 Maria Masnata1,2†Shireen Salem1,3†Aurelie de Rus Jacquet1,2
Maria Masnata1,2†Shireen Salem1,3†Aurelie de Rus Jacquet1,2 Mehwish Anwer1,2
Mehwish Anwer1,2 Francesca Cicchetti1,2,3*
Francesca Cicchetti1,2,3*- 1Centre de Recherche du CHU de Québec, Axe Neurosciences, Québec, QC, Canada
- 2Département de Psychiatrie & Neurosciences, Université Laval, Québec, QC, Canada
- 3Département de Médecine Moléculaire, Université Laval, Québec, QC, Canada
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder characterized by severe motor, cognitive and psychiatric impairments. While motor deficits often confirm diagnosis, cognitive dysfunctions usually manifest early in the disease process and are consistently ranked among the leading factors that impact the patients' quality of life. The genetic component of HD, a mutation in the huntingtin (HTT) gene, is traditionally presented as the main contributor to disease pathology. However, accumulating evidence suggests the implication of the microtubule-associated tau protein to the pathogenesis and therefore, proposes an alternative conceptual framework where tau and mutant huntingtin (mHTT) act conjointly to drive neurodegeneration and cognitive dysfunction. This perspective on disease etiology offers new avenues to design therapeutic interventions and could leverage decades of research on Alzheimer's disease (AD) and other tauopathies to rapidly advance drug discovery. In this mini review, we examine the breadth of tau-targeting treatments currently tested in the preclinical and clinical settings for AD and other tauopathies, and discuss the potential application of these strategies to HD.
Huntington's Disease: A Secondary Tauopathy
Tau is an important microtubule-associated protein primarily expressed in neurons and known to mediate a plethora of cellular functions such as microtubule dynamics, neurite outgrowth, intracellular trafficking and synaptic plasticity (1–3). These activities are tightly regulated by post-translational modifications of tau, including phosphorylation and dephosphorylation (4–6), acetylation (7, 8), glycosylation (9), O-GlcNAcylation (10), nitration (11), sumoylation (12) and truncation (13). However, while phosphorylation is an essential mechanism regulating the biological activities of tau, abnormally hyperphosphorylated tau (p-tau) can form neurofibrillary tangles (NFTs) and/or neuropil threads (NTs) (14) that interfere with these fundamental mechanisms. This has been classically associated with Alzheimer's disease (AD) or with diseases caused by mutations of the tau gene (MAPT) such as Frontotemporal dementia with parkinsonism-17 (15, 16) (Table 1). More recently, similar tau dysregulations have been reported in Huntington's disease (HD) (27, 31, 32, 42, 55, 64, 83, 84) [reviewed in (33, 85)]; a disorder driven by an autosomal dominant pattern of inheritance and caused by a pathological CAG repeat expansion exceeding 35 in exon 1 of the huntingtin (HTT) gene (86) coding for the huntingtin (HTT) protein. This CAG elongation leads to the production of mutant huntingtin (mHTT) (87–90) which confers a cytotoxic activity to this newly formed protein. This includes sequestration of transcription factors, mitochondrial dysfunction, induction of apoptotic cell death and alteration of the ubiquitin-proteasome system (UPS) (88–90).
