 Davide Martino
Davide Martino Isaac Johnson2,3
Isaac Johnson2,3 James F. Leckman
James F. Leckman- 1Department of Clinical Neurosciences & Hotchkiss Brain Institute, University of Calgary, Calgary, AB, Canada
- 2Child Study Center, Yale University, New Haven, CT, United States
- 3Department of Psychiatry, University of California, San Francisco, San Francisco, CA, United States
- 4Departments of Psychiatry, Pediatrics and Psychology, Yale University, New Haven, CT, United States
Objective: The goal of this article is to review the past decade's literature and provide a critical commentary on the involvement of immunological mechanisms in normal brain development, as well as its role in the pathophysiology of Tourette syndrome, other Chronic tic disorders (CTD), and related neuropsychiatric disorders including Obsessive-compulsive disorder (OCD) and Attention deficit hyperactivity disorder (ADHD).
Methods: We conducted a literature search using the Medline/PubMed and EMBASE electronic databases to locate relevant articles and abstracts published between 2009 and 2020, using a comprehensive list of search terms related to immune mechanisms and the diseases of interest, including both clinical and animal model studies.
Results: The cellular and molecular processes that constitute our “immune system” are crucial to normal brain development and the formation and maintenance of neural circuits. It is also increasingly evident that innate and adaptive systemic immune pathways, as well as neuroinflammatory mechanisms, play an important role in the pathobiology of at least a subset of individuals with Tourette syndrome and related neuropsychiatric disorders In the conceptual framework of the holobiont theory, emerging evidence points also to the importance of the “microbiota-gut-brain axis” in the pathobiology of these neurodevelopmental disorders.
Conclusions: Neural development is an enormously complex and dynamic process. Immunological pathways are implicated in several early neurodevelopmental processes including the formation and refinement of neural circuits. Hyper-reactivity of systemic immune pathways and neuroinflammation may contribute to the natural fluctuations of the core behavioral features of CTD, OCD, and ADHD. There is still limited knowledge of the efficacy of direct and indirect (i.e., through environmental modifications) immune-modulatory interventions in the treatment of these disorders. Future research also needs to focus on the key molecular pathways through which dysbiosis of different tissue microbiota influence neuroimmune interactions in these disorders, and how microbiota modification could modify their natural history. It is also possible that valid biomarkers will emerge that will guide a more personalized approach to the treatment of these disorders.
Introduction
Tourette syndrome (TS) is one of the most common neurodevelopmental disorders worldwide, with prevalence estimates ranging between 0.3 and 0.9% between 5 and 18 years of age (1, 2). TS is characterized by tics, i.e., patterned and recurrent, non-rhythmic movements and vocalizations that are partially suppressible with volition. The assessment and management of TS is profoundly influenced by the comorbidity (80–90% of patients) with other neurodevelopmental disorders (3), in particular obsessive-compulsive disorder (OCD) and attention deficit hyperactivity disorder (ADHD) (3). ADHD is the most common co-occurring disorder in TS (50–60% of cases), often pre-dating the onset of tics (4). ADHD comorbidity in TS is a major determinant of impairment of psychosocial and cognitive functioning, self-esteem and quality of life. Whereas, OCD coexists in 10–35% of TS patients, obsessive-compulsive symptoms (OCS) that do not reach, for severity and functional impairment, the threshold of “disorder” are present in up to 90% of TS patients (5, 6). Obsessions are recurrent and persistent thoughts that are unwanted, intrusive and distressing, whereas compulsions are elaborate, rigidly patterned voluntary actions or “mental acts” that may be responses to obsessions and/or executed to relieve a state of anxiety. Both the diagnosis of TS and the comorbidity with ADHD and OCD in the context of TS are more prevalent in males. In addition to comorbidity in the same individual, TS, OCD, and ADHD co-aggregate in families, with higher rates of both OCD/OCS and ADHD in first-degree relatives of TS patients (3).
In line with their familial aggregation, the comorbidity of OCD and ADHD in TS patients appears to be founded in part on genetic grounds. However, the genetic relationship amongst these disorders is complex, and potentially linked to specific sub-phenotypes, which may explain differences in the strength of their pairwise genetic correlation (7, 8). A recent study quantified the genetic sharing from genome-wide association studies across different neuropsychiatric conditions, yielding a significant genetic correlation between TS and OCD, but only a trend toward a correlation between TS and ADHD, and even weaker association between ADHD and OCD (9). Part of the reason for this different degree of genetic sharing is that extra-genetic and epigenetic factors may contribute to shared common pathophysiological mechanisms that support their comorbidity. It is believed that these pathogenic mechanisms ultimately generate abnormalities in the trajectory of maturation of sensory-motor and associative loops of the cortico-basal ganglia circuitry (10). Neuroimaging research in these conditions corroborated theories of accelerated and/or delayed maturation within cortico-basal ganglia and cortico-cortical pathways (11, 12). This abnormal connectivity may reflect anomalies of basic neurodevelopmental processes like synaptic formation and plastic refining, neurogenesis, and neuronal migration. Anomalies in these mechanisms may be induced, at least in part, by a dysfunctional neural-immune crosstalk that stems from problems in the maturation of innate and adaptive immunity [recently reviewed by (13)], in particular the colonization and maturation of immune cells that reside in the CNS, e.g., microglia (Figure 1).
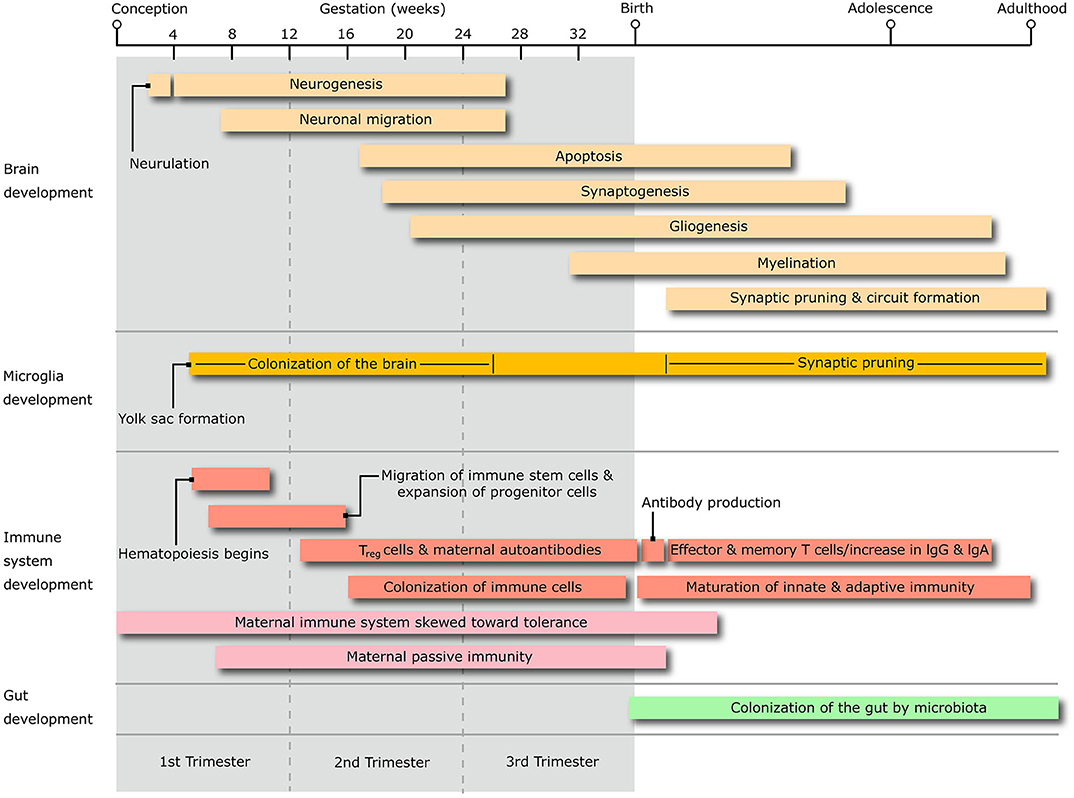
Figure 1. The diagram shows the timing of the salient stages of central nervous system development, alongside that of microglia, immune system and microbiota colonization of the gastrointestinal system, throughout the whole life span from conception to adulthood. Treg, T regulatory lymphocytes; IgG, immunoglobulin G; IgA, Immunoglobulin A.
Autism spectrum disorder (ASD) represents a “model” childhood-onset neurodevelopmental disorder in which genetic and environmental exposures in neural/immune development have been investigated during all stages (prenatal, perinatal, and early postnatal) of brain development. Maternal immune activation (MIA) rodent and non-human primate models of ASD helped elucidating the immune effects on neuronal gene dysregulation and behavioral phenotypes (14, 15). Human studies of ASD have generated hypotheses on the immune-modifying environmental exposures and immunological pathways potentially responsible for the atypical neural-immune crosstalk during different developmental stages (16). Genetic and environmental factors may, independently or interactively, contribute to a direct effect of cytokines and other immune effector molecules on neural cell progenitors, as well as to an abnormal trajectory of maturation of microglia, the multifunctional CNS-resident immune cell type (17).
MIA models have also been important in demonstrating a link between these processes and behavioral and cognitive phenotypes of human ASD (18) and, to a lesser extent, ADHD. Despite their comorbidity rate, though, advances on the immunobiology of TS, OCD, and ADHD have occurred at different pace, probably due to their difference in prevalence, phenotypic heterogeneity, and face and construct validity of representative animal models. Different immunological triggers of maternal-fetal and post-natal immune activation, e.g., infections, autoimmunity, stress, and microbiota constituents, have also been explored with different intensity across these three conditions. Moreover, most human studies exploring the “dysimmune” hypothesis in OCD and TS sought to identify parallel immunopathogenic mechanisms between these chronic, neurodevelopmental disorders and the distinct spectrum of pediatric acute neuropsychiatric syndromes (e.g., PANS), which manifest with OCS and other behavioral and cognitive features, including tics. The PANS spectrum differs from classical, neurodevelopmental OCD and TS in phenomenology, natural history, familial aggregation, prognosis and, probably, susceptibility to immune-based therapies, as extensively reviewed elsewhere (19–22). Therefore, whether mechanistic findings from animal and human studies of acute, putatively immune-mediated, forms of OCS, tics and ADHD-like symptoms can be translated as relevant to the pathophysiology of TS, OCD, and ADHD remains subject for debate.
The main objective of our review is to summarize the key advances in knowledge of risk factors, peripheral and CNS markers of immune dysregulation in TS, OCD, and ADHD (Figure 2). Topics include: their degree of comorbidity with immune-mediated illnesses, the contribution of representative model systems to the conceptualization of neural-immune crosstalk, and the pivotal mechanisms through which dysfunctional neural-immune crosstalk throughout development can contribute to the onset and natural history of these highly overlapping disorders.
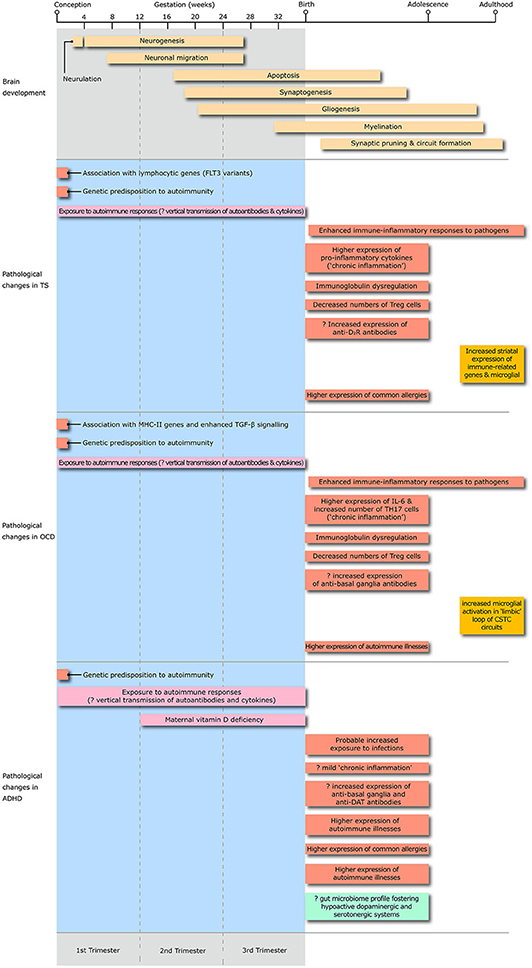
Figure 2. The diagram illustrates the the timing of exposure to risk factors for immune dysregulation, peripheral and central changes in immune-inflammatory responses, changes in microglial activation and alteration in the gut microbiome profile throughout the whole life span from conception to adulthood in Tourette syndrome, obsessive-compulsive disorder (OCD) and attention deficit hyperactivity disorder (ADHD). Treg, T regulatory lymphocytes; D2R, D2 dopamine receptor; IL, interleukin; TH17, T-helper 17 lymphocytes; CSTC, cortico-striato-thalamo-cortical; DAT, dopamine transporter.
Risk Factors for Immune Dysregulation
Genetic Predisposition
The genetic basis of immune dysregulation in immune-mediated diseases is known to be polygenic, supporting a differential expression of gene products functionally interacting within complex mechanistic pathways. Despite important knowledge advances regarding this “immunological” interactome for several autoimmune and neurodevelopmental illnesses (23, 24), this area remains under-explored and poorly understood in TS, OCD, and ADHD. The association of TS with genetic variants predisposing to immune dysregulation was explored by very few studies to date. A single study awaiting replication reported the association between tics and a single nucleotide polymorphism (SNP) of the TNF gene (−308 A/G) coding for the pro-inflammatory cytokine tumor necrosis factor (TNF-α), which controls its transcription and has been linked to atopic dermatitis, asthma, and Graves' disease (25). Summary-level data from genome-wide association studies (GWAS) demonstrated a positive genetic correlation between TS and allergy (26), which may contribute to the comorbidity between TS and allergic illnesses, reviewed below. A recent analysis of genome-wide data from 3,581 TS individuals and 7,682 ancestry-matched controls identified an association of TS with a Lymphocytic gene set, driven by variants in FLT3 (27). This gene is critical for neuro-immune interactions, and its inhibition can alleviate peripheral neuropathic pain, a chronic neuro-immune condition.
A handful of studies produced interesting signals of an association between OCD and genes that are relevant to the development and control of the immune system. In particular, major histocompatibility class (MHC)-II molecules are crucial to the development and homeostasis of neural-immune crosstalk. An exon-focused GWAS identified MHC locus polymorphisms as top signals (28), whereas a case-control study comparing 144 early-onset OCD with general population samples found a significantly higher frequency of alleles comprising HLA-DRB1*04 in OCD (29). A Dutch population-based study (30) reported a significant association between OCD and RFXANK, a gene encoding another MHC-II protein, linked to an immunodeficiency disorder (bare lymphocyte syndrome 2). However, direct gene-gene interactions/pathways or gene-environment interactions relevant to OCD remain unknown. In the attempt to explore protein-protein interaction networks through the identification of de novo SNPs, Cappi et al. (31), highlighted enriched transforming growth factor-β (TGF-β) and glucocorticoid receptor signaling. TGF-β signaling controls immune homeostasis, direction of lymphocyte differentiation and aspects of embryonic development including neuronal migration and synapse formation. Two potentially high relevant genes highlighted by their analyses included WWP1, that codes for a protein inhibiting transcriptional activity induced by TGF-β, and SMAD4, coding for a signal transduction protein activated by TGF-β signaling during CNS proliferation and differentiation.
The evidence for genetic and epigenetic contribution to immune dysregulation in ADHD is also still preliminary. Independent genomic analyses showed an association between ADHD and pathways implicated in gene expression during development and immune-inflammatory regulation, which is shared with major depressive disorder (32, 33). The epidemiological association of ADHD and immune-mediated diseases is supported by correlational data from previous GWASs, that highlighted a modest genetic correlation with psoriasis, rheumatoid arthritis and susceptibility to ear infections and tuberculosis (26). An epigenome-wide association study (34) that meta-analyzed results from three ADHD cohorts revealed six differentially methylated regions in the MHC, including schizophrenia-associated C4A and C4B genes, from blood specimens of two cohorts. Larger, longitudinal studies should confirm these findings and exclude reverse causality (i.e., trait-induced DNA methylation variation). Finally, an exploratory gene expression regulation analysis through the expression of 13 microRNAs (35) found that all the differentially expressed genes in children with ADHD were involved in immune functions, including complement cascade and B-cell receptor signaling.
In summary, there is initial evidence of association with genetic variants modulating systemic immune regulation and immune mechanisms linked to neural development (e.g., TGF-β signaling) in OCD and ADHD. Evidence from GWASs also shows genetic correlation between TS, ADHD and several immune-mediated diseases.
Pre- and Perinatal Exposures
Familial co-aggregation of TS, OCD, and ADHD with autoimmune diseases may indicate genetic predisposition to immune dysregulation, which, for maternal history, might be complicated by enhanced intrauterine immune-inflammatory mechanisms, e.g., vertical autoantibody transmission or dysregulated cytokine environment. A Swedish National Patient Register study (36) reported that mothers, fathers and siblings of individuals with TS were significantly more likely to have any autoimmune disorder, with risk increased by 40, 31, and 17%, respectively. They also found significantly increased risk of autoimmune diseases in first-degree relatives of OCD probands, with risk increased by 17% in mothers, 8% in fathers, and 16% in siblings. Recent anecdotal evidence suggests a link between maternal history of thyroid autoimmunity and an atypical, acute presentation of chronic neurodevelopmental disorders, including ASD, TS, and OCD (37). These associations need to be confirmed in larger clinical samples.
ADHD is also associated with increased frequency of maternal history of autoimmune illnesses and infections. A population study using Danish National Registers (38) identified a 12% increase of maternal, but not paternal, history of type 1 diabetes, autoimmune hepatitis, psoriasis, ankylosing spondylitis, and thyrotoxicosis. A stronger association was detected using longitudinal Norwegian registers (39), with increased risks ranging between 20% for maternal hypothyroidism and 80% for maternal multiple sclerosis, but not for non-immune conditions like hypertension. The association with hypothyroidism, however, might depend on maternal hypothyroxinemia rather than thyroid peroxidase antibodies (40).
Studies exploring the risk of ADHD in the offspring of women with gestational infections yielded noteworthy methodological heterogeneity and inconsistent results. Following reports (41, 42) of an association with viral rash or respiratory infections, Mann and McDermott (43) and Silva et al. (44) reported a 29–37% increase in risk of ADHD in the offspring of women with gestational bacterial or fungal genitourinary infection using record-linkage datasets. Evidence from the Danish National Birth Cohort reported an increase in risk associated with genitourinary infection (33–60%) from the third to the eighth month of pregnancy (45). However, sibling-comparison analyses conducted using Swedish National Registers (46) suggested that this association could spuriously result from unmeasured familial confounding. Similar findings came from a Canadian population-based retrospective cohort study that used sibling-comparison analyses to demonstrate lack of association between prenatal antibiotic exposure and ADHD risk ADHD in children (47).
An association of maternal vitamin D deficiency—predisposing to immune dysregulation- and ADHD in offspring was documented by population birth cohorts. A study pooling five Spanish birth cohorts (48) showed that ADHD severity in the offspring at ages 4–5 decreased by 11% for the inattention scale and by 12% for the hyperactivity-impulsivity scale per 10 ng/ml increment of 25(OH)D3 plasma concentration at 13 weeks of gestation. A subsequent population study using Finnish registers (49) detected a 53% increase of ADHD risk in the offspring of women at the highest quintile of maternal 25(OH)D levels compared to those at the lowest quintile. Maternal C-reactive protein serum levels during early gestation were, instead, not associated with risk of ADHD in the offspring (50).
