.0000313377.49555.2e
56. Rae-Grant AD, Wong C, Bernatowicz R, Fox RJ. Observations on the brain vasculature in multiple sclerosis: a historical perspective. Mult Scler Relat Disord. (2014) 3:156–62. doi: 10.1016/j.msard.2013.08.005
57. Hammond KE, Metcalf M, Carvajal L, Okuda DT, Srinivasan R, Vigneron D, et al. Quantitative in vivo magnetic resonance imaging of multiple sclerosis at 7 Tesla with sensitivity to iron. Ann Neurol. (2008) 64:707–13. doi: 10.1002/ana.21582
58. Sinnecker T, Clarke MA, Meier D, Enzinger C, Calabrese M, de Stefano N, et al. Evaluation of the central vein sign as a diagnostic imaging biomarker in multiple sclerosis. JAMA Neurol. (2019) 76:1446–56. doi: 10.1001/jamaneurol.2019.2478
59. Yates RL, Esiri MM, Palace J, Jacobs B, Perera R, DeLuca GC. Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann Neurol. (2017) 82:259–70. doi: 10.1002/ana.24997
60. Keegan M, Konig F, McClelland R, Bruck W, Morales Y, Bitsch A, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet. (2005) 366:579–82. doi: 10.1016/S0140-6736(05)67102-4
61. Faissner S, Nikolayczik J, Chan A, Hellwig K, Gold R, Yoon MS, et al. Plasmapheresis and immunoadsorption in patients with steroid refractory multiple sclerosis relapses. J Neurol. (2016) 263:1092–8. doi: 10.1007/s00415-016-8105-4
62. Zettl UK, Hartung HP, Pahnke A, Brueck W, Benecke R, Pahnke J. Lesion pathology predicts response to plasma exchange in secondary progressive MS. Neurology. (2006) 67:1515–6. doi: 10.1212/01.wnl.0000240067.03948.68
63. Bornstein MB. A tissue-culture approach to demyelinative disorders. Natl Cancer Inst Monogr. (1963) 11:197–214.
64. Seil FJ. Central and peripheral neurite growth patterns in tissue culture. Neurosci Lett. (1976) 2:115–9. doi: 10.1016/0304-3940(76)90001-X
65. Seil FJ. Tissue culture studies of demyelinating disease: a critical review. Ann Neurol. (1977) 2:345–55. doi: 10.1002/ana.410020417
66. Grundke-Iqbal I, Bornstein MB. Multiple sclerosis: serum gamma globulin and demyelination in organ culture. Neurology. (1980) 30(7 Pt 1):749–54. doi: 10.1212/WNL.30.7.749
67. Elliott C, Lindner M, Arthur A, Brennan K, Jarius S, Hussey J, et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain. (2012) 135(Pt 6):1819–33. doi: 10.1093/brain/aws105
68. Tanaka Y, Tsukada N, Koh CS, Yanagisawa N. Anti-endothelial cell antibodies and circulating immune complexes in the sera of patients with multiple sclerosis. J Neuroimmunol. (1987) 17:49–59. doi: 10.1016/0165-5728(87)90030-0
69. Trojano M, Defazio G, Ricchiuti F, de Salvia R, Livrea P. Serum IgG to brain microvascular endothelial cells in multiple sclerosis. J Neurol Sci. (1996) 143:107–13. doi: 10.1016/S0022-510X(96)00184-0
70. Long Y, Gao C, Qiu W, Hu X, Peng F, Lu Z. Antibodies target microvessels in neuromyelitis optica and multiple sclerosis patients. Neurol Res. (2013) 35:922–9. doi: 10.1179/1743132813Y.0000000229
71. Shimizu F, Tasaki A, Sano Y, Ju M, Nishihara H, Oishi M, et al. Sera from remitting and secondary progressive multiple sclerosis patients disrupt the blood-brain barrier. PLoS ONE. (2014) 9:e92872. doi: 10.1371/journal.pone.0092872
72. Hausser-Kinzel S, Weber MS. The role of B cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front Immunol. (2019) 10:201. doi: 10.3389/fimmu.2019.00201
73. Sabatino JJ Jr, Zamvil SS, Hauser SL. B-cell therapies in multiple sclerosis. Cold Spring Harb Perspect Med. (2019) 9:e032037. doi: 10.1101/cshperspect.a032037
74. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. (2006) 180:63–70. doi: 10.1016/j.jneuroim.2006.06.029
75. Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol. (2016) 3:166–79. doi: 10.1002/acn3.293