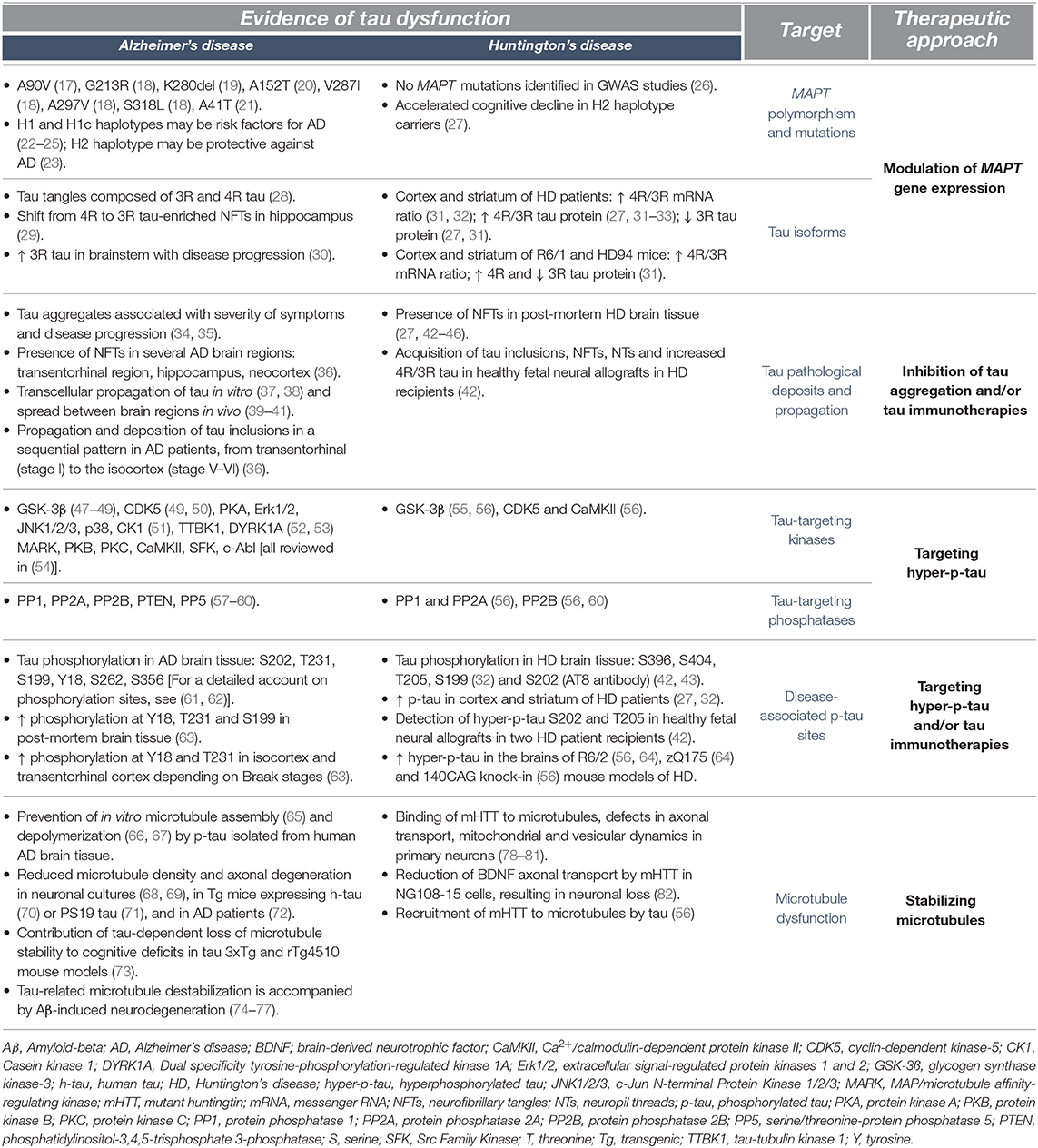
Table 1. Tau pathology and therapeutic strategies in AD and HD.
While the diagnosis of HD is based on motor features (typically chorea), patients exhibit early and progressive cognitive impairments that impact activities of daily living along with psychiatric disturbances that can evolve to frank psychosis (91, 92). Notably, carriers of the H2 MAPT haplotype allegedly experience more rapid cognitive decline than those with an H1 haplotype (27). This is of particular interest since the MAPT haplotype has been proposed as a risk factor for other neurodegenerative disorders such as AD, Parkinson's disease (PD) and PD-associated dementia (22, 23, 93). Along these lines, data collected by Positron Emission Tomography (PET) have allowed to establish correlations between tau and cognitive decline, with tau deposits more closely related with cognitive dysfunction in AD patients than amyloid β (Aβ) (94). Furthermore, both PET and cerebrospinal fluid (CSF) measures of tau, but not Aβ, have been linked to worsening cognition in AD (95). Similarly, the CSF of HD patients contains increasing levels of total tau (t-tau) with disease progression, which correlate with a decline in motor and cognitive functions (96). While a couple of studies have found discrepancies between the levels of CSF t-tau and cognitive decline (96, 97), a correlation between CSF t-tau and mHTT has been reported (97).
In agreement with the concept that HD meets the criteria of a secondary tauopathy is the fact that the cardinal features of tauopathies—misfolding, hyperphosphorylation, NFTs and NTs—have all been identified in post-mortem brain tissue derived from HD patients (27, 43–46, 98–100) [reviewed in (33, 85)]. For example, an increased 4R/3R tau isoform ratio has been observed in HTT mutation carriers (31, 32) at late disease stages (3 and 4) (32). In particular, nuclear rod-like tau deposits composed of the 4R tau isoform are more abundant in striatal and cortical tissues of HD patients, while they are virtually undetectable in the brains of control individuals (31). Both abnormal p-tau and mHTT aggregates can be located within neurons (27), although they rarely colocalize (98) or co-precipitate in HD brain homogenates (31).