Cumulatively, familial co-aggregation of autoimmunity may influence genetic and prenatal risk of TS, OCD, and ADHD in the offspring. The higher risk associated with maternal autoimmunity and vitamin D deficiency in pregnancy suggests an additive or multiplicative effect of autoimmunity through interaction of genetic and vertical risk transmission. However, the questionable association between gestational genitourinary infections and ADHD in offspring highlights the risk of confounding from unobserved factors shared within families in population studies.
Postnatal Exposures
The influence of immunogenic triggers, primarily infections and stress, on the development and clinical course of TS, OCD, and ADHD is incompletely understood. A body of evidence supports the enhancement of immune-inflammatory responses toward common pathogens in TS and OCD. The occurrence of tics in acute neuropsychiatric syndromes associated with Group A streptococcus (GAS) pharyngotonsillitis offered a rationale to cross-sectional clinical studies that revealed stronger anti-streptococcal antibody responses in these patients across different ages (51–54). Retrospective population cohort data from US and Denmark support a 35–59% increase in risk for a diagnosis of tic disorders and GAS exposure in the past year (55–57), although one similar study from the UK did not confirm this association (58). A similar rise in antibody responses was observed for obligate intracellular bacteria like Chlamydia trachomatis and Mycoplasma pneumoniae (59). Population data from Taiwan showed a 24% increase in risk of tic incidence in individuals with non-CNS enterovirus infection (60). Danish health registers data indicated that individuals with a non-streptococcal throat infection have a 25% increased risk of tic disorders, whereas those requiring hospitalization for infection and anti-infective treatment manifest a higher than 300% increase (57, 61). This evidence demonstrates greater risk of different common infections in individuals who will go on to develop TS, even if it cannot be considered proof of causative relationship. On the other hand, there is no evidence from clinic-based data supporting a temporal association between onset or clinical worsening of tics and a recent exposure to GAS in the context of TS (62–64). A recent prospective cohort study of 715 children with TS from different European countries did not detect any association between recent GAS exposure and clinically relevant exacerbations of tics (65).
Healthcare population registries provide some support to an association between OCD and prior infections, particularly with GAS pharyngeal infections. A GAS throat infection in the year prior to symptom onset was associated with a 76% increase in risk of developing OCD in a US health insurance claims database (56), but this finding was not confirmed by UK record-linkage primary care data (58). A more recent study of multiple Danish national healthcare registries (57) reported that a previous record of one or more positive rapid antigen diagnostic tests for GAS was associated with a 51% significantly higher risk of OCD diagnosis, independent on age at first positive test, compared to individuals without a streptococcal test. Importantly, individuals with a non-streptococcal throat infection also had a 28% significantly higher OCD risk. As with TS, smaller prospective clinic-based studies failed to show an association between exacerbation of OCS severity and recent GAS throat infections (62, 63, 66). None of the prospective clinic-based studies that explored the association between GAS exposure and changes in tic or OCS severity analyzed with accuracy the specific risk of different modalities of exposure (“colonization” vs. “infection”). This notwithstanding, the negative results of the largest of these studies focusing on tic severity (65) suggest that any association between GAS exposure and at least tic exacerbations is unlikely.
As for TS, an association of OCD with other pathogens cannot be excluded. Apart from case reports of concurrent Mycoplasma pneumoniae or varicella zoster virus infections (67, 68) in OCD, an association with a 2.5 to 4-fold increased seroprevalence of antibodies to T. gondii (69) has been reported (70, 71). At best, however, this anecdotal evidence suggests a proneness to comorbidity with a broad range of infections, similar to what observed for TS. There is also limited knowledge on whether the characteristics of tics and OCS may differ depending on prior/current exposure to infections. A recent report showed that young OCD patients with higher frequency of self-reported ear or throat infections have increased severity of cleaning/contamination-related symptoms (72), although an altered perception of their medical history and subsequent over-reporting might also explain this finding.
Psychosocial stressors predict short-term future severity of tics, OCS and depressive symptoms in TS, and have an established mechanistic link with immune-inflammatory responses. A clinic-based prospective study observed a multiplicative interaction between psychosocial stress and GAS infections as predictors of future tic severity (73). Immune-inflammatory responses to infections and activation of the hypothalamus-pituitary-adrenal (HPA) axis during stress responses might influence each other, contributing to tic exacerbation, but the neurobiological basis of this process needs to be elucidated (74).
Very pre-term infants exhibit an increased risk of ADHD symptoms during childhood following exposure to neonatal infections (75) and systemic inflammation, the latter expressed by high concentrations of neurotrophic proteins (76). Service- and population-based studies suggested an increase in future ADHD symptomatology in children with prior history of bacterial meningitis (77) and Enterovirus encephalitis (78), although the possibility of neuropsychological sequelae following direct neural tissue insult is high. ADHD behavioral patterns might contribute to increased chances of contact with pathogens (79), especially those with intrafamilial spread like polyomaviruses (80). Scandinavian national register studies provide the strongest evidence for increased risk of ADHD outcome in children exposed to infections requiring hospitalization [by 109% in (61)] and/or anti-infective treatment [by 56% in (61), and 10–60% for antibiotic exposure throughout the first 2 years of life in (81)]. Overall, there is supportive evidence of an association between prior infectious exposures in early childhood and ADHD diagnosis or symptoms, although these epidemiological studies do not yet constitute a clear proof of causality.
Immune-Inflammatory Markers (see Table 1)
TS
Even with some discrepancies, peripheral immune responses in TS are skewed toward pro-inflammatory mechanisms in most cross-sectional observational studies. Longitudinal observation showed covariation of TNF-α and interleukin (IL)-12 plasma levels and tic/OCS severity (82), regardless of medical treatment or concurrent infections. A subsequent study reported decreased TNF-α and soluble IL-1 receptor antagonist levels (83). A positive correlation between tic severity and IL-2 levels was reported in one study (84), whereas another reported increased IL-2 and IL-12 circulating levels only in TS patients with comorbid OCD (85). Further reports documented higher circulating levels of IL-6, IL-1β, IL-17 (86), and neopterin, a pteridine synthesized by the monocyte-macrophage cell lineage and linked to cell-mediated TH1 pro-inflammatory responses (62, 87).
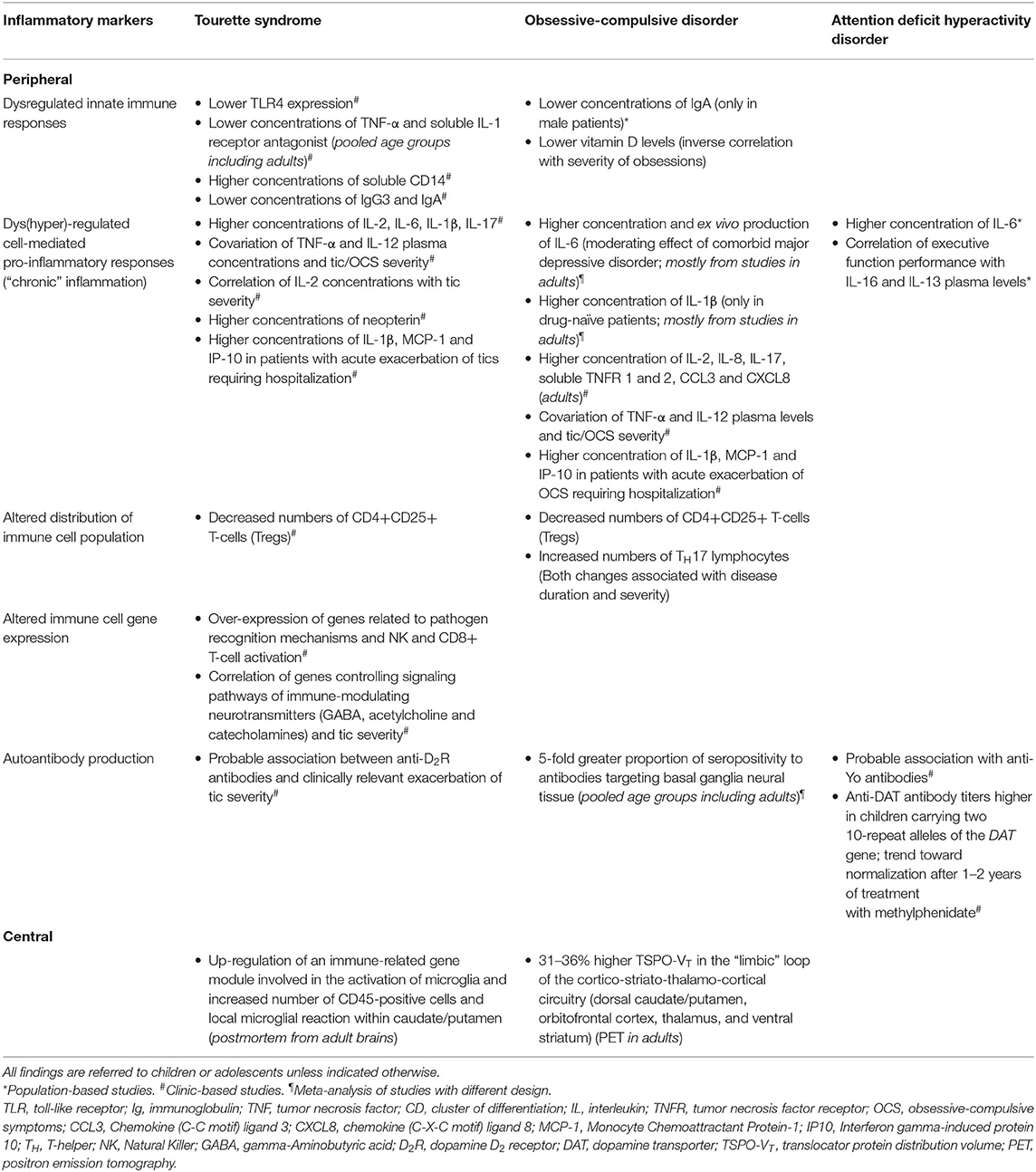
Table 1. Summary of findings on peripheral and central inflammatory markers in Tourette syndrome, obsessive-compulsive disorder and attention deficit hyperactivity disorder.
An increased susceptibility to infections and, perhaps, autoimmune processes might be supported by defective immune mechanisms (innate and adaptive) that protect against infections and autoimmunity under physiological conditions. Weidinger et al. (88) documented an impairment of innate responses in a cross-sectional study that compared 33 TS patients to 31 healthy subjects, showing lower receptor expression of toll-like receptor 4 (TLR4) after stimulation with lipopolysaccharide (LPS), and higher levels of soluble Cluster Differentiation (CD)14. These results could indicate inhibition of monocyte responses to LPS, suggesting defective innate responses to pathogens that would activate a vicious cycle of increased susceptibility to infections and sustain chronic inflammation. Immunoglobulin (Ig) synthesis may be dysregulated (reduced IgG3 plasma level) in TS patients (84). This Ig subclass promotes the classical complement cascade and pathogen elimination; hence, its downregulation might be related to higher infection exposure and overactive systemic inflammation. Finally, overactivation of immune responses in TS may be facilitated by lower numbers of CD4+CD25+ T-cells (Treg) in the periphery (89). Treg cells develop during physiological T-cell maturation in the thymus, survive in the periphery for constant monitoring of self-antigens, and prevent autoimmune responses.
Transcriptomic studies on peripheral blood mononuclear cells revealed different gene expression patterns throughout different developmental periods, with TS patients over-expressing genes related to pathogen recognition mechanisms before age 9, and over-expressing genes involved in Natural Killer (NK) and CD8+ T-cell activation during puberty. Furthermore, tic severity correlated with the expression of genes that control signaling pathways of immune-modulating neurotransmitters involved in tic generation in the CNS, like GABA, acetylcholine, and catecholamines (90–92). Increased expression of the DRD5 gene in lymphocytes might contribute to decreased Treg numbers (93). However, these gene expression studies were performed on small clinical samples and provided correlational, but not causative, evidence.
Dysregulated systemic immune responses can promote the development of autoimmunity. However, the nature and severity of autoimmune processes in TS, and their role in the pathogenesis of tic disorders, remains unclear. Earlier investigations of autoantibodies in TS have used antigen-denaturing methods (94, 95) or immunoprecipitation techniques (96) that may detect non-specific antibody reactivities, or indirect immunofluorescence that does not allow the characterization of the target antigens. Independent reports using denaturing techniques could not demonstrate a significant titer rise of antibodies proposed as relevant in putative post-streptococcal disorders like Sydenham's chorea (97, 98).
Qualitative and quantitative measurement of IgG cell surface binding to live cells expressing candidate self-antigens is much more accurate to detect antigen-antibody reactivities occurring in vivo. Earlier studies applying immunofluorescence-based live-cell assays to explore circulating antibodies in TS patients demonstrated antibodies binding to the surface of rat striatal neurons or neuroblastoma cell lines (99, 100). This finding, however, was not replicated by a subsequent study that applied fluorescence-activated cell sorting (FACS) to live differentiated cells from the SH-SY5Y cell line (101). Dale et al. (102) demonstrated the presence of anti-D2 dopamine receptor (D2R) antibodies in the serum of 4 of 44 TS patients, using FACS applied on human embryonic kidney (HEK) cells transfected with a commercial form of the D2R; none of these 44 patients were positive for anti-D1 dopamine receptor antibodies. Very recently, Addabbo et al. (103) tested anti-D2R antibody reactivity in sera from 137 children with TS at a baseline time point and at time points coinciding with and following a clinically relevant tic exacerbation. These authors used a similar protocol to the one used by Dale et al. (102), but adopting a transfection method on Chinese Hamster Ovary (CHO)-K1 cells (102). Anti-D2R antibodies were detected in 6.6% of patients at baseline; an additional 9.8% of patients seroconverted during the peri-exacerbation time period, independently from new concurrent GAS infections. Although this does not demonstrate a causal link between anti-D2R antibodies and tics, it supports the implication of autoimmunity and concurrent behavioral-immunological fluctuations in TS. An earlier cross-sectional report on 51 patients with TS did not detect any specific serum autoantibodies associated with encephalopathies (targeting LGI1, CASPR2, NMDAR, AMPA1/2, GABAB1/B2) (104). More recently, Baglioni et al. (105) investigated the same patients tested by Addabbo et al. (103) plus a cohort of unaffected siblings of these patients, screening for autoantibodies associated with established encephalopathies (antibodies targeting NMDAR, CASPR2, LGI1, AMPAR, and GABAAR). Using live cell-based assays applied to live rat hippocampal neurons, only two individuals (one patient, one sibling) tested weakly positive for anti-NMDAR antibodies, indicating unlikely association with these pathogenic antibodies. Another report from the Hannover group applied indirect immunofluorescence to HEK cells to confirm the absence of these autoantibodies in the cerebrospinal fluid (CSF) of 20 adults with TS (106). Due to the relative paucity of postmortem data, the presence of autoantibodies in the brain of TS patients has not been systematically explored. Moreover, unlike for autism, the presence of circulating serum anti-fetal brain antibodies has not been investigated in mothers of children with TS.
There is initial evidence of enduring inflammatory changes in the neural tissue of patients with TS. Neuroinflammatory patterns may reveal direct and indirect links between immune-mediated processes and altered neuronal/synaptic maturation and functioning. Earlier studies (107, 108) that adopted a microarray transcriptomics approach on very small samples of adult TS postmortem brains documented a significant putaminal increase in the expression of genes coding for IL-2, IL-2 receptor β, and monocyte chemotactic factor-1. These results seemed to suggest a transcriptomics pattern in keeping with signaling pathways promoting T-cell and microglial proliferation and activation, increased blood-brain barrier permeability, and neural stem cell differentiation.
A subsequent RNA sequencing transcriptomics study of post mortem tissue from the caudate/putamen of nine TS subjects and nine age- and sex-matched control individuals [5 males and 4 females per group; (109)] provided more robust evidence of microglial involvement in this condition. These authors detected 309 down-regulated and 822 up-regulated genes, and 17 gene co-expression modules, identified with data-driven gene network analysis, in the striatum of TS brains. The top-scoring up-regulated module included immune-related genes involved in the activation of microglia. Consistent with this, they observed an increase in CD45-positive cells and local microglial reaction within caudate/putamen. This up-regulated “immune-related” module did not overlap with “neuronal” and “astrocyte” modules comprising striatal interneuron-related transcripts and transcripts related to cell adhesion and astrocyte-related metabolic pathways, respectively. The limited sample size did not allow the exploration of sex-dependent effects on the association between “immune-related” gene expression and TS. Even considering the long interval between onset and specimen collection, this finding suggests that immune and neuronal events co-occur, in part independently, in the brain of TS patients. It also provides preliminary, but compelling, evidence that the expression of molecular pathways associated with microglial maturation and functioning is involved in TS, adding this disorder to the spectrum of neurodevelopmental disorders linked to microglial dysfunction.
OCD
The vast majority of studies investigating peripheral immunity in OCD focused on cross-sectional adult clinical samples, with large heterogeneity across studies. Therefore, an association between specific immune effector molecules and OCD remains unconfirmed. Meta-analytic estimates are available for the serum levels of six different cytokines that were explored in 31 studies (110). Similar to TS, the existing studies provide modest support to the overactivity of pro-inflammatory mechanisms. However, the heterogeneity of study populations and the intrinsic complexity of the OCD spectrum across different age groups indicate that immune dysregulation can be detected only after adjustment for potential confounding variables, a consideration that could be applied also to studies of TS patients. For example, co-morbid major depression moderates the association between OCD and raised IL-6 serum levels. Increased ex vivo production of IL-6 by LPS-stimulated macrophages could be detected including exclusively studies that enrolled adult patients without other behavioral comorbidities and gender- and age-matched control participants. The exposure to medications like SSRIs could also confound the association between OCD and cytokine levels: for instance, a significant increase in IL-1β serum levels was observed only including in the meta-analysis studies that enrolled drug-naïve patients. Meta-analyses and more recent observational studies (111) related to serum levels of other cytokines (TNFα, IL-4, IL-10, interferon [IFN]-γ) failed to demonstrate an association with OCD. The same study observed correlations with specific executive function performances in the OCD patient group. Finally, other cytokines were explored only in isolated or smaller studies, and therefore require replication. These studies reported raised serum levels of IL-2, IL-8, IL-17, soluble TNF receptors 1 and 2, and chemokines like CCL3 and CXCL8. Like for TS, these findings are overall consistent with a possible state of low-grade inflammation and propensity to exacerbate after exposure to appropriate triggers, e.g., infections. Acute exacerbation of OCD and tics requiring hospitalization was also associated with raised peripheral levels of IL-1β, MCP-1, and IP-10 (112).
A predisposition to autoimmune and post-infectious organ-specific immune responses could be related to IgA deficiency and altered distribution of different immune cell types. A retrospective study using electronic health records of 206 children and 1,024 adults with OCD showed that pediatric OCD patients have a 93–98% greater chance of manifesting serum IgA deficiency than children with ASD and anxiety disorders, but not higher than children with TS, and comparable to that of children diagnosed with celiac disease. Interestingly, this difference was significant only in males (113). Another controlled clinic-based study on 99 youth with OCD and 46 healthy volunteers showed higher frequencies of T-helper (Th)17 cells and lower percentages of Tregs in OCD (114). These changes became more marked as disease duration and severity increased and were independent of the comorbidity profile or exposure to psychoactive substances, suggesting the possibility of progressive immune dysregulation in OCD. Longitudinal studies should confirm whether immune dysregulation has indeed a dynamic pattern in this condition. Interestingly, autoreactive Th17 cells triggered grooming behaviors in a mouse model of experimental autoimmune encephalomyelitis (115). An isolated report identified lower vitamin D levels and inverse correlation between 25OH-D3 and severity of obsessive thoughts (116).