76. Studer V, Rossi S, Motta C, Buttari F, Centonze D. Peripheral B cell depletion and central proinflammatory cytokine reduction following repeated intrathecal administration of rituximab in progressive multiple sclerosis. J Neuroimmunol. (2014) 276:229–31. doi: 10.1016/j.jneuroim.2014.08.617
77. Varrin-Doyer M, Pekarek KL, Spencer CM, Bernard CC, Sobel RA, Cree BA, et al. Treatment of spontaneous EAE by laquinimod reduces Tfh, B cell aggregates, and disease progression. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e272. doi: 10.1212/NXI.0000000000000272
78. Ceronie B, Jacobs BM, Baker D, Dubuisson N, Mao Z, Ammoscato F, et al. Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells. J Neurol. (2018) 265:1199–209. doi: 10.1007/s00415-018-8830-y
79. Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20(+) B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics. (2017) 14:835–41. doi: 10.1007/s13311-017-0557-4
80. Fraussen J, de Bock L, Somers V. B cells and antibodies in progressive multiple sclerosis: contribution to neurodegeneration and progression. Autoimmun Rev. (2016) 15:896–9. doi: 10.1016/j.autrev.2016.07.008
81. Myhr KM, Torkildsen O, Lossius A, Bo L, Holmoy T. B cell depletion in the treatment of multiple sclerosis. Expert Opin Biol Ther. (2019) 19:261–71. doi: 10.1080/14712598.2019.1568407
82. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
83. Kaschka WP, Theilkaes L, Eickhoff K, Skvaril F. Disproportionate elevation of the immunoglobulin G1 concentration in cerebrospinal fluids of patients with multiple sclerosis. Infect Immun. (1979) 26:933–41. doi: 10.1128/IAI.26.3.933-941.1979
84. Greve B, Magnusson CG, Melms A, Weissert R. Immunoglobulin isotypes reveal a predominant role of type 1 immunity in multiple sclerosis. J Neuroimmunol. (2001) 121:120–5. doi: 10.1016/S0165-5728(01)00436-2
85. Garcia-Merino A, Persson MA, Ernerudh J, Diaz-Gil JJ, Olsson T. Serum and cerebrospinal fluid antibodies against myelin basic protein and their IgG subclass distribution in multiple sclerosis. J Neurol neurosurg Psychiatry. (1986) 49:1066–70. doi: 10.1136/jnnp.49.9.1066
86. Salier JP, Glynn P, Goust JM, Cuzner ML. Distribution of nominal and latent IgG (Gm) allotypes in plaques of multiple sclerosis brain. Clin Exp Immunol. (1983) 54:634–40.
87. Gaiser CN, Johnson MJ, de Lange G, Rassenti L, Cavalli-Sforza LL, Steinman L. Susceptibility to multiple sclerosis associated with an immunoglobulin gamma 3 restriction fragment length polymorphism. J Clin Invest. (1987) 79:309–13. doi: 10.1172/JCI112801
88. Buck D, Albrecht E, Aslam M, Goris A, Hauenstein N, Jochim A, et al. Genetic variants in the immunoglobulin heavy chain locus are associated with the IgG index in multiple sclerosis. Ann Neurol. (2013) 73:86–94. doi: 10.1002/ana.23749
89. Trend S, Jones AP, Cha L, Byrne SN, Geldenhuys S, Fabis-Pedrini MJ, et al. Higher serum immunoglobulin G3 levels may predict the development of multiple sclerosis in individuals with clinically isolated syndrome. Front Immunol. (2018) 9:1590. doi: 10.3389/fimmu.2018.01590
Keywords: multiple sclerosis, antibody, oligoclonal bands, immunoglobulin G, cytotoxicity, cerebrospinal fluid, serum, B cells
Citation: Yu X, Graner M, Kennedy PGE and Liu Y (2020) The Role of Antibodies in the Pathogenesis of Multiple Sclerosis. Front. Neurol. 11:533388. doi: 10.3389/fneur.2020.533388
Received: 07 February 2020; Accepted: 08 September 2020;
Published: 20 October 2020.
Edited by:
Roberta Magliozzi, University of Verona, ItalyReviewed by:
Yue Wang, AstraZeneca, United StatesMakbule Senel, Ulm University Medical Center, Germany
Copyright © 2020 Yu, Graner, Kennedy and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Yu, eGlhb2xpLnl1QGN1YW5zY2h1dHouZWR1