Collectively, these findings suggest an association between altered tau biology and HD pathology. However, whether tau impairments have a causative effect on the manifestation of certain aspects of the disease, such as cognitive decline, has yet to be established. A closer look at the evidence of tau dysfunction in HD allows us to explore rather uncharted territories in therapeutic development for this condition. Taking advantage of the discoveries and therapeutics designed to attenuate tau dysfunction in AD (101), as a significant number of preclinical studies and clinical trials have already been initiated, may indeed prove to be useful in HD as well. There is a broad diversity of approaches (Figure 1), which include decreasing tau phosphorylation, inhibiting tau aggregation and reducing pathological forms of tau using microtubule stabilizing compounds, immunotherapies or silencing of the MAPT gene (Figure 1), which could all serve treatment purposes. In the following sections, we present the multiple therapeutic approaches to target tau, describe the treatments that have reached clinical trials and discuss their potential application to HD.
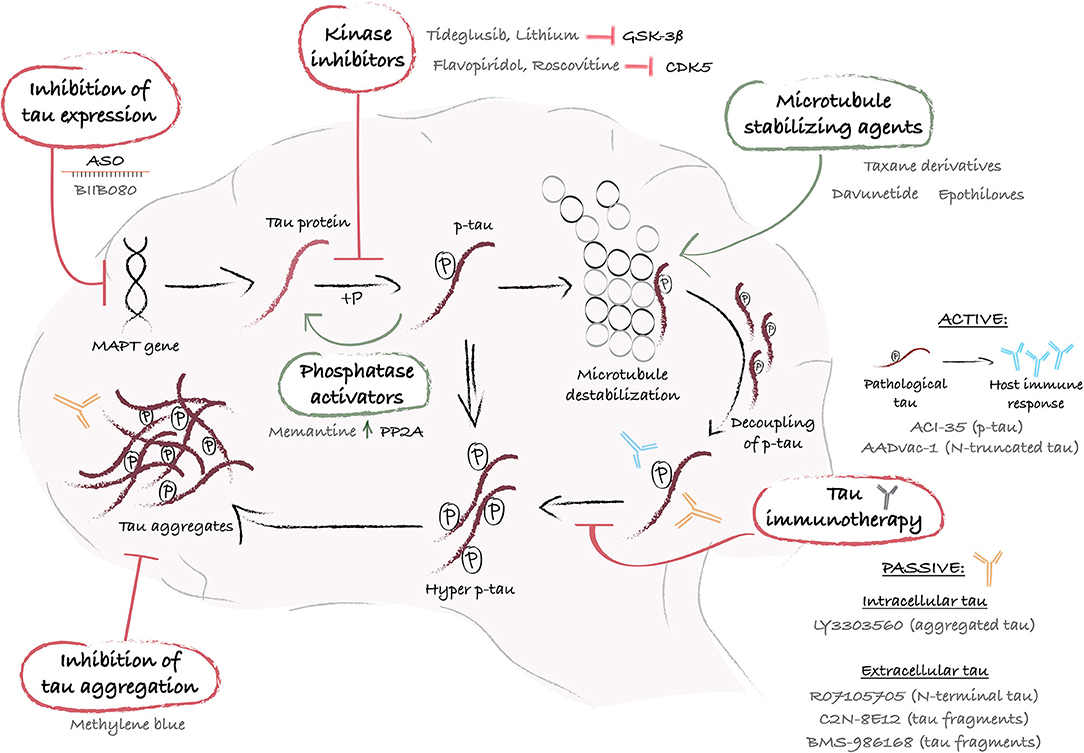
Figure 1. Schematic representation of mechanistic interventions using tau-targeting therapies. The MAPT gene encodes for the protein tau, which undergoes post-translational phosphorylation and dephosphorylation that regulate its affinity for microtubules and ensure its functional role as a microtubule stabilizer. When tau undergoes hyperphosphorylation, generally via an abnormal stimulus, it loses its affinity for microtubules and accumulates into the cytoplasm or exits the cells. When released from the microtubules, intracellular hyperphosphorylated tau proteins self-assemble into aggregates of increasing complexity, and ultimately produce large pathological aggregates and NFTs. At each step of the process, drug targets have been identified and a variety of therapeutic approaches have been designed and tested in preclinical and/or clinical studies. A first strategy leverages the use of ASO (e.g. BIIB080) to control MAPT gene expression and reduce tau synthesis. Alternatively, pathological tau hyperphosphorylation can be reversed with kinase inhibitors (e.g. Tideglusib or lithium) or phosphatases activators (e.g. Memantine), and tau loss of function can be counterbalanced with microtubule stabilizers (e.g. Taxane derivatives, Davunetide, or Epothilones). Furthermore, immunization-based strategies offer an interesting approach to sequester and degrade p-tau before aggregation, using peptides that mimic a specific p-tau amino acid sequence (e.g. ACl-35 or AADvac-1), active or passive immunization to target either intracellular (e.g. LY3303560) or extracellular tau (e.g. RO7105705, C2N-8E12 or BMS-986168). Lastly, tau aggregation inhibitors, such as Methylene blue, provide an additional means by which to eliminate tau aggregates and restore cellular health. ASO, antisense oligonucleotide; CDK5, cyclin-dependent kinase-5; GSK-3ß, glycogen synthase kinase-3; hyper p-tau, hyperphosphorylated tau; MAPT, microtubule-associated protein tau; P, phosphate; PP2A, protein phosphatase 2A; p-tau, phosphorylated tau.