The characterization of Pediatric Autoimmune Neuropsychiatric Associated with Streptococcal Infection PANDAS; (117) has fostered the exploration of immunopathogenic mechanisms involving autoimmune processes and the role of autoantibodies (100, 118, 119) in the whole spectrum of OCD. These antibodies target D1 and D2 dopamine receptors, tubulin (120), and lysoganglioside (121), and were found to activate calcium calmodulin-dependent protein kinase II (CaMKII) in a SKNSH human neuronal cell line (121). These reactivities were proposed as markers of acute post-streptococcal OCS, targeting dopaminergic neurotransmission within the cortico-basal ganglia circuitry (122, 123). However, the reliability of this autoimmune panel (aka Cunningham panel) is debated, and the relevance of these autoantibodies to chronic OCD remains to be demonstrated. Initial data indicate that antibodies targeting striatal cholinergic interneurons may also play a role in PANDAS pathogenesis (19, 124).
Antineuronal antibodies were explored also in youth and adults with “garden variety” OCD. Earlier studies applying denaturing protocols of enzyme-linked immunosorbent assay and Western immunoblotting reported discrepant results on antibody reactivities to basal ganglia or other human brain tissue specimens from serum or CSF samples of patients with typical OCD (125–128). Other reports showed greater anti-brain antibody reactivity only in OCS in the context of PANDAS, but not in other children with OCD, compared to healthy subjects (129, 130). In an attempt to resolve this discrepancy, a systematic review, and meta-analysis of seven case-control studies cumulatively exploring 844 participants with primary OCD reported a significantly 5-fold greater proportion of seropositivity to antibodies targeting basal ganglia tissue compared to different control groups, and independent of demographics, disease characteristics, immunostaining method, study quality, publication type or publication bias, and sample size (131). The results of this meta-analysis still need to be taken with caution, given that there was a substantial variability of the accuracy of the different immunostaining assays used across the selected studies. Importantly, it remains undemonstrated whether the antigenic targets of these anti-basal ganglia antibodies are expressed on the cell surface and could therefore be central to a pathological mechanism affecting neuronal cells.
Similar to TS, the direct evidence of brain inflammatory changes in OCD is extremely limited. The link between a peripheral low inflammation state and neuroinflammation remains undemonstrated, as two independent studies that measured cytokine levels in the CSF of OCD patients reported discrepant results. To the best of our knowledge, evidence of activation of inflammatory pathways, either direct (based on immunohistochemistry or cytopathology) or indirect (based on transcriptomics) is lacking from postmortem brain tissue of OCD patients. The only evidence in this respect came from a positron emission tomography (PET) study that used a second-generation radiotracer binding the translocator protein TSPO (132). The density of this protein on microglia increases following activation and is expressed by its distribution volume (VT). Twenty OCD patients in their third decade of age (11 women) and 20 age-matched healthy volunteers were compared in TSPO-VT using the N-(2-(2-fluoroethoxy) benzyl)-N-(4-phenoxypyridin-3-yl) acetamide ([18F]FEPPA) PET tracer. The SNP rs6971 of the TSPO gene was analyzed as a nuisance factor because it may influence the binding to this radiotracer (high-affinity homozygotes vs. mixed-affinity binding heterozygotes). TSPO-VT was significantly 31–36% higher in OCD patients throughout the whole “limbic” loop of the cortico-striato-thalamo-cortical circuitry, i.e., in the dorsal caudate/putamen, orbitofrontal cortex, thalamus, and ventral striatum. Milder increases were reported in the anterior cingulate, medial prefrontal, ventrolateral prefrontal, insular, temporal cortical regions, and hippocampus. Interestingly, TSPO-VT in the orbitofrontal cortex significantly positively correlated with greater distress associated with preventing compulsive behaviors. These findings provide the first direct evidence of neuroinflammatory changes in the brain circuits associated with OCD and indicate that activated microglia (and related potentially harmful M1 responses) may be present in adults with this condition, even many years following onset. This study obviously does not indicate a causative link, but rather a strong evidence of correlation, between elevated TSPO and OCD, which needs to be taken with caution also due to the other potential cellular pathomechanisms associated with TSPO elevation (e.g., cholesterol translocation from outer to inner mitochondrial membranes).
ADHD
Despite the observed association with immune-mediated illnesses, there has been limited research on systemic immune regulatory mechanisms in ADHD. Most reports showed lack of significant differences of circulating levels of pro- and anti-inflammatory cytokines, as well as of other chronic immune activation markers, e.g., markers of the tryptophan-kynurenine pathway, of neurotrophic markers of glial integrity, e.g., S100-B protein, and of immunomodulating molecules like vitamin D3 in both pediatric (133–137) and adult (138, 139) populations. The only cytokine reported as significantly increased in ADHD in two independent studies has been IL-6 (140, 141). At the same time, the observation that raised circulating concentrations of pro-inflammatory cytokines, including IL-6, in the first 2 weeks of life of preterm infants are associated with attention difficulties at 2 years of age (142), and the correlation of executive function performance with IL-16 and IL-13 plasma levels in ADHD children (134), suggests an influence of mild chronic inflammation on the development of prefronto-subcortical connections related to attention and executive functions (143).
Autoantibody markers in ADHD have been investigated by a handful of studies during the past decade. Two studies reported higher immunoreactivity for anti-Purkinje cell antibodies in ADHD (144, 145). The association of ADHD with antibodies against basal ganglia homogenate (146) and dopamine transporter [DAT; (147)] was reported only by isolated studies. Basal anti-DAT antibody titers were higher in ADHD children carrying two 10-repeat alleles of the DAT gene, and tended to normalization after 1–2 years of treatment with methylphenidate. The presence of high serum anti-DAT antibodies supports hypermethylation at CpG1 position on the DAT gene as an epigenetic marker of ADHD severity (148).
Overall, the evidence of enhanced immune-inflammatory mechanisms in ADHD provides modest support in favor of overactive systemic immune mechanisms only in younger patients. Finally, an important knowledge gap is characterized by the complete lack of studies directly evaluating CNS inflammatory changes in ADHD.
Comorbidity With Immune-Mediated Illnesses (Table 2)
TS
Whereas, secondary tics may occur in autoimmune conditions like Sydenham's (or rheumatic) chorea, a systematic review by Perez-Vigil et al. (149) highlighted the dearth of investigations on autoimmune comorbidities in TS. A population-based investigation using the Swedish National Patient Register that assessed the association between TS and 40 autoimmune diseases (36) observed increased risk for comorbidity with Hashimoto's thyroiditis, celiac disease, scarlet fever, type 1 diabetes mellitus, and psoriasis (Table 2).
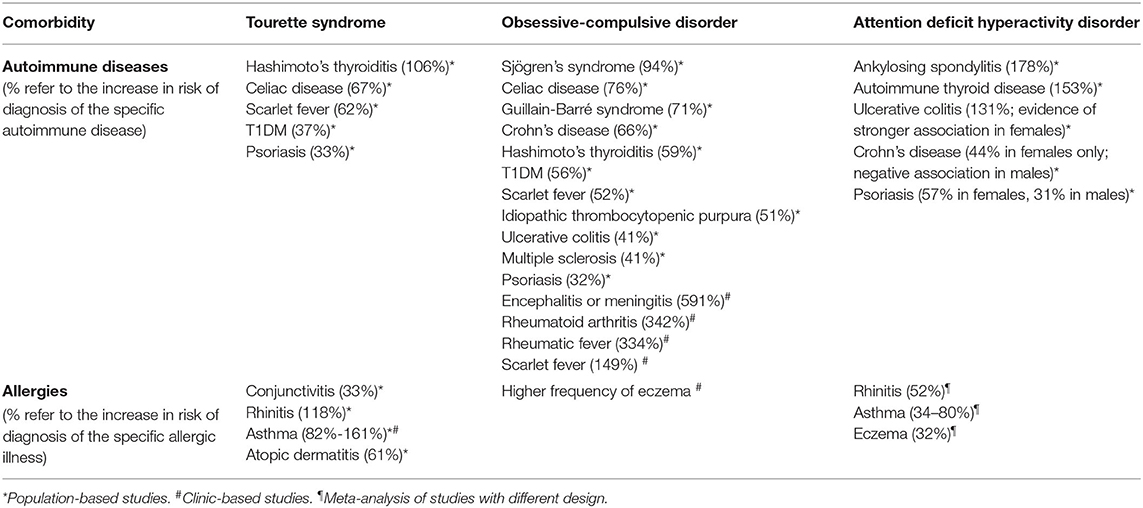
Table 2. Summary of findings on immunological comorbidity in Tourette syndrome, obsessive-compulsive disorder and attention deficit hyperactivity disorder.
On the other hand, an association between TS and allergies is supported by a more conspicuous body of evidence. Retrospective, case-control population studies of Taiwanese national health insurance datasets (150, 151) reported higher risk of conjunctivitis, rhinitis, asthma, and atopic dermatitis in individuals diagnosed with TS. A higher age- and sex-adjusted comorbidity rate for asthma in TS was also documented through the Canadian Community Health Survey (152). Administering a structured questionnaire to a clinic-based sample of 32 TS patients, Yuce et al. (153) found that more than half of these children presented with allergies, rhinitis being the most common. Evidence of a genetic basis for this comorbidity is increasing, as reported in the Genetic predisposition section of this review. Moreover, a predisposition to dysfunctional histamine receptor-mediated signaling in TS suggests a link between type I IgE-mediated hypersensitivity mechanisms and tic generation (154). At the same time, exposure to stressors may facilitate both a surge of tic severity and a flare-up of allergies. New studies should appraise to what degree the association between TS and allergies is supported by concurrent anxiety or depressive disorders, also known to be associated with allergies. Finally, a negative effect on tic severity of anti-allergic pharmacological treatments, steroids in particular, has not been ruled out definitively (155).
OCD
A potential association between OCD and autoimmune diseases was summarized in the systematic review by Pérez-Vigil et al. (149), which included 74 studies on patients with OCD across all age groups. OCS were confirmed to be common manifestations of Sydenham's chorea. A possible association between a diagnosis of OCD/OCS and a diagnosis of multiple sclerosis or systemic lupus erythematosus was observed, but firm conclusions were precluded by methodological limitations of the included studies, e.g., sample size limitations, involvement of unblinded assessors, use of sub-optimal self-report measures, and lack of control groups. In a Swedish population-based cohort study (36), individuals with OCD were 43% more likely to have any comorbid autoimmune disease compared to individuals without OCD. The increase in comorbidity rate was strongest for Sjogren's syndrome, followed by celiac disease, Guillain-Barré syndrome, Crohn's disease, Hashimoto's thyroiditis, type 1 diabetes mellitus, scarlet fever, idiopathic thrombocytopenic purpura, ulcerative colitis, multiple sclerosis, and psoriasis (Table 2). This study could not confirm the association with Sydenham's chorea due to power limitations. A potential limitation of this work is the likely under-representation of OCD patients who do not seek medical help. In relation to this, Westwell-Roper et al. (72) published the largest to date clinic-based survey that estimated the lifetime prevalence of immune-related diseases using self-report medical questionnaires from 1,401 youth with OCD and 1,045 of their first-degree relatives enrolled in the OCD Collaborative Genetics Association Study. This descriptive study was limited by the lack of a control population, reliance on retrospective self-report and post-hoc evaluation of questionnaires that did not screen for all autoimmune comorbidities. Nevertheless, it reported a higher than expected prevalence of scarlet fever, rheumatic fever, rheumatoid arthritis, encephalitis or meningitis in both probands and relatives, independent of OCD status, but not of other immunological disorders. The latter finding is probably due to power limitations and the well-known rarity of autoimmune conditions in children. The reasons of the observed association between OCD and post-streptococcal illnesses should be searched also in shared genetic and/or environmental factors, as well as in dysregulation of mucosal immunity involving the oropharynx.
There is very limited evidence of a relationship between OCD and allergies. A small clinic-based study reported greater frequency of positive skin prick tests in 26 OCD patients compared to control subjects, but not greater eosinophil counts of IgE levels (153). In this study, eczema was significantly over-represented in OCD.
ADHD
The past decade has consolidated the notion that ADHD is characterized by comorbidity with allergic and autoimmune illnesses. Cross-sectional and longitudinal studies based on clinical services and population health registries have been systematically reviewed and meta-analyzed by two independent groups. In their systematic review, Schans et al. (156) observed that most of the selected studies reported a statistically significant positive association between atopic diseases and ADHD in pediatric populations, suggesting that atopic subjects have a 30–50% greater likelihood of receiving a diagnosis of ADHD. Following meta-analyses of nine selected studies, these authors reported an overall weighted odds ratio of 1.34 (95% CI 1.24–1.44) for asthma, of 1.32 (95% CI 1.20–1.45) for atopic eczema, and of 1.52 (95% CI 1.43–1.63) for allergic rhinitis. Interestingly, they noticed that study heterogeneity was low for the association with eczema and asthma, but substantial for rhinitis. In their systematic review on more than 61,000 children (about 8,000 of whom were ADHD patients), Miyazaki et al. (157) selected a lower number of studies (n = 5), despite a similar methodological design to the review by Schans et al. apart from an apparently more restrictive study selection criteria for allergy and ADHD ascertainment. Overall, the quality of the evidence was rated as low, yet confirming an 80% increase in risk for ADHD in children with asthma. They also highlighted a greater study heterogeneity for the association between ADHD and rhinitis, dermatitis and conjunctivitis. Finally, they reported lack of association between food allergy and ADHD. In line with the evidence summarized above, one study has interestingly reported that greater numbers of atopic comorbidities are significantly related to a greater risk of developing ADHD (158). Individuals with a dual diagnosis of ADHD and chronic tics exhibited a stronger association with the coexistence of multiple allergic illnesses than patients with ADHD without tics (159). Following the two metaanalyses summarized above, Chang et al. (160) used the Taiwan National Health Insurance Research Database to report a higher risk of developing asthma, atopic dermatitis, allergic rhinitis, and allergic conjunctivitis among 20,170 unaffected siblings of ADHD patients compared to >80,000 matched control individuals, with an overall risk increase ranging between 10 and 19%. The relationship between ADHD and allergies is likely to be complex and multifactorial. ADHD has been related to general hypersensitivity to environmental stimuli (161), whereas other authors support a mediational role of inflammatory cytokines and stress to explain this association (143). Larger prospective cohort studies will shed more light on the complex link between allergies and ADHD.
Population-based cohort studies across all age groups, leveraging on the richness of national health registries in Taiwan, Denmark and Norway, have recently provided support for increased prevalence of autoimmune diseases in ADHD. Comparing 8,201 individuals with ADHD to > 36,000 age- and gender-matched control individuals from the Taiwan National Health Insurance Research Database (162), ADHD patients exhibited greater prevalence of ankylosing spondylitis (OR = 2.78), ulcerative colitis (OR = 2.31), and autoimmune thyroid disease (OR = 2.53). Another population-based cohort study of almost one million individuals (>23,000 of whom diagnosed with ADHD) using Danish National Health Registers (38) identified an incidence rate ratio of 1.24 (95% CI 1.10–1.40) for autoimmune diseases in ADHD individuals compared to non-ADHD individuals. The associations between autoimmune diseases and ADHD may be sex-specific. Using Norwegian national registries, Hegvik et al. (163) conducted a cross-sectional study of a cohort of more than 2.5 million individuals, in which the authors could demonstrate sex-based associations between diagnosis of ADHD and different autoimmune diseases. Increased odds for psoriasis were seen in both females and males with ADHD, although the adjusted odds ratio (aOR) was higher in females (1.57 vs. 1.31), with a highly significant interaction of this association with sex. For inflammatory bowel diseases, the association was either observed only in females (ulcerative colitis) or found to be positive in females and negative in males (Crohn's disease). The lack of association with other common autoimmune diseases may depend also on the limited capacity of this study to capture associations with autoimmune diseases of later onset. The observed effect of sex on the relationship between ADHD and autoimmune diseases is intriguing and could be related to sex-specific developmental patterns of immune and neural functioning, as commented in the last section of this review. Moreover, genetic variants associated with the modulation of the risk for ADHD and autoimmunity may be pleiotropic, exhibiting sex-specific associations in opposite directions, as shown for inflammatory bowel diseases and ADHD.
Insight From Animal Models (Table 3)
Compulsive and repetitive behaviors in rodent and non-human primate models including overgrooming, hoarding and perseverative responding are typically attributed face validity for human OCD. Alongside genetic models targeting proteins involved in synaptogenesis and endocrine models expressing oxytocin and estrogen deficiencies (189), several models primarily involving immune-mediated mechanisms have been proposed in the past decade.
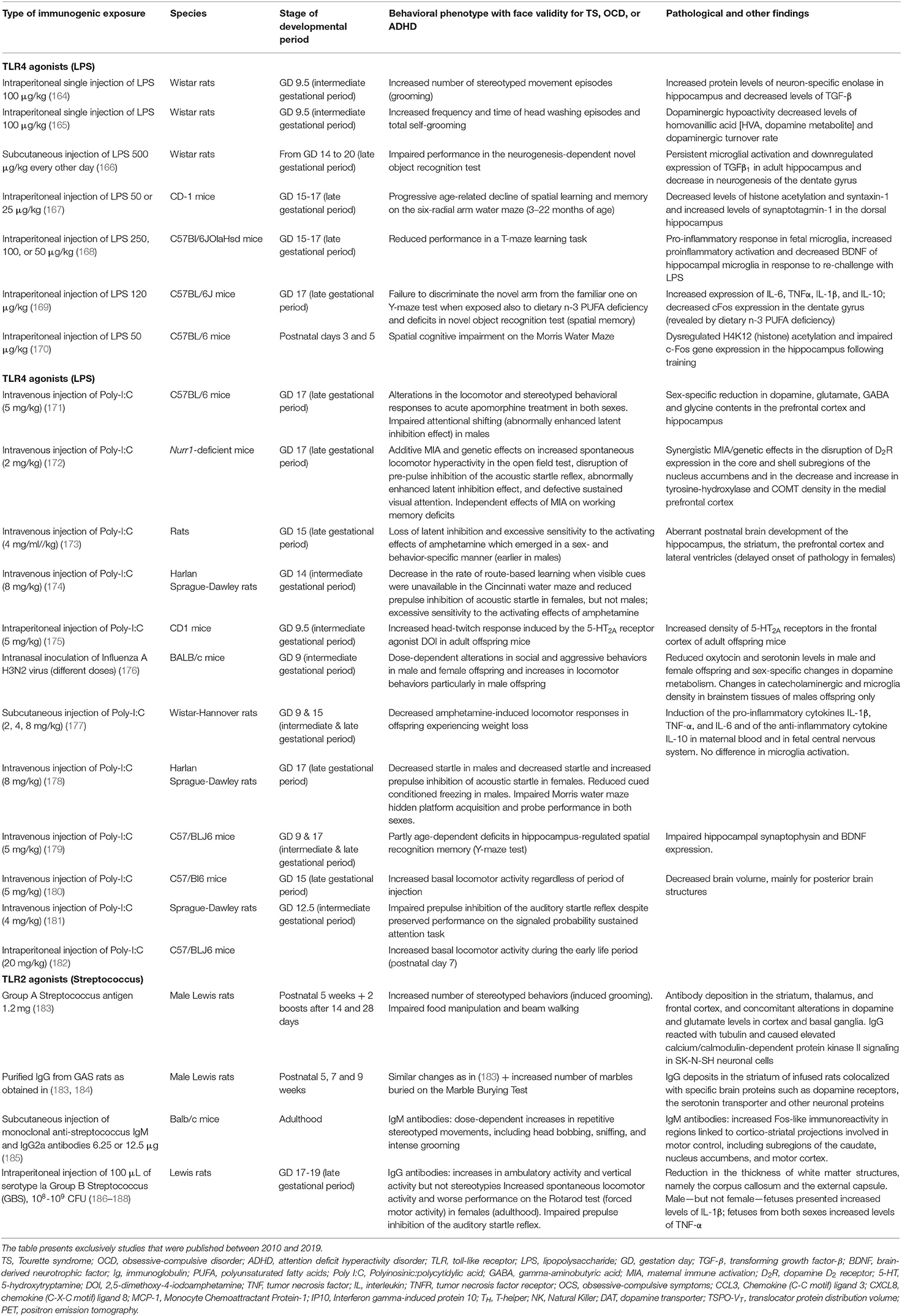
Table 3. Summary of findings on maternal immune activation models with a behavioral phenotype exhibiting face validity for compulsions, tics, hyperactivity and cognitive abnormalities observed in attention deficit hyperactivity disorder.
The first category of these models comprises immunogenic models, e.g., MIA with exposure to LPS, a cell wall component of Gram-negative bacteria, or Polyinosinic:polycytidylic acid (Poly I:C), a synthetic analog of viral double-stranded RNA, or postnatal exposure to GAS antigens (190). Prenatal exposure (mouse) to 100 μg/kg i.p. LPS at gestational day 9 (164, 165) led to increased frequency of overgrooming behaviors or head washing, increased number of buried marbles in the Marble Burying Test (MBT), and increased repetitive behaviors in the Open Field Test, the latter representative of overanxious behavior. In one of these studies, prenatal LPS exposure was associated with increased dopamine levels in the hypothalamus during the adult period (165). Prenatal exposure (mouse) to 5 mg/kg Poly I:C at gestational day 9 (175) led to increased head-twitch responses induced by the 5-hydroxytryptamine (5-HT)2A/C receptor agonist 2,5-dimethoxy-4-iodoamphetamine. Interestingly, the behavioral effects of Poly I:C during gestation could vary depending on the gestational period, as a subsequent study using the same exposure dose i.v. at gestational day 17 (late pregnancy) decreased food hoarding (179). Postnatal exposure (rat) to 1.2 mg s.c. of GAS exposure at 5, 7, and 9 postnatal weeks led to overgrooming and increased number of buried marbles on the MBT (183, 184), the latter behavior being prevented by ampicillin. The exposure of adult BALB/c mice to GAS monoclonal IgM also caused overgrooming and increased head bobbing stereotypies (185). From a mechanistic point of view, post-streptococcal models yielded changes in neurotransmitter systems, including the dopaminergic (increased dopamine levels and D1 and D2 receptor expression in the medial and prefrontal cortex and entopeduncular nucleus), serotonergic (lower activity and turnover in medial frontal cortex and cerebellum, prevention of the behavioral phenotype with paroxetine), and glutamate/glutamine (higher levels in the striatum). IgG from GAS exposed animals targeted serotonin and dopamine receptors and the serotonin transporter (184).
The second category of immune-mediated models of OCD comprises genetically manipulated models that involve cell types with immune-modulating function, e.g., microglia. One of the key functions of microglia is to survey the tissue microenvironment, orchestrating the reaction to tissue injury through release of pro-inflammatory effector molecules and phagocytosis of apoptotic debris. These microglial functions modulate also synaptic density and refinement. Loss of function of a particular subset of microglia expressing the transcription factor Hoxb8, which accounts for one third of all the brain adult microglia, causes overgrooming behavior and hair pulling leading to coat loss in mice (191). A more recent study has demonstrated that this phenotype may be associated with anxiety-like behavior and enhanced stress response (raised cortisol levels and pupil fight-or-flight response) in Hoxb8 KO female mice around the onset of sexual maturity (postnatal 6–8th week), but not in Hoxb8 KO male animals (192). Blocking estrogens with trilostane or ovariectomy could block coat loss, overgrooming, anxiety-like behaviors and enhanced stress response in Hoxb8 KO female animals, whereas the administration of 17β-estradiol and progesterone in Hoxb8 KO male animals replicated the anxiogenic phenotype. This fascinating work suggests a direct interaction between this subset of microglia and synaptic developmental trajectories controlled by sex hormones, which might have important effects on the organization of stress responses and, albeit still unexplored, immune responses. The link between microglia and OCD is supported also by the microglia-specific Grn KO model, in which the lack of progranulin is associated with increased internalization of synaptic terminals by microglia, particularly in the ventral thalamus, and with excessive grooming at adulthood (193, 194).
Several MIA models are associated with behavioral, cognitive, and neuroanatomical phenotypes that model human ADHD. However, caution is needed when interpreting this evidence due to heterogeneity of study design (195). Two important sources of heterogeneity include the gestational timepoints of the immunological insult, and the environmental setting in which behavioral outcomes are measured (home cage vs. behavioral testing apparatus). Moreover, most of the behavioral outcomes in the offspring were measured in adulthood, as opposed to during the pre- or peri-pubescent period.
The administration of the TLR4 agonist LPS during late gestation (gestational days 14–17) to pregnant rodent dams produced a cognitive phenotype characterized by spatial learning impairment assessed using the Morris-Water (196), the Six-Radial Arm Water (167), or the T-Maze (168) and impairment on the neurogenesis-dependent Novel Object Recognition Test for recognition memory (166, 197). These cognitive defects were associated with selective overactivation of hippocampal microglia (166, 168) and could be exacerbated by polyunsaturated fatty acid deficiency (169). Interestingly, a similar model can induce long-term microglial activation and astrogliosis also in the amygdala (198). Finally, LPS injection during the neonatal period can also be associated with spatial learning deficits in adulthood (170).
MIA models associated with TLR2 agonists like inactivated Group B Streptococcus have also been found to generate sex-dependent behavioral responses in the offspring with onset around late puberty, whereby male offspring manifest hyposocial, autistic-like behavioral traits and impaired processing of sensory information, and female offspring show hyperlocomotion and social disinhibition (186, 187, 199). This behavioral sexual dimorphism is associated with sexual differences also in innate immune responses (188).
MIA models using viral TLR3 agonists like Poly I:C administered in most studies during late gestation have produced similar behavioral and cognitive phenotypes (180, 200), albeit with exceptions (177, 181), and not always associated with morphological or functional changes of microglia (201). Increased hyperactivity, sensorimotor gating deficits, and attentional shifting and sustained attention impairments were reported from a “dual” model in which Poly I:C administration was given to pregnant dams carrying a heterozygous deletion of Nurr1 (172). This phenotype was associated with multiple neurotransmitter abnormalities in the prefrontal cortex and ventral striatum. Other reports demonstrated offspring sex-specific responses within Poly I:C or influenza A MIA models (171, 174, 178), with greater hyperlocomotion in males (176, 182), and females exhibiting a later onset of prefrontal cortex and hippocampal volume decreases, attentional deficit, and metamphetamine-induced hyperlocomotion (173). Similar male specificity to locomotor hyperactivity was observed also in prenatal stress models (202).
Juvenile spontaneously hypertensive rats, a metabolic/endocrine model of ADHD, show systemically increased inflammatory cytokines associated with decreased medial prefrontal cortical volume and up-regulation of D2 dopamine receptors. These changes tended to normalize during maturation, in conjunction with compensatory elevation of steroid hormones (203).
Finally, proteomics using induced pluripotent stem cells (iPSC)-derived neurons and microglia from individuals with TS and related disorders will continue to push the field toward better understanding of disease pathophysiology which hopefully will contribute to the future development of therapeutics against specific targets (204, 205).
Association With Specific Gut Microbiota Profiles
Evidence for a role of the microbiota-gut-brain axis in neural development and behavior is constantly increasing, and has been extensively reviewed (206–208). The influence of symbiotic microorganisms could start even in utero via penetration of maternal microbial molecules through the placental barrier, as recent reviewed by Ganal-Vonarburg et al. (209). Immune-based ASD rodent models display imbalanced gut microbiota composition (dysbiosis) and gut barrier dysfunction (“leaky gut”), associated with abnormal social behavior (210, 211). Microbiota composition dysbiosis remains, however, underinvestigated in ADHD and fundamentally unexplored in TS and OCD.
To date, most of the studies that explored the gut microbiome in ADHD have resorted to 16S rRNA amplicon sequencing. This methodological approach allows a snapshot of the taxonomic composition of intestinal microbial colonies (212). However, there is large heterogeneity across studies with respect to sample size and adjustment for several confounding factors, particularly exposure to stimulants, dietary habits, exposure to pro- and antibiotics, and perinatal factors (delivery, early feeding). Moreover, the presence of gastrointestinal dysfunction, which is strongly associated with gut microbiota composition, remains uncertain in ADHD. Index measures of gut microbiota diversity provided discrepant results (213–215). Despite these discrepancies, some differences between ADHD and normotypical youth with respect to relative abundance of gut flora at a genus level have been replicated. Among these, the decreased abundance in Faecalibacterium spp. yielded also a negative correlation with ADHD severity scores (215, 216). This genus is associated with anti-inflammatory skewing of immune responses and its decreased abundance is observed in allergies. However, this finding needs to be verified adjusting for dietary habits, as certain diets like the long-term Mediterranean are associated with higher abundance in Faecalibacterium spp. Increased abundance in the Bifidobacterium genus was found in adolescents/adults with ADHD (217) and, although remaining a controversial finding (218), was reversed by micronutrient supplementation in a recent randomized controlled trial (RCT) (219). The largest study that used 16S rRNA amplicon sequencing investigated adolescents/adults with ADHD and identified increased relative abundance of different, potentially interacting, Ruminococcaceae genera, and positive correlation with inattentive symptoms (218). The same group performed an exploratory fecal matter transplantation study of stool specimens from ADHD patients into germ-free mice, generating anxiety-like behaviors (220).
Next generation “shotgun” (metagenomic) sequencing has been applied to a small study of ADHD youth (216). Interestingly, pathway analyses of gene functional annotations revealed abnormalities of metabolic pathways associated with dopaminergic and serotonergic systems, consistent with their hypofunctionality. Additional indirect evidence supporting a role of microbiota in ADHD comes from the observation of greater odds of ADHD among boys with prenatal exposure to dog pets (221), known to influence microbiota composition within familial clusters. Moreover, Slykerman et al. (222) reported an increased risk of ADHD in children exposed to antibiotics in the first 6 months of life, which may represent a surrogate of early infectious exposure, but also a source of gut microbiota imbalance.
The investigation of gut microbiota ecology and its association with pathways modulating the neural-immune crosstalk during development is in its dawning age in the context of TS and OCD. This influence was investigated only in PANS/PANDAS, applying conventional 16S rRNA-based gut microbiome metagenomics to a cohort of 30 patients and a control population. This study revealed gut dysbiosis characterized by an increased abundance of Bacteroidetes especially in younger patients, with a negative correlation between genera of the Firmicutes phylum and anti-streptolysin O titers (223). Pharmacological animal models expressing compulsive behaviors revealed an association between the behavioral phenotype and changes in gut microbial communities. Chronic quinpirole injections in rats induced locomotor sensitization and compulsive checking associated with changes in abundance of Firmicutes, predominantly Lachnospiraceae and Ruminococcaceae (224). Another study demonstrated different gut microbiota composition between the naturally occurring obsessive-compulsive and the normal phenotypes of deer mice (225). A systematic investigation of the gut microenvironment in patients with OCD or TS has not been published yet. Interestingly, a RCT of probiotic treatment with Lactobacillus and Bifidobacterium species in adults with OCD is currently ongoing (https://clinicaltrials.gov/ct2/show/NCT02334644).
Responsiveness to Immune-Based Therapies
The therapeutic efficacy of immune-modulatory and anti-inflammatory treatments in TS, OCD, and ADHD has been interrogated by a small number of studies. The use of non-specific immune-modifying approaches such as intravenous immunoglobulins (IVIg) and plasma exchange in the PANS/PANDAS group is supported only by one underpowered RCT as well as uncontrolled case series (226). However, previous attempts to use IVIg in an unselected population of TS patients has not been beneficial, whereas evidence of the efficacy of IVIg and plasma exchange in unselected clinical samples of OCD and ADHD patients is lacking.
A handful of studies have documented the potential effectiveness of anti-inflammatory and antibiotic drugs in decreasing OCS. None of these studies has, however, explored the actual mechanism underlying this potential therapeutic effect. A RCT of 50 outpatients with moderate to severe OCD underwent 10 weeks of treatment with either celecoxib (200 mg bid) or placebo as adjuvant to fluvoxamine (227). A significant effect for “time x treatment” interaction on the Y-BOCS total scores [F(1.38, 66.34) = 6.91, p = 0.005] was reported, consistent with a more rapid response to celecoxib group than to placebo group (p < 0.001), and no difference in adverse events between the two arms.
A small RCT compared cefdinir, a beta-lactam antibiotic promoting glutamate transporter GLT1 expression and enhancing complement-mediated immunity, to placebo over the course of 30 days in 20 subjects with recent onset OCD and/or tics (228). Only a trend was observed for tic severity improvement (44.4% showing at least a 25% reduction in YGTSS), whereas there was no significant difference in OCS severity despite a slightly larger decrease on the CY-BOCS score with cefdinir. This study was underpowered to detect clinically relevant and statistically significant severity changes for both types of symptoms.
A more recent study randomized 102 patients with moderate-to-severe OCD (medication-free for 6 weeks prior to the study) to receive minocycline 100 mg twice per day or placebo for 10 weeks in addition to fluvoxamine 100 mg/day for the first 4 weeks and then 200 mg/day for the rest of the trial (229). There was a significantly greater rate (31.9%) of partial and complete responses in the minocycline group (>35% reduction in Y-BOCS score, p < 0.001), with similar frequency of adverse events. Apart from exerting neuroprotective effects decreasing glutamate-induced neurotoxicity, minocycline regulates nitric oxide, TNFα and IL-1β release and showed some benefit in improving schizophrenia, depressive and autistic symptoms. A previous open-label trial by Rodriguez et al. (230) showed that minocycline augmentation may not improve OCD in all adult OCD patients, but only in those with early onset OCD (45%) and with primary hoarding.
Potentially interesting immunotherapy approaches include antibodies or antibody components targeting specific cytokines, such as IL-6, as these may exert relevant effects on neural/immune crosstalks hindering cytokine peripheral receptors from crossing the blood-brain barrier and dampening inflammatory cascades in the periphery.
Pathophysiological Considerations on Immune Dysregulation in TS, OCD, and ADHD and Questions for Future Research
MIA has become a prominent pathophysiological model for a large series of neurodevelopmental disorders, including ASD, schizophrenia, and bipolar disorder. Even if supported by a substantially smaller body of evidence, it is becoming increasingly convincing that MIA could act as an early hit and a “disease primer” also in the development of OCD, ADHD, and probably also TS. The advancing field of iPSC-derived cellular models has fostered progress toward the development of 3D-brain organoids, which may shed important light on physiological early brain development and basic mechanisms of neurodevelopment-related disorders (205). Interestingly, an organoid model of dorsal forebrain exhibited an organization of cell types that recapitulated in part the developmental trajectory and circuit functionality of the developing human brain (231). This field might offer unprecedented insight into the physiology and pathophysiology of neural network formation (232), including the role of microglia on synaptic formation and refining, as well as on gene-environment interactions during brain development.
Infectious MIA models (LPS- or poly [I:C]-induced) are associated with increased rate of repetitive behaviors modeling compulsions (e.g., overgrooming, head washing), complex tics (head twitch responses secondary to 5-HT2A/C receptor agonists), and hyperactivity/anxious behavior. In a proportion of these studies, the gestational period of the immune insult and the sex of the offspring are important mediators. With respect to cognitive phenotype, spatial exploratory changes were observed following mid-gestational (day 9) exposure to MIA in rodent models, whereas learning changes and dopamine-derived motor behavioral changes have been linked to late (day 16–17) gestational insults. With respect to repetitive behaviors, on the other hand, compulsion-like and tic-like behaviors were observed more frequently after mid-gestational immune activation, whereas hyperactivity and over-anxious behaviors could be provoked mostly by late gestational immunological insults. More exploration is necessary to understand the exact immunological pathways that mediate the effect of these insults on synaptic formation, synaptic pruning, and neural migration at different gestational periods. Moreover, we are only beginning the understand the correlation between cognitive/behavioral phenotypes and gestational timing of the insult. Greater clarity will be achieved overcoming the marked methodological heterogeneity across MIA experimental protocols exhibited by studies to date (233). Likewise, there are initial signals of sex-dependent responses in the offspring, which differ in age of onset during postnatal life (older in female offspring), behavioral phenotype (e.g., higher frequency of hyperlocomotion behavior in female offspring), and even maturation of innate immune responses.
The mechanisms through which MIA primes behavior through synaptic refinement of developing neural networks, as well as the development of immune responses, are still under investigation. At a molecular level, the production of pro-inflammatory cytokines like IL-1β, TNF-α, IL-17-α, and IL-6 has classically been considered as a major culprit of region-specific neuropathologic alterations producing long-lasting effects in adult offspring. Although these cytokines may be produced by different neural cell types, including neurons and astrocytes, their production by microglia, alongside growth factors and oxidative stress markers, has emerged as pivotal in the genesis of neural and immune abnormalities linked to different neurodevelopmental disorders. Microglia can respond to MIA insults even since its neural cell progenitor stage of development (yolk sac macrophage stage) around day 10 of gestation. Remarkably, immunological priming of early microglia can set transcriptomic signatures in these cells that may last until adulthood. It is therefore intriguing to hypothesize that the transcriptomic profile detected post mortem in brains of TS patients (109), or the activated microglial pattern observed using PET in young adults with OCD (132), might have been predetermined, at least in part, already since the intrauterine period. The initial evidence in favor of an age-dependent variability of MIA effects in the offspring can also be explained with the highly dynamic functional pattern of microglial activity during the course of development (234, 235). The central role of microglia in abnormal neural-immune development in the context of OCD is strongly supported by the overgrooming phenotype exhibited by mice lacking one of the most abundant sets of microglia (Hoxb8-positive), or by mice lacking the microglia-specific Grn gene. The first of these two transgenic models provides further support to the estrogen-dependence of microglial effects on the development of overgrooming, anxiety-like behaviors and enhanced stress responses. This sexual dimorphism is of exceptional interest, as it may point to sex-specific mechanisms in the microglial modulation of neural-immune developmental abnormalities in OCD and related conditions. The maturation and functional activation of microglial populations follow sex-determined neurodevelopmental patterns. Sexually dimorphic responses to immune-activating triggers (e.g., Poly I:C) are recognized with respect to microglial density, morphology, and transcriptional profile. Moreover, this sexual dimorphism appears to be long-lasting, as the female offspring of MIA-associated pregnancies display greater microglial activation in adulthood compared to male offspring. This is intriguing at the light of an aging-progressive decrease of the male predominance in the sex ratio observed in both TS and OCD. Finally, there is limited evidence of dual-hit processes associated with MIA in ADHD, TS and OCD. An exception is the interaction between MIA and Nurr1 genotype demonstrated in a poly(I:C) model of ADHD, with associated abnormalities in the prefronto-striatal pathways.
At difference from infectious MIA models, the effect of non-infectious causes of MIA (prenatal stress, maternal diet/obesity, microbiome) await to be investigated in the context of TS, OCD, and ADHD. Microglial activation, with changes in morphology and cytokine production, and migration of GABAergic progenitor migration were reported in the offspring of prenatal stress (restraint/bright light) MIA models of ASD (236), with predilection for the female offspring. The effect of maternal diet and obesity has also been explored in the context of an ASD-like phenotype, and linked to decrease in oxytocin-producing cells in the paraventricular nucleus and decreased synaptic plasticity in the ventral tegmental area (237).