Therapeutic Strategies to Target Pathological Forms of Tau
Targeting Tau Hyperphosphorylation
Tau function depends on its phosphorylation state. Hyperphosphorylation generates negatively charged repulsive forces which impede functional interactions with microtubules, leading to their destabilization and subsequent cell death (5, 6, 102). Recent studies have reported an increase in p-tau levels in the cortex and striatum of post-mortem brain tissue derived from HD patients (27, 32). More specifically, hyperphosphorylation of tau has been detected at 5 distinct epitopes—S396, S404, T205, S202, and S199 residues—(32, 42, 43), and S202 and T205 were found in neuronal inclusions (27) (Table 1). The association of tau hyperphosphorylation in various forms of tauopathies (4, 103, 104), as well as in HD, suggests that approaches aiming at restoring normal levels of p-tau could improve disease outcome (Figure 1); approaches that have already been extensively tested in AD models (105–108). One of the strategies to reduce p-tau levels is to modulate the activity of tau-targeting kinases and phosphatases. The physiological function of the tau protein is facilitated by the orchestrated activity of these enzymes through phosphorylation and dephosphorylation at the threonine and serine residues (109). A number of small molecule inhibitors of protein kinases or activators of phosphatases have been studied in pre-clinical and clinical settings and are presented in the following section.
Kinase Inhibitors
Small molecules, which have the ability to reduce tau hyperphosphorylation, have been among the first tau-targeted treatments developed for AD. This evolved from the evidence that kinases, such as cyclin-dependent-like kinase 5 (CDK5) and glycogen synthase kinase 3 beta (GSK-3β), are altered in patients and animal models of the disease (49, 110–114) (Table 1). In a transgenic (Tg) HD mouse model that expresses exon 1 of the human HTT gene, with approximately 125 CAG repeats (R6/2) (115), a knock-in chimeric HD mouse model, that expresses a human HTT exon 1/ mouse Htt with 140 CAG repeats (KI140) (116), as well as in patients, the levels and activity of CDK5 and GSK-3β have been found to be dysregulated (55, 56), suggesting that redirecting kinase inhibitors levels/activity could potentially abrogate tau hyperphosphorylation observed in HD.
CDK5 inhibitors, such as Flavopiridol (Alvocidib) and Roscovitine (Seliciclib), have been tested in AD preclinical studies. Flavopiridol efficiently inhibits CDK5 and improves synaptic plasticity as well as motor behavior in mice injected with Aβ oligomers (117). Roscovitine (Seliciclib) prevents tau phosphorylation in a Niemann-Pick Type C disease mouse model (118), which is characterized by CDK5 dysfunction that triggers tau hyperphosphorylation (119). However, CDK5 inhibitors have yet to be tested in the clinical setting and further investigation is therefore needed to determine if they can, in effect, ameliorate tau-associated pathology in either, or both, AD and HD.