Human studies offer limited information on the role of MIA in the pathogenesis of these neurodevelopmental disorders. The association between infections during pregnancy remains unexplored in TS and OCD, whereas data on ADHD carry known limitations coming from national registry datasets, first of all the lack of serial serologic measurements to prevent biases in the ascertainment of the exposure. The lack of birth seasonality in OCD, TS, and ADHD indirectly does not speak in favor of an effect of maternal infection on the risk for these disorders in the offspring (238). The effect of stressors in pregnancy also remains underexplored in these conditions. There is, nevertheless, well-established evidence that stressors cause overproduction of proinflammatory cytokines by immune cells in response to immunostimulants, and alterations in the HPA axis, which may also influence the development of immune responses in the offspring, especially T and B cell proliferation, NK cell cytotoxicity, and cytokine receptor density (239, 240).
The involvement of mechanisms occurring at the placental interface in the pathoogenesis of TS, OCD and ADHD also remains unexplored. MIA models based on Poly I:C demonstrated the activity of maternal immune cells in the placenta, particularly in association with increased IL-6 production at the maternal-fetal interface. The placenta might also modulate stress responses induced by prenatal stress-induced MIA, mediating the metabolism of glucocorticoids into inactive metabolites (240).
Human studies that explored the physiological profile of immune-inflammatory responses and the clinical profile of immunological comorbidities in patients with TS, OCD, and ADHD consistently indicate the long-term persistence of abnormally enhanced responses to exogenous (pathogens, allergens) and endogenous (self-antigens targeted by autoimmune processes) triggers. The limited evidence of dysregulated expression of immune effector molecules (e.g., cytokines) in individuals with established ADHD is at odds with the robust evidence of comorbidity of allergies and autoimmune diseases in this condition, and is probably due to the lack of adequately powered clinical observational studies. An abnormal immune priming, probably acting on a combination of predisposing genomic factors and possibly promoted by epigenetic modifications, may contribute to altering the process of maturation of specific neural networks, as well as of both innate and adaptive immune regulatory mechanisms. The resulting outcome of this pathophysiological substrate is likely to be the long-lasting co-existence of behavioral and cognitive deficits and hypersensitive innate and adaptive responses to antigens or allergens of sufficient immunogenic potency. Our critical review of the literature suggests that there is insufficient evidence to support the hypothesis that immunogenic triggers cause or predict the short- or medium-term worsening of tics, OCS, attention/executive deficits, or hyperactivity/impulsivity. In the case of tics, there is actually mounting evidence supporting the lack of association between common immunogenic triggers e.g., GAS infections and exacerbations of tics. Alternatively, it seems more likely that enhanced immune-inflammatory responses toward pathogens could be epiphenomenal to tics and related symptoms, as an increase in immune responses targeting GAS and other pathogens appears as longstanding in these patients (64, 241). As mentioned above, this could stem from a vulnerability deriving from genetic background and abnormal immune priming in sensitive developmental periods. At the same time, the contribution of a “reverse causation” mechanism, whereby behavioral patterns that are especially characteristic of the ADHD spectrum could increase the likelihood of contacts with pathogens or exposure to certain allergens.
The involvement of the microbiota-gut-brain axis in the neuroimmune connection that influences network maturation trajectories in TS, OCD, and ADHD is still unchartered territory that awaits to be explored. The limited evidence we have available today suggests the presence of a few differences in relative abundance of microbiota constituents between ADHD and neurotypical individuals, and a possible influence on neurotransmitter pathways of the microbiome profiling. Future research should employ germ-free versions of animal models of TS, OCD, and ADHD in order to investigate the contribution of the microbiome to sexually dimorphic characteristics of neural development and maturation of microglia in these disorders. It would also be very interesting to explore the production of bacterial metabolites that are known to steer microglial function, e.g., short chain fatty acids and tryptophan-derived ligands, across different age groups in patients with TS, OCD and ADHD. Metagenomic and metabolomic studies related to the microbiome profiling are warranted to discover the most relevant metabolic pathways influenced by gut microbiota in these disorders, and design mechanism-driven microbiota manipulation interventions.
Behavioral therapies based on habit reversal training or exposure-response prevention constitute first-line interventions in the management of tics and obsessive-compulsive symptoms. However, little is known on their potential impact on the regulation of immune responses in patients with TS and OCD. There is increasing evidence suggesting an effect of cognitive-behavioral therapy (CBT) on immune markers in other psychiatric conditions. In major depressive disorder (MDD), a greater reduction of pro-inflammatory markers (TLR-4 RNA and protein and NF-κβ RNA) during CBT was associated with greater clinical improvement (242). Moreover, baseline levels of pro-inflammatory cytokines (IL-12, IL-1β) may predict the response of depressive symptoms to CBT in youth with bipolar disorder or MDD (243). Interestingly, a similar effect of decreased plasma chemokine levels was reported for internet-based CBT in patients with mild-moderate depression (244). Finally, other psychotherapeutic approaches still under-investigated in these conditions, e.g., mindfulness meditation, appear promising for the modulation of inflammation and cell-mediated immunity (245). It would be interesting to explore whether immune-based mechanisms contribute to the efficacy of comprehensive-behavioral intervention for tics (CBIT) or ERP in TS and OCD patients.
Closing Remarks
Neural development is an enormously complex and dynamic process. Immunological pathways are intimately involved in several very early neurodevelopmental processes including the formation and refinement of neural circuits. Hyper-reactivity of systemic immune pathways and neuroinflammation may contribute to the natural fluctuations of the core behavioral features of tic disorders and related conditions including OCD and ADHD (Box 1). While the genetic relationship amongst these disorders is complex, and potentially linked to specific sub-phenotypes, it is clear that early environmental risk factors can influence the course and severity of these disorders (Box 1). Future research is needed to gain a deeper understanding of these gene-environment interactions and how they set the stage from a neurodevelopmental perspective for the emergence of these disorders. which may explain differences in the strength of their pairwise genetic correlation (Box 2). Likewise, future research also needs to focus on the key molecular pathways through which dysbiosis of different tissue microbiota influence neuroimmune interactions in these disorders, and how microbiota modification could impact their natural history. Efforts need to be made to establish valid biomarkers that will guide a personalized approach to the treatment of these conditions.
Box 1. Summary key points on the relationship between immune-related mechanisms and Tourette syndrome (TS), obsessive-compulsive disorder (OCD), and attention deficit hyperactivity disorder (ADHD).
• Initial research demonstrates an association between OCD and ADHD and genes that are implicated in immune system function
• Several immune-mediated diseases also have genetic correlation with TS and ADHD
• The data investigating a potential correlation between postnatal Group A Streptococcus (GAS) or non-streptococcal infections and the development of tic disorders or obsessive-compulsive symptoms remains mixed
• There is evidence for an association between significant infectious exposure in early childhood and the development of ADHD symptoms
• Direct evidence of pro-inflammatory overactivity in the brain in TS and OCD is modest and limited
• There is preliminary but compelling evidence to indicate that dysfunction in microglial maturation and functioning is implicated in TS
• There is limited, strong evidence for a correlation between the activation of microglia and OCD
• In younger patients with ADHD, there is modest evidence supporting the overactivity of systemic inflammatory mechanisms
• TS and ADHD have been associated with an increased risk for multiple autoimmune disorders and allergic conditions
• OCD has been associated with an increased risk for multiple autoimmune disorders, but has not been associated with an increased risk for allergic conditions
• Patients with ADHD demonstrate some evidence of alterations in the gut microbiome, whereas this topic has been studied only minimally in TS and OCD
• Direct and indirect immune-modulatory interventions aimed at treating TS, OCD, and ADHD have only been studied to a limited degree
• Maternal immune activation is likely involved in the early development of OCD, ADHD, and probably also TS
• There is insufficient evidence that immunogenic triggers influence short- or medium-term worsening in tic symptoms, obsessive compulsive symptoms, or symptoms of inattention or hyperactivity
Box 2. Questions that should be addressed by future research projects exploring the relationship between immune-related mechanisms and Tourette syndrome (TS), obsessive-compulsive disorder (OCD), and attention deficit hyperactivity disorder (ADHD).
• What are the key genetic and epigenetic factors that influence a dysfunctional neural-immune crosstalk during development in TS, OCD, and ADHD?
• Can induced pluripotent stem cells-derived cellular models and 3D-brain organoids help recapitulating the dysfunction of microglia interaction with other neural cell types that may occur during development in individuals with TS, OCD, and ADHD?
• Do sex-specific alterations of microglia development contribute to the sexual dimorphism observed in the pathological manifestations of TS, OCD, and ADHD?
• What are the key molecular pathways by which changes in microbiota play a role in neuroimmune interactions in TS, OCD, and ADHD? Is the development of microglia the critical physiological process influenced by symbiotic microbiota in the pathogenesis of TS, OCD, and ADHD?
• Will immune-modifying treatments targeting specific molecular pathways be able to counteract abnormal neurodevelopmental trajectories in TS, OCD, and ADHD?
• Will microbiota manipulation strategies targeting specific molecular pathways be able to counteract abnormal neurodevelopmental trajectories in TS, OCD, and ADHD?
• Will the analysis of multivariate datasets, incorporating metagenomics, metabolomics, “inflammasomics,” gastrointestinal, and CNS physiomarkers, pay off in accelerating the discovery of key mechanistic pathways to prevent and modify disease course in TS, OCD, and ADHD?
Author Contributions
DM and JL contributed to conception and design of this review. DM prepared the initial draft of the manuscript. Each of the authors then critically reviewed and revised the manuscript before approving for submission the final version of the article.
Funding
While preparing this article, two of the authors relied on salary support and discretionary funds from the University of Calgary (DM) and Yale University (JL). IJ was completing his medical school training at Yale University and he received no financial support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Scharf JM, Miller LL, Gauvin CA, Alabiso J, Mathews CA, Ben-Shlomo Y. Population prevalence of Tourette syndrome: a systematic review and meta-analysis. Mov Disord. (2015) 30:221–8. doi: 10.1002/mds.26089
2. Robertson MM, Eapen V, Singer HS, Martino D, Scharf JM, Paschou P, et al. Gilles de la Tourette syndrome. Nat Rev Dis Primers. (2017) 3:16097. doi: 10.1038/nrdp.2016.97
3. Hirschtritt ME, Lee PC, Pauls DL, Dion Y, Grados MA, Illmann C, et al. Lifetime prevalence, age of risk, and genetic relationships of comorbid psychiatric disorders in Tourette syndrome. JAMA Psychiatry. (2015) 72:325–33. doi: 10.1001/jamapsychiatry.2014.2650
4. Martino D, Ganos C, Pringsheim TM. Tourette syndrome and chronic tic disorders: the clinical spectrum beyond Tics. Int Rev Neurobiol. (2017) 134:1461–90. doi: 10.1016/bs.irn.2017.05.006
5. Kurlan R, Como PG, Miller B, Palumbo D, Deeley C, Andresen EM, et al. The behavioral spectrum of tic disorders: a community-based study. Neurology. (2002) 59:414–20. doi: 10.1212/WNL.59.3.414
6. Leckman JF, King RA, Bloch MH. Clinical features of Tourette syndrome and Tic disorders. J Obsessive Compuls Relat Disord. (2014) 3:372–9. doi: 10.1016/j.jocrd.2014.03.004
7. Darrow SM, Hirschtritt ME, Davis LK, Illmann C, Osiecki L, Grados M, et al. Identification of two heritable cross-disorder endophenotypes for Tourette syndrome. Am J Psychiatry. (2017) 174:387–96. doi: 10.1176/appi.ajp.2016.16020240
8. Hirschtritt ME, Darrow SM, Illmann C, Osiecki L, Grados M, Sandor P, et al. Genetic and phenotypic overlap of specific obsessive-compulsive and attention-deficit/hyperactive subtypes with Tourette syndrome. Psychol Med. (2018) 48:279–93. doi: 10.1017/S0033291717001672
9. Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. Analysis of shared heritability in common disorders of the brain. Science. (2018) 360:eaap8757. doi: 10.1126/science.aap8757
10. Cortese S. The neurobiology and genetics of Attention-Deficit/Hyperactivity Disorder (ADHD): what every clinician should know. Eur J Paediatr Neurol. (2012) 16:422–33. doi: 10.1016/j.ejpn.2012.01.009
11. Wen H, Liu Y, Rekik I, Wang S, Chen Z, Zhang J, et al. Combining disrupted and discriminative topological properties of functional connectivity networks as neuroimaging biomarkers for accurate diagnosis of early Tourette syndrome children. Mol Neurobiol. (2018) 55:3251–69. doi: 10.1007/s12035-017-0519-1
12. Nielsen AN, Gratton C, Church JA, Dosenbach NUF, Black KJ, Petersen SE, et al. Atypical functional connectivity in Tourette syndrome differs between children and adults. Biol Psychiatry. (2020) 87:164–73. doi: 10.1016/j.biopsych.2019.06.021
13. Park JE, Jardine L, Gottgens B, Teichmann SA, Haniffa M. Prenatal development of human immunity. Science. (2020) 368:600–3. doi: 10.1126/science.aaz9330
14. Bauman MD, Van De Water J. Translational opportunities in the prenatal immune environment: promises and limitations of the maternal immune activation model. Neurobiol Dis. (2020) 141:104864. doi: 10.1016/j.nbd.2020.104864
15. Haddad FL, Patel SV, Schmid S. Maternal immune activation by Poly I:C as a preclinical model for neurodevelopmental disorders: a focus on autism and schizophrenia. Neurosci Biobehav Rev. (2020) 113:546–67. doi: 10.1016/j.neubiorev.2020.04.012
16. Hafizi S, Tabatabaei D, Lai MC. Review of clinical studies targeting inflammatory pathways for individuals with autism. Front Psychiatry. (2019) 10:849. doi: 10.3389/fpsyt.2019.00849
17. Gumusoglu SB, Stevens HE. Maternal inflammation and neurodevelopmental programming: a review of preclinical outcomes and implications for translational psychiatry. Biol Psychiatry. (2019) 85:107–21. doi: 10.1016/j.biopsych.2018.08.008
18. Bergdolt L, Dunaevsky A. Brain changes in a maternal immune activation model of neurodevelopmental brain disorders. Prog Neurobiol. (2019) 175:1–19. doi: 10.1016/j.pneurobio.2018.12.002
19. Frick LR, Rapanelli M, Jindachomthong K, Grant P, Leckman JF, Swedo S, et al. Differential binding of antibodies in PANDAS patients to cholinergic interneurons in the striatum. Brain Behav Immun. (2018) 69:304–11. doi: 10.1016/j.bbi.2017.12.004
20. Leon J, Hommer R, Grant P, Farmer C, D'souza P, Kessler R, et al. Longitudinal outcomes of children with pediatric autoimmune neuropsychiatric disorder associated with streptococcal infections (PANDAS). Eur Child Adolesc Psychiatry. (2018) 27:637–43. doi: 10.1007/s00787-017-1077-9
21. Gromark C, Harris RA, Wickstrom R, Horne A, Silverberg-Morse M, Serlachius E, et al. Establishing a pediatric acute-onset neuropsychiatric syndrome clinic: baseline clinical features of the pediatric acute-onset neuropsychiatric syndrome Cohort at Karolinska Institutet. J Child Adolesc Psychopharmacol. (2019) 29:625–33. doi: 10.1089/cap.2018.0127
22. Baj J, Sitarz E, Forma A, Wroblewska K, Karakula-Juchnowicz H. Alterations in the nervous system and gut microbiota after beta-hemolytic Streptococcus Group A infection-characteristics and diagnostic criteria of PANDAS recognition. Int J Mol Sci. (2020) 21:1476. doi: 10.3390/ijms21041476
23. Marson A, Housley WJ, Hafler DA. Genetic basis of autoimmunity. J Clin Invest. (2015) 125:2234–41. doi: 10.1172/JCI78086
24. Benros ME, Mortensen PB. Role of infection, autoimmunity, atopic disorders, and the immune system in schizophrenia: evidence from epidemiological and genetic studies. Curr Top Behav Neurosci. (2020) 44:141–59. doi: 10.1007/7854_2019_93
25. Keszler G, Kruk E, Kenezloi E, Tarnok Z, Sasvari-Szekely M, Nemoda Z. Association of the tumor necrosis factor−308 A/G promoter polymorphism with Tourette syndrome. Int J Immunogenet. (2014) 41:493–8. doi: 10.1111/iji.12147
26. Tylee DS, Sun J, Hess JL, Tahir MA, Sharma E, Malik R, et al. Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data. Am J Med Genet B Neuropsychiatr Genet. (2018) 177:641–57. doi: 10.1002/ajmg.b.32652
27. Tsetsos F, Yu D, Sul JH, Huang AY, Illmann C, Osiecki L, et al. Synaptic processes and immune-related pathways implicated in Tourette syndrome. medRXiv. (2020).
28. Teixeira AL, Rodrigues DH, Marques AH, Miguel EC, Fontenelle LF. Searching for the immune basis of obsessive-compulsive disorder. Neuroimmunomodulation. (2014) 21:152–8. doi: 10.1159/000356554
29. Rodriguez N, Morer A, Gonzalez-Navarro EA, Gasso P, Boloc D, Serra-Pages C, et al. Human-leukocyte antigen class II genes in early-onset obsessive-compulsive disorder. World J Biol Psychiatry. (2019) 20:352–8. doi: 10.1080/15622975.2017.1327669
30. Den Braber A, Zilhao NR, Fedko IO, Hottenga JJ, Pool R, Smit DJ, et al. Obsessive-compulsive symptoms in a large population-based twin-family sample are predicted by clinically based polygenic scores and by genome-wide SNPs. Transl Psychiatry. (2016) 6:e731. doi: 10.1038/tp.2015.223
31. Cappi C, Brentani H, Lima L, Sanders SJ, Zai G, Diniz BJ, et al. Whole-exome sequencing in obsessive-compulsive disorder identifies rare mutations in immunological and neurodevelopmental pathways. Transl Psychiatry. (2016) 6:e764. doi: 10.1038/tp.2016.30
32. Zayats T, Athanasiu L, Sonderby I, Djurovic S, Westlye LT, Tamnes CK, et al. Genome-wide analysis of attention deficit hyperactivity disorder in Norway. PLoS ONE. (2015) 10:e0122501. doi: 10.1371/journal.pone.0122501
33. De Jong S, Newhouse SJ, Patel H, Lee S, Dempster D, Curtis C, et al. Immune signatures and disorder-specific patterns in a cross-disorder gene expression analysis. Br J Psychiatry. (2016) 209:202–8. doi: 10.1192/bjp.bp.115.175471
34. Van Dongen J, Zilhao NR, Sugden K, Consortium B, Hannon EJ, Mill J, et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder symptoms in adults. Biol Psychiatry. (2019) 86:599–607. doi: 10.1016/j.biopsych.2019.02.016
35. Nuzziello N, Craig F, Simone M, Consiglio A, Licciulli F, Margari L, et al. Integrated analysis of microRNA and mRNA expression profiles: an attempt to disentangle the complex interaction network in attention deficit hyperactivity disorder. Brain Sci. (2019) 9:288. doi: 10.3390/brainsci9100288
36. Mataix-Cols D, Frans E, Perez-Vigil A, Kuja-Halkola R, Gromark C, Isomura K, et al. A total-population multigenerational family clustering study of autoimmune diseases in obsessive-compulsive disorder and Tourette's/chronic tic disorders. Mol Psychiatry. (2018) 23:1652–8. doi: 10.1038/mp.2017.215
37. Jones HF, Ho ACC, Sharma S, Mohammad SS, Kothur K, Patel S, et al. Maternal thyroid autoimmunity associated with acute-onset neuropsychiatric disorders and global regression in offspring. Dev Med Child Neurol. (2019) 61:984–8. doi: 10.1111/dmcn.14167
38. Nielsen PR, Benros ME, Dalsgaard S. Associations between autoimmune diseases and attention-deficit/hyperactivity disorder: a Nationwide study. J Am Acad Child Adolesc Psychiatry. (2017) 56:234–40 e231. doi: 10.1016/j.jaac.2016.12.010
39. Instanes JT, Halmoy A, Engeland A, Haavik J, Furu K, Klungsoyr K. Attention-deficit/hyperactivity disorder in offspring of mothers with inflammatory and immune system diseases. Biol Psychiatry. (2017) 81:452–9. doi: 10.1016/j.biopsych.2015.11.024
40. Modesto T, Tiemeier H, Peeters RP, Jaddoe VW, Hofman A, Verhulst FC, et al. Maternal mild thyroid hormone insufficiency in early pregnancy and attention-deficit/hyperactivity disorder symptoms in children. JAMA Pediatr. (2015) 169:838–45. doi: 10.1001/jamapediatrics.2015.0498
41. Arpino C, Marzio M, D'argenzio L, Longo B, Curatolo P. Exanthematic diseases during pregnancy and attention-deficit/hyperactivity disorder (ADHD). Eur J Paediatr Neurol. (2005) 9:363–5. doi: 10.1016/j.ejpn.2005.05.001
42. Pineda DA, Palacio LG, Puerta IC, Merchan V, Arango CP, Galvis AY, et al. Environmental influences that affect attention deficit/hyperactivity disorder: study of a genetic isolate. Eur Child Adolesc Psychiatry. (2007) 16:337–46. doi: 10.1007/s00787-007-0605-4
43. Mann JR, Mcdermott S. Are maternal genitourinary infection and pre-eclampsia associated with ADHD in school-aged children? J Atten Disord. (2011) 15:667–73. doi: 10.1177/1087054710370566
44. Silva D, Colvin L, Hagemann E, Bower C. Environmental risk factors by gender associated with attention-deficit/hyperactivity disorder. Pediatrics. (2014) 133:e14–22. doi: 10.1542/peds.2013-1434
45. Werenberg Dreier J, Nybo Andersen AM, Hvolby A, Garne E, Kragh Andersen P, Berg-Beckhoff G. Fever and infections in pregnancy and risk of attention deficit/hyperactivity disorder in the offspring. J Child Psychol Psychiatry. (2016) 57:540–8. doi: 10.1111/jcpp.12480
46. Ginsberg Y, D'onofrio BM, Rickert ME, Class QA, Rosenqvist MA, Almqvist C, et al. Maternal infection requiring hospitalization during pregnancy and attention-deficit hyperactivity disorder in offspring: a quasi-experimental family-based study. J Child Psychol Psychiatry. (2019) 60:160–8. doi: 10.1111/jcpp.12959
47. Hamad AF, Alessi-Severini S, Mahmud S, Brownell M, Kuo IF. Prenatal antibiotic exposure and risk of attention-deficit/hyperactivity disorder: a population-based cohort study. CMAJ. (2020) 192:E527–E535. doi: 10.1503/cmaj.190883
48. Morales E, Julvez J, Torrent M, Ballester F, Rodriguez-Bernal CL, Andiarena A, et al. Vitamin D in pregnancy and attention deficit hyperactivity disorder-like symptoms in childhood. Epidemiology. (2015) 26:458–65. doi: 10.1097/EDE.0000000000000292
49. Sucksdorff M, Brown AS, Chudal R, Surcel HM, Hinkka-Yli-Salomäki S, Cheslack-Postava K, et al. Maternal vitamin D levels and the risk of offspring attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. (2019). doi: 10.1016/j.jaac.2019.11.021. [Epub ahead of print].
50. Chudal R, Brown AS, Gyllenberg D, Hinkka-Yli-Salomaki S, Sucksdorff M, Surcel HM, et al. Maternal serum C-reactive protein (CRP) and offspring attention deficit hyperactivity disorder (ADHD). Eur Child Adolesc Psychiatry. (2020) 29:239–47. doi: 10.1007/s00787-019-01372-y
51. Cardona F, Orefici G. Group A streptococcal infections and tic disorders in an Italian pediatric population. J Pediatr. (2001) 138:71–5. doi: 10.1067/mpd.2001.110325
52. Morshed SA, Parveen S, Leckman JF, Mercadante MT, Bittencourt Kiss MH, Miguel EC, et al. Antibodies against neural, nuclear, cytoskeletal, and streptococcal epitopes in children and adults with Tourette's syndrome, Sydenham's chorea, and autoimmune disorders. Biol Psychiatry. (2001) 50:566–77. doi: 10.1016/S0006-3223(01)01096-4
53. Muller N, Kroll B, Schwarz MJ, Riedel M, Straube A, Lutticken R, et al. Increased titers of antibodies against streptococcal M12 and M19 proteins in patients with Tourette's syndrome. Psychiatry Res. (2001) 101:187–93. doi: 10.1016/S0165-1781(01)00215-3
54. Ebrahimi Taj F, Noorbakhsh S, Ghavidel Darestani S, Shirazi E, Javadinia S. Group A beta-hemolytic streptococcal infection in children and the resultant neuro-psychiatric disorder; a cross sectional study; Tehran, Iran. Basic Clin Neurosci. (2015) 6:38–43.
55. Mell LK, Davis RL, Owens D. Association between streptococcal infection and obsessive-compulsive disorder, Tourette's syndrome, and tic disorder. Pediatrics. (2005) 116:56–60. doi: 10.1542/peds.2004-2058
56. Leslie DL, Kozma L, Martin A, Landeros A, Katsovich L, King RA, et al. Neuropsychiatric disorders associated with streptococcal infection: a case-control study among privately insured children. J Am Acad Child Adolesc Psychiatry. (2008) 47:1166–72. doi: 10.1097/CHI.0b013e3181825a3d
57. Orlovska S, Vestergaard CH, Bech BH, Nordentoft M, Vestergaard M, Benros ME. Association of streptococcal throat infection with mental disorders: testing key aspects of the PANDAS hypothesis in a nationwide study. JAMA Psychiatry. (2017) 74:740–6. doi: 10.1001/jamapsychiatry.2017.0995
58. Schrag A, Gilbert R, Giovannoni G, Robertson MM, Metcalfe C, Ben-Shlomo Y. Streptococcal infection, Tourette syndrome, and OCD: is there a connection? Neurology. (2009) 73:1256–63. doi: 10.1212/WNL.0b013e3181bd10fd
59. Krause D, Matz J, Weidinger E, Wagner J, Wildenauer A, Obermeier M, et al. Association between intracellular infectious agents and Tourette's syndrome. Eur Arch Psychiatry Clin Neurosci. (2010) 260:359–63. doi: 10.1007/s00406-009-0084-3
60. Tsai CS, Yang YH, Huang KY, Lee Y, Mcintyre RS, Chen VC. Association of Tic disorders and enterovirus infection: a Nationwide Population-Based Study. Medicine. (2016) 95:e3347. doi: 10.1097/MD.0000000000003347
61. Kohler-Forsberg O, Petersen L, Gasse C, Mortensen PB, Dalsgaard S, Yolken RH, et al. A nationwide study in denmark of the association between treated infections and the subsequent risk of treated mental disorders in children and adolescents. JAMA Psychiatry. (2019) 76:271–9. doi: 10.1001/jamapsychiatry.2018.3428
62. Luo F, Leckman JF, Katsovich L, Findley D, Grantz H, Tucker DM, et al. Prospective longitudinal study of children with tic disorders and/or obsessive-compulsive disorder: relationship of symptom exacerbations to newly acquired streptococcal infections. Pediatrics. (2004) 113:e578–585. doi: 10.1542/peds.113.6.e578
63. Leckman JF, King RA, Gilbert DL, Coffey BJ, Singer HS, Dure LST, et al. Streptococcal upper respiratory tract infections and exacerbations of tic and obsessive-compulsive symptoms: a prospective longitudinal study. J Am Acad Child Adolesc Psychiatry. (2011) 50:108–18 e103. doi: 10.1016/j.jaac.2010.10.011
64. Martino D, Chiarotti F, Buttiglione M, Cardona F, Creti R, Nardocci N, et al. The relationship between group A streptococcal infections and Tourette syndrome: a study on a large service-based cohort. Dev Med Child Neurol. (2011) 53:951–7. doi: 10.1111/j.1469-8749.2011.04018.x
65. Martino D, Schrag A, Anastasiou Z, et al. Group A Streptococcus and exacerbations of chronic tic disorders: a large prospective cohort study. submitted (2020).
66. Kurlan R, Johnson D, Kaplan EL, Tourette Syndrome Study G. Streptococcal infection and exacerbations of childhood tics and obsessive-compulsive symptoms: a prospective blinded cohort study. Pediatrics. (2008) 121:1188–97. doi: 10.1542/peds.2007-2657
67. Ercan TE, Ercan G, Severge B, Arpaozu M, Karasu G. Mycoplasma pneumoniae infection and obsessive-compulsive disease: a case report. J Child Neurol. (2008) 23:338–40. doi: 10.1177/0883073807308714
68. Yaramis A, Herguner S, Kara B, Tatli B, Tuzun U, Ozmen M. Cerebral vasculitis and obsessive-compulsive disorder following varicella infection in childhood. Turk J Pediatr. (2009) 51:72–5.
69. Miman O, Mutlu EA, Ozcan O, Atambay M, Karlidag R, Unal S. Is there any role of Toxoplasma gondii in the etiology of obsessive-compulsive disorder? Psychiatry Res. (2010) 177:263–5. doi: 10.1016/j.psychres.2009.12.013
70. Flegr J, Horacek J. Toxoplasma-infected subjects report an Obsessive-Compulsive Disorder diagnosis more often and score higher in Obsessive-Compulsive Inventory. Eur Psychiatry. (2017) 40:82–7. doi: 10.1016/j.eurpsy.2016.09.001
71. Akaltun I, Kara SS, Kara T. The relationship between Toxoplasma gondii IgG antibodies and generalized anxiety disorder and obsessive-compulsive disorder in children and adolescents: a new approach. Nord J Psychiatry. (2018) 72:57–62. doi: 10.1080/08039488.2017.1385850
72. Westwell-Roper C, Williams KA, Samuels J, Bienvenu OJ, Cullen B, Goes FS, et al. Immune-related comorbidities in childhood-onset obsessive compulsive disorder: lifetime prevalence in the obsessive compulsive disorder collaborative genetics association study. J Child Adolesc Psychopharmacol. (2019) 29:615–24. doi: 10.1089/cap.2018.0140
73. Lin H, Williams KA, Katsovich L, Findley DB, Grantz H, Lombroso PJ, et al. Streptococcal upper respiratory tract infections and psychosocial stress predict future tic and obsessive-compulsive symptom severity in children and adolescents with Tourette syndrome and obsessive-compulsive disorder. Biol Psychiatry. (2010) 67:684–91. doi: 10.1016/j.biopsych.2009.08.020
74. Martino D, Macerollo A, Leckman JF. Neuroendocrine aspects of Tourette syndrome. Int Rev Neurobiol. (2013) 112:239–79. doi: 10.1016/B978-0-12-411546-0.00009-3
75. Rand KM, Austin NC, Inder TE, Bora S, Woodward LJ. neonatal infection and later neurodevelopmental risk in the very preterm infant. J Pediatr. (2016) 170:97–104. doi: 10.1016/j.jpeds.2015.11.017
76. Allred EN, Dammann O, Fichorova RN, Hooper SR, Hunter SJ, Joseph RM, et al. Systemic inflammation during the first postnatal month and the risk of attention deficit hyperactivity disorder characteristics among 10 year-old Children Born Extremely Preterm. J Neuroimmune Pharmacol. (2017) 12:531–43. doi: 10.1007/s11481-017-9742-9
77. Hadzic E, Sinanovic O, Memisevic H. Is bacterial meningitis a risk factor for developing attention deficit hyperactivity disorder. Isr J Psychiatry Relat Sci. (2017) 54:54–7.
78. Chou IC, Lin CC, Kao CH. Enterovirus encephalitis increases the risk of attention deficit hyperactivity disorder: a Taiwanese Population-based Case-control Study. Medicine. (2015) 94:e707. doi: 10.1097/MD.0000000000000707
79. Bekdas M, Tufan AE, Hakyemez IN, Tas T, Altunhan H, Demircioglu F, et al. Subclinical immune reactions to viral infections may correlate with child and adolescent diagnosis of attention-deficit/hyperactivity disorder: a preliminary study from Turkey. Afr Health Sci. (2014) 14:439–45. doi: 10.4314/ahs.v14i2.21
80. Karachaliou M, Chatzi L, Roumeliotaki T, Kampouri M, Kyriklaki A, Koutra K, et al. Common infections with polyomaviruses and herpesviruses and neuropsychological development at 4 years of age, the Rhea birth cohort in Crete, Greece. J Child Psychol Psychiatry. (2016) 57:1268–76. doi: 10.1111/jcpp.12582
81. Lavebratt C, Yang LL, Giacobini M, Forsell Y, Schalling M, Partonen T, et al. Early exposure to antibiotic drugs and risk for psychiatric disorders: a population-based study. Transl Psychiatry. (2019) 9:317. doi: 10.1038/s41398-019-0653-9
82. Leckman JF, Katsovich L, Kawikova I, Lin H, Zhang H, Kronig H, et al. Increased serum levels of interleukin-12 and tumor necrosis factor-alpha in Tourette's syndrome. Biol Psychiatry. (2005) 57:667–73. doi: 10.1016/j.biopsych.2004.12.004
83. Matz J, Krause DL, Dehning S, Riedel M, Gruber R, Schwarz MJ, et al. Altered monocyte activation markers in Tourette's syndrome: a case-control study. BMC Psychiatry. (2012) 12:29. doi: 10.1186/1471-244X-12-29
84. Bos-Veneman NG, Olieman R, Tobiasova Z, Hoekstra PJ, Katsovich L, Bothwell AL, et al. Altered immunoglobulin profiles in children with Tourette syndrome. Brain Behav Immun. (2011) 25:532–8. doi: 10.1016/j.bbi.2010.12.003
85. Gabbay V, Coffey BJ, Guttman LE, Gottlieb L, Katz Y, Babb JS, et al. A cytokine study in children and adolescents with Tourette's disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2009) 33:967–71. doi: 10.1016/j.pnpbp.2009.05.001
86. Cheng YH, Zheng Y, He F, Yang JH, Li WB, Wang ML, et al. Detection of autoantibodies and increased concentrations of interleukins in plasma from patients with Tourette's syndrome. J Mol Neurosci. (2012) 48:219–24. doi: 10.1007/s12031-012-9811-8
87. Hoekstra PJ, Anderson GM, Troost PW, Kallenberg CG, Minderaa RB. Plasma kynurenine and related measures in tic disorder patients. Eur Child Adolesc Psychiatry. (2007) 16 Suppl 1:71–7. doi: 10.1007/s00787-007-1009-1
88. Weidinger E, Krause D, Wildenauer A, Meyer S, Gruber R, Schwarz MJ, et al. Impaired activation of the innate immune response to bacterial challenge in Tourette syndrome. World J Biol Psychiatry. (2014) 15:453–8. doi: 10.3109/15622975.2014.907503
89. Kawikova I, Leckman JF, Kronig H, Katsovich L, Bessen DE, Ghebremichael M, et al. Decreased numbers of regulatory T cells suggest impaired immune tolerance in children with tourette syndrome: a preliminary study. Biol Psychiatry. (2007) 61:273–8. doi: 10.1016/j.biopsych.2006.06.012
90. Tian Y, Gunther JR, Liao IH, Liu D, Ander BP, Stamova BS, et al. GABA- and acetylcholine-related gene expression in blood correlate with tic severity and microarray evidence for alternative splicing in Tourette syndrome: a pilot study. Brain Res. (2011) 1381:228–36. doi: 10.1016/j.brainres.2011.01.026
91. Tian Y, Liao IH, Zhan X, Gunther JR, Ander BP, Liu D, et al. Exon expression and alternatively spliced genes in Tourette Syndrome. Am J Med Genet B Neuropsychiatr Genet. (2011) 156B:72–8. doi: 10.1002/ajmg.b.31140
92. Gunther J, Tian Y, Stamova B, Lit L, Corbett B, Ander B, et al. Catecholamine-related gene expression in blood correlates with tic severity in tourette syndrome. Psychiatry Res. (2012) 200:593–601. doi: 10.1016/j.psychres.2012.04.034
93. Ferrari M, Termine C, Franciotta D, Castiglioni E, Pagani A, Lanzi G, et al. Dopaminergic receptor D5 mRNA expression is increased in circulating lymphocytes of Tourette syndrome patients. J Psychiatr Res. (2008) 43:24–9. doi: 10.1016/j.jpsychires.2008.01.014
94. Martino D, Defazio G, Church AJ, Dale RC, Giovannoni G, Robertson MM, et al. Antineuronal antibody status and phenotype analysis in Tourette's syndrome. Mov Disord. (2007) 22:1424–9. doi: 10.1002/mds.21454
95. Martino D, Draganski B, Cavanna A, Church A, Defazio G, Robertson MM, et al. Anti-basal ganglia antibodies and Tourette's syndrome: a voxel-based morphometry and diffusion tensor imaging study in an adult population. J Neurol Neurosurg Psychiatry. (2008) 79:820–2. doi: 10.1136/jnnp.2007.136689
96. Yeh CB, Shui HA, Chu TH, Chen YA, Tsung HC, Shyu JF. Hyperpolarisation-activated cyclic nucleotide channel 4 (HCN4) involvement in Tourette's syndrome autoimmunity. J Neuroimmunol. (2012) 250:18–26. doi: 10.1016/j.jneuroim.2012.05.009
97. Kawikova I, Grady BP, Tobiasova Z, Zhang Y, Vojdani A, Katsovich L, et al. Children with Tourette's syndrome may suffer immunoglobulin A dysgammaglobulinemia: preliminary report. Biol Psychiatry. (2010) 67:679–83. doi: 10.1016/j.biopsych.2009.09.034
98. Morris-Berry CM, Pollard M, Gao S, Thompson C, Tourette Syndrome Study G, Singer HS. Anti-streptococcal, tubulin, and dopamine receptor 2 antibodies in children with PANDAS and Tourette syndrome: single-point and longitudinal assessments. J Neuroimmunol. (2013) 264:106–13. doi: 10.1016/j.jneuroim.2013.09.010
99. Singer HS, Giuliano JD, Hansen BH, Hallett JJ, Laurino JP, Benson M, et al. Antibodies against a neuron-like (HTB-10 neuroblastoma) cell in children with Tourette syndrome. Biol Psychiatry. (1999) 46:775–80. doi: 10.1016/S0006-3223(98)00384-9
100. Kirvan CA, Swedo SE, Snider LA, Cunningham MW. Antibody-mediated neuronal cell signaling in behavior and movement disorders. J Neuroimmunol. (2006) 179:173–9. doi: 10.1016/j.jneuroim.2006.06.017
101. Brilot F, Merheb V, Ding A, Murphy T, Dale RC. Antibody binding to neuronal surface in Sydenham chorea, but not in PANDAS or Tourette syndrome. Neurology. (2011) 76:1508–13. doi: 10.1212/WNL.0b013e3182181090
102. Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TK, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain. (2012) 135:3453–68. doi: 10.1093/brain/aws256
103. Addabbo F, Baglioni V, Schrag A, Schwarz MJ, Dietrich A, Hoekstra PJ, et al. Anti-dopamine D2 receptor antibodies in chronic tic disorders: potential link to fluctuations of tic severity. Dev Med Child Neurol. (2020) 62:1205–12. doi: 10.1111/dmcn.14613
104. Sühs KW, Skripuletz T, Pul R, Alvermann S, Schwenkenbecher P, Stangel M, et al. Gilles de la Tourette syndrome is not linked to contactinassociated protein receptor 2 antibodies. Mol Brain. (2015) 8:62. doi: 10.1186/s13041-015-0154-6
105. Baglioni V, Coutinho E, Menassa DA, Giannoccaro MP, Jacobson L, Buttiglione M, et al. Antibodies to neuronal surface proteins in Tourette syndrome: lack of evidence in a European paediatric cohort. Brain Behav Immun. (2019) 81:665–9. doi: 10.1016/j.bbi.2019.08.008
106. Baumgaertel C, Skripuletz T, Kronenberg J, Stangel M, Schwenkenbecher P, Sinke C, et al. Immunity in gilles de la Tourette-syndrome: results from a cerebrospinal fluid study. Front Neurol. (2019) 10:732. doi: 10.3389/fneur.2019.00732
107. Hong JJ, Loiselle CR, Yoon DY, Lee O, Becker KG, Singer HS. Microarray analysis in Tourette syndrome postmortem putamen. J Neurol Sci. (2004) 225:57–64. doi: 10.1016/j.jns.2004.06.019
108. Morer A, Chae W, Henegariu O, Bothwell AL, Leckman JF, Kawikova I. Elevated expression of MCP-1, IL-2 and PTPR-N in basal ganglia of Tourette syndrome cases. Brain Behav Immun. (2010) 24:1069–73. doi: 10.1016/j.bbi.2010.02.007
109. Lennington JB, Coppola G, Kataoka-Sasaki Y, Fernandez TV, Palejev D, Li Y, et al. Transcriptome analysis of the human striatum in Tourette syndrome. Biol Psychiatry. (2016) 79:372–82. doi: 10.1016/j.biopsych.2014.07.018
110. Cosco TD, Pillinger T, Emam H, Solmi M, Budhdeo S, Matthew Prina A, et al. Immune aberrations in obsessive-compulsive disorder: a systematic review and meta-analysis. Mol Neurobiol. (2019) 56:4751–9. doi: 10.1007/s12035-018-1409-x
111. Karaguzel EO, Arslan FC, Uysal EK, Demir S, Aykut DS, Tat M, et al. Blood levels of interleukin-1 beta, interleukin-6 and tumor necrosis factor-alpha and cognitive functions in patients with obsessive compulsive disorder. Compr Psychiatry. (2019) 89:61–6. doi: 10.1016/j.comppsych.2018.11.013
112. Gariup M, Gonzalez A, Lazaro L, Torres F, Serra-Pages C, Morer A. IL-8 and the innate immunity as biomarkers in acute child and adolescent psychopathology. Psychoneuroendocrinology. (2015) 62:233–42. doi: 10.1016/j.psyneuen.2015.08.017
113. Williams K, Shorser-Gentile L, Sarvode Mothi S, Berman N, Pasternack M, Geller D, et al. Immunoglobulin A dysgammaglobulinemia is associated with pediatric-onset obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. (2019) 29:268–75. doi: 10.1089/cap.2018.0043
114. Rodriguez N, Morer A, Gonzalez-Navarro EA, Serra-Pages C, Boloc D, Torres T, et al. Altered frequencies of Th17 and Treg cells in children and adolescents with obsessive-compulsive disorder. Brain Behav Immun. (2019) 81:608–16. doi: 10.1016/j.bbi.2019.07.022
115. Kant R, Pasi S, Surolia A. Auto-reactive Th17-cells trigger obsessive-compulsive-disorder like behavior in mice with experimental autoimmune encephalomyelitis. Front Immunol. (2018) 9:2508. doi: 10.3389/fimmu.2018.02508
116. Yazici KU, Percinel Yazici I, Ustundag B. Vitamin D levels in children and adolescents with obsessive compulsive disorder. Nord J Psychiatry. (2018) 72:173–8. doi: 10.1080/08039488.2017.1406985
117. Swedo SE, Leonard HL, Garvey M, Mittleman B, Allen AJ, Perlmutter S, et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry. (1998) 155:264–71. doi: 10.1176/ajp.155.2.26410.1176/ajp.155.2.264
118. Ben-Pazi H, Stoner JA, Cunningham MW. Dopamine receptor autoantibodies correlate with symptoms in Sydenham's chorea. PLoS ONE. (2013) 8:e73516. doi: 10.1371/journal.pone.0073516
119. Cox CJ, Zuccolo AJ, Edwards EV, Mascaro-Blanco A, Alvarez K, Stoner J, et al. Antineuronal antibodies in a heterogeneous group of youth and young adults with tics and obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. (2015) 25:76–85. doi: 10.1089/cap.2014.0048
120. Kirvan CA, Cox CJ, Swedo SE, Cunningham MW. Tubulin is a neuronal target of autoantibodies in Sydenham's chorea. J Immunol. (2007) 178:7412–21. doi: 10.4049/jimmunol.178.11.7412
121. Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med. (2003) 9:914–20. doi: 10.1038/nm892
122. Cox CJ, Sharma M, Leckman JF, Zuccolo J, Zuccolo A, Kovoor A, et al. Brain human monoclonal autoantibody from sydenham chorea targets dopaminergic neurons in transgenic mice and signals dopamine D2 receptor: implications in human disease. J Immunol. (2013) 191:5524–41. doi: 10.4049/jimmunol.1102592
123. Cunningham MW, Cox CJ. Autoimmunity against dopamine receptors in neuropsychiatric and movement disorders: a review of Sydenham chorea and beyond. Acta Physiol. (2016) 216:90–100. doi: 10.1111/apha.12614
124. Xu J, Liu R-J, Fahey S, Frick LR, Leckman JF, Vaccarino FM, et al. Antibodies from children with PANDAS bind specifically to striatal cholinergic interneurons and alter their activity. Am J Psychiatry. (2020). doi: 10.1176/appi.ajp.2020.19070698. [Epub ahead of print].
125. Dale RC, Heyman I, Giovannoni G, Church AW. Incidence of anti-brain antibodies in children with obsessive-compulsive disorder. Br J Psychiatry. (2005) 187:314–9. doi: 10.1192/bjp.187.4.314
126. Morer A, Lazaro L, Sabater L, Massana J, Castro J, Graus F. Antineuronal antibodies in a group of children with obsessive-compulsive disorder and Tourette syndrome. J Psychiatr Res. (2008) 42:64–8. doi: 10.1016/j.jpsychires.2006.09.010
127. Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R, Shankar SK. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology. (2009) 34:2489–96. doi: 10.1038/npp.2009.77
128. Nicholson TR, Ferdinando S, Krishnaiah RB, Anhoury S, Lennox BR, Mataix-Cols D, et al. Prevalence of anti-basal ganglia antibodies in adult obsessive-compulsive disorder: cross-sectional study. Br J Psychiatry. (2012) 200:381–6. doi: 10.1192/bjp.bp.111.092007
129. Gause C, Morris C, Vernekar S, Pardo-Villamizar C, Grados MA, Singer HS. Antineuronal antibodies in OCD: comparisons in children with OCD-only, OCD+chronic tics and OCD+PANDAS. J Neuroimmunol. (2009) 214:118–24. doi: 10.1016/j.jneuroim.2009.06.015
130. Maina G, Albert U, Bogetto F, Borghese C, Berro AC, Mutani R, et al. Anti-brain antibodies in adult patients with obsessive-compulsive disorder. J Affect Disord. (2009) 116:192–200. doi: 10.1016/j.jad.2008.11.019
131. Pearlman DM, Vora HS, Marquis BG, Najjar S, Dudley LA. Anti-basal ganglia antibodies in primary obsessive-compulsive disorder: systematic review and meta-analysis. Br J Psychiatry. (2014) 205:8–16. doi: 10.1192/bjp.bp.113.137018
132. Attwells S, Setiawan E, Wilson AA, Rusjan PM, Mizrahi R, Miler L, et al. Inflammation in the neurocircuitry of obsessive-compulsive disorder. JAMA Psychiatry. (2017) 74:833–40. doi: 10.1001/jamapsychiatry.2017.1567
133. Oades RD, Dauvermann MR, Schimmelmann BG, Schwarz MJ, Myint AM. Attention-deficit hyperactivity disorder (ADHD) and glial integrity: S100B, cytokines and kynurenine metabolism–effects of medication. Behav Brain Funct. (2010) 6:29. doi: 10.1186/1744-9081-6-29
134. Oades RD, Myint AM, Dauvermann MR, Schimmelmann BG, Schwarz MJ. Attention-deficit hyperactivity disorder (ADHD) and glial integrity: an exploration of associations of cytokines and kynurenine metabolites with symptoms and attention. Behav Brain Funct. (2010) 6:32. doi: 10.1186/1744-9081-6-32
135. Reinehr T, Langrock C, Hamelmann E, Lucke T, Koerner-Rettberg C, Holtmann M, et al. 25-Hydroxvitamin D concentrations are not lower in children with bronchial asthma, atopic dermatitis, obesity, or attention-deficient/hyperactivity disorder than in healthy children. Nutr Res. (2018) 52:39–47. doi: 10.1016/j.nutres.2018.01.002
136. Saedisomeolia A, Samadi M, Gholami F, Seyedi M, Effatpanah M, Hashemi R, et al. Vitamin D's molecular action mechanism in attention-deficit/ hyperactivity disorder: a review of evidence. CNS Neurol Disord Drug Targets. (2018) 17:280–90. doi: 10.2174/1871527317666180501111627
137. Verlaet AJ, Breynaert A, Ceulemans B, De Bruyne T, Fransen E, Hermans N. Oxidative stress and immune aberrancies in attention-deficit/hyperactivity disorder (ADHD): a case-control comparison. Eur Child Adolesc Psychiatry. (2019) 28, 719–729. doi: 10.1007/s00787-018-1239-4
138. Aarsland TI, Landaas ET, Hegvik TA, Ulvik A, Halmoy A, Ueland PM, et al. Serum concentrations of kynurenines in adult patients with attention-deficit hyperactivity disorder (ADHD): a case-control study. Behav Brain Funct. (2015) 11:36. doi: 10.1186/s12993-015-0080-x
139. Corominas-Roso M, Armario A, Palomar G, Corrales M, Carrasco J, Richarte V, et al. IL-6 and TNF-alpha in unmedicated adults with ADHD: relationship to cortisol awakening response. Psychoneuroendocrinology. (2017) 79:67–73. doi: 10.1016/j.psyneuen.2017.02.017
140. Donfrancesco R, Nativio P, Borrelli E, Giua E, Andriola E, Villa MP, et al. Serum cytokines in paediatric neuropsychiatric syndromes: focus on Attention Deficit Hyperactivity Disorder. Minerva Pediatr. (2016).
141. Darwish AH, Elgohary TM, Nosair NA. Serum interleukin-6 level in children with Attention-Deficit Hyperactivity Disorder (ADHD). J Child Neurol. (2019) 34:61–7. doi: 10.1177/0883073818809831
142. O'shea TM, Joseph RM, Kuban KC, Allred EN, Ware J, Coster T, et al. Elevated blood levels of inflammation-related proteins are associated with an attention problem at age 24 mo in extremely preterm infants. Pediatr Res. (2014) 75:781–7. doi: 10.1038/pr.2014.41
143. Buske-Kirschbaum A, Schmitt J, Plessow F, Romanos M, Weidinger S, Roessner V. Psychoendocrine and psychoneuroimmunological mechanisms in the comorbidity of atopic eczema and attention deficit/hyperactivity disorder. Psychoneuroendocrinology. (2013) 38:12–23. doi: 10.1016/j.psyneuen.2012.09.017
144. Passarelli F, Donfrancesco R, Nativio P, Pascale E, Di Trani M, Patti AM, et al. Anti-Purkinje cell antibody as a biological marker in attention deficit/hyperactivity disorder: a pilot study. J Neuroimmunol. (2013) 258:67–70. doi: 10.1016/j.jneuroim.2013.02.018
145. Donfrancesco R, Nativio P, Di Benedetto A, Villa MP, Andriola E, Melegari MG, et al. Anti-yo antibodies in children with ADHD: first results about serum cytokines. J Atten Disord. (2020) 24:1497–502. doi: 10.1177/1087054716643387
146. Toto M, Margari F, Simone M, Craig F, Petruzzelli MG, Tafuri S, et al. Antibasal ganglia antibodies and antistreptolysin O in noncomorbid ADHD. J Atten Disord. (2015) 19:965–70. doi: 10.1177/1087054712455505
147. Giana G, Romano E, Porfirio MC, D'ambrosio R, Giovinazzo S, Troianiello M, et al. Detection of auto-antibodies to DAT in the serum: interactions with DAT genotype and psycho-stimulant therapy for ADHD. J Neuroimmunol. (2015) 278:212–22. doi: 10.1016/j.jneuroim.2014.11.008
148. Adriani W, Romano E, Pucci M, Pascale E, Cerniglia L, Cimino S, et al. Potential for diagnosis versus therapy monitoring of attention deficit hyperactivity disorder: a new epigenetic biomarker interacting with both genotype and auto-immunity. Eur Child Adolesc Psychiatry. (2018) 27:241–52. doi: 10.1007/s00787-017-1040-9
149. Perez-Vigil A, Fernandez De La Cruz L, Brander G, Isomura K, Gromark C, Mataix-Cols D. The link between autoimmune diseases and obsessive-compulsive and tic disorders: a systematic review. Neurosci Biobehav Rev. (2016) 71:542–62. doi: 10.1016/j.neubiorev.2016.09.025
150. Chang YT, Li YF, Muo CH, Chen SC, Chin ZN, Kuo HT, et al. Correlation of Tourette syndrome and allergic disease: nationwide population-based case-control study. J Dev Behav Pediatr. (2011) 32:98–102. doi: 10.1097/DBP.0b013e318208f561
151. Chen MH, Su TP, Chen YS, Hsu JW, Huang KL, Chang WH, et al. Attention deficit hyperactivity disorder, tic disorder, and allergy: is there a link? A nationwide population-based study. J Child Psychol Psychiatry. (2013) 54:545–51. doi: 10.1111/jcpp.12018
152. Yang J, Hirsch L, Osland S, Martino D, Jette N, Roberts JI, et al. Health status, health related behaviours and chronic health indicators in people with Tourette Syndrome: a Canadian population-based study. Psychiatry Res. (2017) 250:228–33. doi: 10.1016/j.psychres.2017.02.002
153. Yuce M, Guner SN, Karabekiroglu K, Baykal S, Kilic M, Sancak R, et al. Association of Tourette syndrome and obsessive-compulsive disorder with allergic diseases in children and adolescents: a preliminary study. Eur Rev Med Pharmacol Sci. (2014) 18:303–10.
154. Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry. (2012) 71:392–402. doi: 10.1016/j.biopsych.2011.09.034
155. Steele M, Rosner J. A possible link between fluticasone propionate and tics in pediatric asthmatics. Can J Neurol Sci. (2012) 39:851. doi: 10.1017/S031716710001578X
156. Schans JV, Cicek R, De Vries TW, Hak E, Hoekstra PJ. Association of atopic diseases and attention-deficit/hyperactivity disorder: a systematic review and meta-analyses. Neurosci Biobehav Rev. (2017) 74:139–48. doi: 10.1016/j.neubiorev.2017.01.011
157. Miyazaki C, Koyama M, Ota E, Swa T, Mlunde LB, Amiya RM, et al. Allergic diseases in children with attention deficit hyperactivity disorder: a systematic review and meta-analysis. BMC Psychiatry. (2017) 17:120. doi: 10.1186/s12888-017-1281-7
158. Chen MH, Su TP, Chen YS, Hsu JW, Huang KL, Chang WH, et al. Is atopy in early childhood a risk factor for ADHD and ASD? A longitudinal study. J Psychosom Res. (2014) 77:316–21. doi: 10.1016/j.jpsychores.2014.06.006
159. Chen MH, Su TP, Chen YS, Hsu JW, Huang KL, Chang WH, et al. Asthma and attention-deficit/hyperactivity disorder: a nationwide population-based prospective cohort study. J Child Psychol Psychiatry. (2013) 54:1208–14. doi: 10.1111/jcpp.12087
160. Chang TH, Tai YH, Dai YX, Chang YT, Chen TJ, Chen MH. Risk of atopic diseases among siblings of patients with attention-deficit hyperactivity disorder: a Nationwide Population-Based Cohort Study. Int Arch Allergy Immunol. (2019) 180:37–43. doi: 10.1159/000500831
161. Pelsser LM, Buitelaar JK, Savelkoul HF. ADHD as a (non) allergic hypersensitivity disorder: a hypothesis. Pediatr Allergy Immunol. (2009) 20:107–12. doi: 10.1111/j.1399-3038.2008.00749.x
162. Chen MH, Su TP, Chen YS, Hsu JW, Huang KL, Chang WH, et al. Comorbidity of allergic and autoimmune diseases among patients with ADHD. J Atten Disord. (2017) 21:219–27. doi: 10.1177/1087054712474686
163. Hegvik TA, Instanes JT, Haavik J, Klungsoyr K, Engeland A. Associations between attention-deficit/hyperactivity disorder and autoimmune diseases are modified by sex: a population-based cross-sectional study. Eur Child Adolesc Psychiatry. (2018) 27:663–75. doi: 10.1007/s00787-017-1056-1
164. Fortunato JJ, Da Rosa N, Martins Laurentino AO, Goulart M, Michalak C, Borges LP, et al. Effects of omega-3 fatty acids on stereotypical behavior and social interactions in Wistar rats prenatally exposed to lipopolysaccarides. Nutrition. (2017) 35:119–27. doi: 10.1016/j.nut.2016.10.019
165. Kirsten TB, Bernardi MM. Prenatal lipopolysaccharide induces hypothalamic dopaminergic hypoactivity and autistic-like behaviors: repetitive self-grooming and stereotypies. Behav Brain Res. (2017) 331:25–9. doi: 10.1016/j.bbr.2017.05.013
166. Graciarena M, Depino AM, Pitossi FJ. Prenatal inflammation impairs adult neurogenesis and memory related behavior through persistent hippocampal TGFbeta1 downregulation. Brain Behav Immun. (2010) 24:1301–9. doi: 10.1016/j.bbi.2010.06.005
167. Li XW, Cao L, Wang F, Yang QG, Tong JJ, Li XY, et al. Maternal inflammation linearly exacerbates offspring age-related changes of spatial learning and memory, and neurobiology until senectitude. Behav Brain Res. (2016) 306:178–96. doi: 10.1016/j.bbr.2016.03.011
168. Schaafsma W, Basterra LB, Jacobs S, Brouwer N, Meerlo P, Schaafsma A, et al. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol Dis. (2017) 106:291–300. doi: 10.1016/j.nbd.2017.07.017
169. Labrousse VF, Leyrolle Q, Amadieu C, Aubert A, Sere A, Coutureau E, et al. Dietary omega-3 deficiency exacerbates inflammation and reveals spatial memory deficits in mice exposed to lipopolysaccharide during gestation. Brain Behav Immun. (2018) 73:427–40. doi: 10.1016/j.bbi.2018.06.004
170. Peng L, Zhu M, Yang Y, Weng Y, Zou W, Zhu X, et al. Neonatal lipopolysaccharide challenge induces long-lasting spatial cognitive impairment and dysregulation of hippocampal histone acetylation in mice. Neuroscience. (2019) 398:76–87. doi: 10.1016/j.neuroscience.2018.12.001
171. Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. (2010) 35:2462–78. doi: 10.1038/npp.2010.129
172. Vuillermot S, Joodmardi E, Perlmann T, Ogren SO, Feldon J, Meyer U. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J Neurosci. (2012) 32:436–51. doi: 10.1523/JNEUROSCI.4831-11.2012
173. Piontkewitz Y, Arad M, Weiner I. Tracing the development of psychosis and its prevention: what can be learned from animal models. Neuropharmacology. (2012) 62:1273–89. doi: 10.1016/j.neuropharm.2011.04.019
174. Vorhees CV, Graham DL, Braun AA, Schaefer TL, Skelton MR, Richtand NM, et al. Prenatal immune challenge in rats: altered responses to dopaminergic and glutamatergic agents, prepulse inhibition of acoustic startle, and reduced route-based learning as a function of maternal body weight gain after prenatal exposure to poly IC. Synapse. (2012) 66:725–37. doi: 10.1002/syn.21561
175. Holloway T, Moreno JL, Umali A, Rayannavar V, Hodes GE, Russo SJ, et al. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: role of maternal immune system. J Neurosci. (2013) 33:1088–98. doi: 10.1523/JNEUROSCI.2331-12.2013
176. Miller VM, Zhu Y, Bucher C, Mcginnis W, Ryan LK, Siegel A, et al. Gestational flu exposure induces changes in neurochemicals, affiliative hormones and brainstem inflammation, in addition to autism-like behaviors in mice. Brain Behav Immun. (2013) 33:153–63. doi: 10.1016/j.bbi.2013.07.002
177. Missault S, Van Den Eynde K, Vanden Berghe W, Fransen E, Weeren A, Timmermans JP, et al. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun. (2014) 42:138–46. doi: 10.1016/j.bbi.2014.06.013
178. Vorhees CV, Graham DL, Braun AA, Schaefer TL, Skelton MR, Richtand NM, et al. Prenatal immune challenge in rats: effects of polyinosinic-polycytidylic acid on spatial learning, prepulse inhibition, conditioned fear, and responses to MK-801 and amphetamine. Neurotoxicol Teratol. (2015) 47:54–65. doi: 10.1016/j.ntt.2014.10.007
179. Giovanoli S, Engler H, Engler A, Richetto J, Feldon J, Riva MA, et al. Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl Psychiatry. (2016) 6:e772. doi: 10.1038/tp.2016.38
180. Da Silveira VT, Medeiros DC, Ropke J, Guidine PA, Rezende GH, Moraes MF, et al. Effects of early or late prenatal immune activation in mice on behavioral and neuroanatomical abnormalities relevant to schizophrenia in the adulthood. Int J Dev Neurosci. (2017) 58:1–8. doi: 10.1016/j.ijdevneu.2017.01.009
181. Bates V, Maharjan A, Millar J, Bilkey DK, Ward RD. Spared motivational modulation of cognitive effort in a maternal immune activation model of schizophrenia risk. Behav Neurosci. (2018) 132:66–74. doi: 10.1037/bne0000230
182. Missig G, Mokler EL, Robbins JO, Alexander AJ, Mcdougle CJ, Carlezon WA Jr. Perinatal immune activation produces persistent sleep alterations and epileptiform activity in male mice. Neuropsychopharmacology. (2018) 43:482–91. doi: 10.1038/npp.2017.243
183. Brimberg L, Benhar I, Mascaro-Blanco A, Alvarez K, Lotan D, Winter C, et al. Behavioral, pharmacological, and immunological abnormalities after streptococcal exposure: a novel rat model of Sydenham chorea and related neuropsychiatric disorders. Neuropsychopharmacology. (2012) 37:2076–87. doi: 10.1038/npp.2012.56
184. Lotan D, Benhar I, Alvarez K, Mascaro-Blanco A, Brimberg L, Frenkel D, et al. Behavioral and neural effects of intra-striatal infusion of anti-streptococcal antibodies in rats. Brain Behav Immun. (2014) 38:249–62. doi: 10.1016/j.bbi.2014.02.009
185. Zhang D, Patel A, Zhu Y, Siegel A, Zalcman SS. Anti-streptococcus IgM antibodies induce repetitive stereotyped movements: cell activation and co-localization with Fcalpha/mu receptors in the striatum and motor cortex. Brain Behav Immun. (2012) 26:521–33. doi: 10.1016/j.bbi.2012.01.005
186. Allard MJ, Bergeron JD, Baharnoori M, Srivastava LK, Fortier LC, Poyart C, et al. A sexually dichotomous, autistic-like phenotype is induced by Group B Streptococcus maternofetal immune activation. Autism Res. (2017) 10:233–45. doi: 10.1002/aur.1647
187. Allard MJ, Brochu ME, Bergeron JD, Sebire G. Hyperactive behavior in female rats in utero-exposed to group B Streptococcus-induced inflammation. Int J Dev Neurosci. (2018) 69:17–22. doi: 10.1016/j.ijdevneu.2018.06.005
188. Allard MJ, Giraud A, Segura M, Sebire G. Sex-specific maternofetal innate immune responses triggered by group B Streptococci. Sci Rep. (2019) 9:8587. doi: 10.1038/s41598-019-45029-x
189. Grados M, Prazak M, Saif A, Halls A. A review of animal models of obsessive-compulsive disorder: a focus on developmental, immune, endocrine and behavioral models. Expert Opin Drug Discov. (2016) 11:27–43. doi: 10.1517/17460441.2016.1103225
190. Mora S, Martin-Gonzalez E, Flores P, Moreno M. Neuropsychiatric consequences of childhood group A streptococcal infection: a systematic review of preclinical models. Brain Behav Immun. (2020) 86:53–62. doi: 10.1016/j.bbi.2019.02.027
191. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. (2010) 141:775–85. doi: 10.1016/j.cell.2010.03.055
192. Trankner D, Boulet A, Peden E, Focht R, Van Deren D, Capecchi M. A microglia sublineage protects from sex-linked anxiety symptoms and obsessive compulsion. Cell Rep. (2019) 29:791–9 e793. doi: 10.1016/j.celrep.2019.09.045
193. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. (2016) 165:921–35. doi: 10.1016/j.cell.2016.04.001
194. Krabbe G, Minami SS, Etchegaray JI, Taneja P, Djukic B, Davalos D, et al. Microglial NFkappaB-TNFalpha hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc Natl Acad Sci USA. (2017) 114:5029–34. doi: 10.1073/pnas.1700477114
195. Dunn GA, Nigg JT, Sullivan EL. Neuroinflammation as a risk factor for attention deficit hyperactivity disorder. Pharmacol Biochem Behav. (2019) 182:22–34. doi: 10.1016/j.pbb.2019.05.005
196. Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. (2005) 48:903–17. doi: 10.1016/j.neuropharm.2004.12.023
197. Graciarena M, Roca V, Mathieu P, Depino AM, Pitossi FJ. Differential vulnerability of adult neurogenesis by adult and prenatal inflammation: role of TGF-beta1. Brain Behav Immun. (2013) 34:17–28. doi: 10.1016/j.bbi.2013.05.007
198. O'loughlin E, Pakan JMP, Yilmazer-Hanke D, Mcdermott KW. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J Neuroinflammation. (2017) 14:212. doi: 10.1186/s12974-017-0981-8
199. Bergeron JD, Deslauriers J, Grignon S, Fortier LC, Lepage M, Stroh T, et al. White matter injury and autistic-like behavior predominantly affecting male rat offspring exposed to group B streptococcal maternal inflammation. Dev Neurosci. (2013) 35:504–15. doi: 10.1159/000355656
200. Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. (2006) 59:546–54. doi: 10.1016/j.biopsych.2005.07.031
201. Giovanoli S, Notter T, Richetto J, Labouesse MA, Vuillermot S, Riva MA, et al. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J Neuroinflammation. (2015) 12:221. doi: 10.1186/s12974-015-0437-y
202. Bronson SL, Bale TL. Prenatal stress-induced increases in placental inflammation and offspring hyperactivity are male-specific and ameliorated by maternal antiinflammatory treatment. Endocrinology. (2014) 155:2635–46. doi: 10.1210/en.2014-1040
203. Kozlowska A, Wojtacha P, Rowniak M, Kolenkiewicz M, Huang ACW. ADHD pathogenesis in the immune, endocrine and nervous systems of juvenile and maturating SHR and WKY rats. Psychopharmacology. (2019) 236:2937–58. doi: 10.1007/s00213-019-5180-0
204. Vaccarino FM, Stevens HE, Kocabas A, Palejev D, Szekely A, Grigorenko EL, et al. Induced pluripotent stem cells: a new tool to confront the challenge of neuropsychiatric disorders. Neuropharmacology. (2011) 60:1355–63. doi: 10.1016/j.neuropharm.2011.02.021
205. Hoffmann A, Ziller M, Spengler D. Progress in iPSC-based modeling of psychiatric disorders. Int J Mol Sci. (2019) 20:4896. doi: 10.3390/ijms20194896
206. Wang Y, Wang Z, Wang Y, Li F, Jia J, Song X, et al. The gut-microglia connection: implications for central nervous system diseases. Front Immunol. (2018) 9:2325. doi: 10.3389/fimmu.2018.02325
207. Abdel-Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. Microbiome-microglia connections via the gut-brain axis. J Exp Med. (2019) 216:41–59. doi: 10.1084/jem.20180794
208. Cryan JF, O'riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. (2019) 99:1877–2013. doi: 10.1152/physrev.00018.2018
209. Ganal-Vonarburg SC, Hornef MW, Macpherson AJ. Microbial-host molecular exchange and its consequences in early mammalian life. Science. (2020) 368:604–7. doi: 10.1126/science.aba0478
210. Matta SM, Hill-Yardin EL, Crack PJ. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav Immun. (2019) 79:75–90. doi: 10.1016/j.bbi.2019.04.037
211. Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA, et al. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron. (2019) 101:246–59 e246. doi: 10.1016/j.neuron.2018.11.018
212. Lacorte E, Gervasi G, Bacigalupo I, Vanacore N, Raucci U, Parisi P. A systematic review of the microbiome in children with neurodevelopmental disorders. Front Neurol. (2019) 10:727. doi: 10.3389/fneur.2019.00727
213. Jiang HY, Zhang X, Yu ZH, Zhang Z, Deng M, Zhao JH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. (2018) 104:130–6. doi: 10.1016/j.jpsychires.2018.07.007
214. Prehn-Kristensen A, Zimmermann A, Tittmann L, Lieb W, Schreiber S, Baving L, et al. Reduced microbiome alpha diversity in young patients with ADHD. PLoS ONE. (2018) 13:e0200728. doi: 10.1371/journal.pone.0200728
215. Wang LJ, Yang CY, Chou WJ, Lee MJ, Chou MC, Kuo HC, et al. Gut microbiota and dietary patterns in children with attention-deficit/hyperactivity disorder. Eur Child Adolesc Psychiatry. (2020) 29:287–97. doi: 10.1007/s00787-019-01352-2
216. Wan L, Ge WR, Zhang S, Sun YL, Wang B, Yang G. Case-control study of the effects of gut microbiota composition on neurotransmitter metabolic pathways in children with attention deficit hyperactivity disorder. Front Neurosci. (2020) 14:127. doi: 10.3389/fnins.2020.00127
217. Aarts E, Ederveen THA, Naaijen J, Zwiers MP, Boekhorst J, Timmerman HM, et al. Gut microbiome in ADHD and its relation to neural reward anticipation. PLoS ONE. (2017) 12:e0183509. doi: 10.1371/journal.pone.0183509
218. Szopinska-Tokov J, Dam S, Naaijen J, Konstanti P, Rommelse N, Belzer C, et al. Investigating the gut microbiota composition of individuals with attention-deficit/hyperactivity disorder and association with symptoms. Microorganisms. (2020) 8:406. doi: 10.3390/microorganisms8030406
219. Stevens AJ, Purcell RV, Darling KA, Eggleston MJF, Kennedy MA, Rucklidge JJ. Human gut microbiome changes during a 10 week Randomised Control Trial for micronutrient supplementation in children with attention deficit hyperactivity disorder. Sci Rep. (2019) 9:10128. doi: 10.1038/s41598-019-46146-3
220. Tengeler AC, Dam SA, Wiesmann M, Naaijen J, Van Bodegom M, Belzer C, et al. Gut microbiota from persons with attention-deficit/hyperactivity disorder affects the brain in mice. Microbiome. (2020) 8:44. doi: 10.1186/s40168-020-00816-x
221. Cassidy-Bushrow AE, Sitarik AR, Johnson-Hooper TM, Phillips JM, Jones K, Johnson CC, et al. Prenatal pet keeping and caregiver-reported attention deficit hyperactivity disorder through preadolescence in a United States birth cohort. BMC Pediatr. (2019) 19:390. doi: 10.1186/s12887-019-1719-9
222. Slykerman RF, Coomarasamy C, Wickens K, Thompson JMD, Stanley TV, Barthow C, et al. Exposure to antibiotics in the first 24 months of life and neurocognitive outcomes at 11 years of age. Psychopharmacology. (2019) 236:1573–82. doi: 10.1007/s00213-019-05216-0
223. Quagliariello A, Del Chierico F, Russo A, Reddel S, Conte G, Lopetuso LR, et al. Gut microbiota profiling and gut-brain crosstalk in children affected by pediatric acute-onset neuropsychiatric syndrome and pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Front Microbiol. (2018) 9:675. doi: 10.3389/fmicb.2018.00675
224. Jung TD, Jung PS, Raveendran L, Farbod Y, Dvorkin-Gheva A, Sakic B, et al. Changes in gut microbiota during development of compulsive checking and locomotor sensitization induced by chronic treatment with the dopamine agonist quinpirole. Behav Pharmacol. (2018) 29:211–24. doi: 10.1097/FBP.0000000000000363
225. Scheepers IM, Cryan JF, Bastiaanssen TFS, Rea K, Clarke G, Jaspan HB, et al. Natural compulsive-like behaviour in the deer mouse (Peromyscus maniculatus bairdii) is associated with altered gut microbiota composition. Eur J Neurosci. (2020) 51:1419–27. doi: 10.1111/ejn.14610
226. Williams KA, Swedo SE, Farmer CA, Grantz H, Grant PJ, D'souza P, et al. Randomized, controlled trial of intravenous immunoglobulin for pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. J Am Acad Child Adolesc Psychiatry. (2016) 55:860–7 e862. doi: 10.1016/j.jaac.2016.06.017
227. Shalbafan M, Mohammadinejad P, Shariat SV, Alavi K, Zeinoddini A, Salehi M, et al. Celecoxib as an adjuvant to fluvoxamine in moderate to severe obsessive-compulsive disorder: a double-blind, placebo-controlled, randomized trial. Pharmacopsychiatry. (2015) 48:136–40. doi: 10.1055/s-0035-1549929
228. Murphy TK, Parker-Athill EC, Lewin AB, Storch EA, Mutch PJ. Cefdinir for recent-onset pediatric neuropsychiatric disorders: a pilot randomized trial. J Child Adolesc Psychopharmacol. (2015) 25:57–64. doi: 10.1089/cap.2014.0010
229. Esalatmanesh S, Abrishami Z, Zeinoddini A, Rahiminejad F, Sadeghi M, Najarzadegan MR, et al. Minocycline combination therapy with fluvoxamine in moderate-to-severe obsessive-compulsive disorder: A placebo-controlled, double-blind, randomized trial. Psychiatry Clin Neurosci. (2016) 70:517–26. doi: 10.1111/pcn.12430
230. Rodriguez CI, Bender JJr, Marcus SM, Snape M, Rynn M, Simpson HB. Minocycline augmentation of pharmacotherapy in obsessive-compulsive disorder: an open-label trial. J Clin Psychiatry. (2010) 71:1247–9. doi: 10.4088/JCP.09l05805blu
231. Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. (2019) 570:523–7. doi: 10.1038/s41586-019-1289-x
232. Mansour AA, Goncalves JT, Bloyd CW, Li H, Fernandes S, Quang D, et al. An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol. (2018) 36:432–41. doi: 10.1038/nbt.4127
233. Smolders S, Notter T, Smolders SMT, Rigo JM, Brone B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun. (2018) 73:51–65. doi: 10.1016/j.bbi.2018.06.001
234. Cowan M, Petri WA Jr. Microglia: immune regulators of neurodevelopment. Front Immunol. (2018) 9:2576. doi: 10.3389/fimmu.2018.02576
235. Lebovitz Y, Ringel-Scaia VM, Allen IC, Theus MH. Emerging developments in microbiome and microglia research: implications for neurodevelopmental disorders. Front Immunol. (2018) 9:1993. doi: 10.3389/fimmu.2018.01993
236. Gumusoglu SB, Fine RS, Murray SJ, Bittle JL, Stevens HE. The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav Immun. (2017) 65:274–83. doi: 10.1016/j.bbi.2017.05.015
237. Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa-Mattioli M. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell. (2016) 165:1762–75. doi: 10.1016/j.cell.2016.06.001
238. Atladottir HO, Parner ET, Schendel D, Dalsgaard S, Thomsen PH, Thorsen P. Variation in incidence of neurodevelopmental disorders with season of birth. Epidemiology. (2007) 18:240–5. doi: 10.1097/01.ede.0000254064.92806.13
239. Howerton CL, Bale TL. Prenatal programing: at the intersection of maternal stress and immune activation. Horm Behav. (2012) 62:237–42. doi: 10.1016/j.yhbeh.2012.03.007
240. Minakova E, Warner BB. Maternal immune activation, central nervous system development and behavioral phenotypes. Birth Defects Res. (2018) 110:1539–50. doi: 10.1002/bdr2.1416
241. Bombaci M, Grifantini R, Mora M, Reguzzi V, Petracca R, Meoni E, et al. Protein array profiling of tic patient sera reveals a broad range and enhanced immune response against Group A Streptococcus antigens. PLoS ONE. (2009) 4:e6332. doi: 10.1371/journal.pone.0006332
242. Kéri S, Szabó C, Kelemen O. Expression of Toll-Like Receptors in peripheral blood mononuclear cells and response to cognitive-behavioral therapy in major depressive disorder. Brain Behav Immun. (2014) 40:235–43. doi: 10.1016/j.bbi.2014.03.020
243. Pearlstein JG, Staudenmaier PJ, West AE, Geraghty S, Cosgrove VE. Immune response to stress induction as a predictor of cognitive-behavioral therapy outcomes in adolescent mood disorders: a pilot study. J Psychiatr Res. (2020) 120:56–63. doi: 10.1016/j.jpsychires.2019.10.012
244. Romero-Sanchiz P, Nogueira-Arjona R, Araos P, et al. Variation in chemokines plasma concentrations in primary care depressed patients associated with Internet-based cognitive-behavioral therapy. Sci Rep. (2020) 10:1078. doi: 10.1038/s41598-020-57967-y
Keywords: Tourette, obsessive-compulsive disorder, attention deficit hyperactivity disorder, immunology, neuroinflammation, microbiome, microglia, neural organoids
Citation: Martino D, Johnson I and Leckman JF (2020) What Does Immunology Have to Do With Normal Brain Development and the Pathophysiology Underlying Tourette Syndrome and Related Neuropsychiatric Disorders? Front. Neurol. 11:567407. doi: 10.3389/fneur.2020.567407
Received: 29 May 2020; Accepted: 20 August 2020;
Published: 16 September 2020.
Edited by:
Takashi Morishita, Fukuoka University, JapanReviewed by:
Joseph McGuire, Johns Hopkins University, United StatesAtsushi Ishii, Fukuoka University, Japan
Copyright © 2020 Martino, Johnson and Leckman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James F. Leckman, amFtZXMubGVja21hbkB5YWxlLmVkdQ==