GSK-3β inhibitors form another class of compounds, which have been tested in preclinical and clinical contexts. Among the GSK-3β inhibitors, Tideglusib and lithium have shown improvements in AD Tg mouse models that express either four familial AD mutations (FTDP-17 G272V, P301L, and R406W, referred to as VLW mice and VLW mice overexpressing GSK-3β) (120, 121), three familial AD mutations (APP Swedish, MAPT P301L and PSEN1 M146V) (3xTg-AD) (122), AD-related mutations (APP Swedish crossed with Tau VLW) (108) or the MAPT mutation P301L alone (123). Tideglusib and lithium considerably reduce phosphorylation and aggregation of tau, decrease neuroinflammation and neuronal death and improve learning and memory abilities (108, 120–123). Both drugs have also been tested in phase I and II clinical trials for AD with overall positive results, demonstrating safety, tolerability and improved cognition in comparison to placebo-treated individuals (124) [reviewed in (125)]. The therapeutic potential of lithium has further been evaluated in the R6/2 mouse model and in HD patients, and studies have reported encouraging results (126–128) [reviewed in (129)]. Lithium was found to regulate HD-associated pathological processes such as glutamate excitotoxicity, altered levels of neurotrophic and growth factors and transcriptional dysregulation (130–133) [reviewed in (129)]. For example, lithium increased levels of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in several brain regions of an animal model of mania generated with ouabain or wild-type animals, as well as in wild-type cultured neurons (133–136). Furthermore, exposure to lithium mediated significant transcriptional changes connected to signal transduction (e.g., mTOR and Wnt-related signaling) in the corpus callosum of wild-type rats (137), as well as transcription factors and genes related to metabolism in the substantia nigra pars compacta of parkinsonian mice (induced by the neurotoxin MPTP) (138). Clinical studies showed that motor abnormalities were not improved in most HD patients treated with lithium, but some reports suggest an amelioration of cognitive disturbances and mood disorder (139, 140). The positive outcomes on non-motor impairments suggest a potential benefit of lithium treatment in HD individuals, especially for patients in early stages of disease, when cognitive and psychiatric symptoms are predominant. However, it is unclear whether the administration of lithium diminishes GSK-3β activity and p-tau levels, as conflicting findings on GSK-3β levels in post-mortem tissues of HD patients have been reported. Indeed, GSK-3β levels and activity have been shown to be intrinsically decreased in the striatum and cortex of HD individuals (83, 141), while increased GSK-3β levels were measured in the hippocampus (55). These discrepancies may reflect a dynamic molecular response at different disease stages, or suggest the differential alteration of GSK-3β levels in specific brain regions. Evaluation of p-tau in the CSF or imaging with p-tau PET tracers could help identify whether the beneficial effects of lithium are directly related to a reduction of p-tau levels.
Phosphatase Activators
Since tau phosphorylation is a reversible process, triggering tau dephosphorylation at serine and threonine residues could reduce hyperphosphorylation. Phosphatases [described extensively in (57)] catalyze the interconversion reactions of tau, from phosphorylated to dephosphorylated states, thereby regulating the degree of tau phosphorylation. Several studies have demonstrated that the activity of protein phosphatase 2A (PP2A) and serine/threonine-protein phosphatase (PP2B) is decreased in AD mouse models and patients (58, 142) [reviewed in (59)], as well as in both Tg and knock-in HD mouse models (56, 64, 143) (Table 1). The Tg mouse line used in the aforementioned studies was the R6/1, which expresses the exon 1 of the human HTT gene containing approximately 114 CAG repeats (115) and the R6/2 model, described above. The knock-in mouse lines discussed above were KI140 and zQ175, which respectively express human HTT exon 1 with approximately 140 and 188 CAG repeats inserted in the mouse Htt (116, 144). Furthermore, PP2A is an important phosphatase known to regulate the activity of the GSK-3β and other kinases implicated in tau pathological modifications (145), and dysregulation of PP2A activity leads to cellular dysfunction including cytoskeletal alterations, impairment of synaptic function and tau mislocalization (146–149). Activation of the phosphatase PP2A has been shown to reduce abnormal phosphorylation of tau in the brain and ameliorate AD pathology (150, 151), suggesting that PP2A phosphatase activators could likely restore physiological levels of tau phosphorylation in patients.
Memantine is a small molecule that has been found to reverse tau hyperphosphorylation. It enhances PP2A activity, improves neuronal viability (152) and reduces glutamate excitotoxicity in GABAergic neurons by acting as a N-methyl-D-aspartate (NMDA) receptor antagonist (153). Furthermore, Memantine mitigates lipopolysaccharide-induced neurodegeneration in mixed rat primary cultures by attenuating the microglial inflammatory response (154). It therefore appears that Memantine is a multifunctional molecule that acts on several cell types and on tau-independent molecular targets, although additional studies are needed to determine if tau directly promotes the beneficial effects of Memantine. Clinical studies indicated that Memantine improves attention, agitation, delusion, global well-being, daily functions and independence in patients with mild cognitive decline (106, 107). There are 87 Memantine clinical trials listed on clinicaltrials.gov, all aiming to treat dementia associated with neurodegenerative diseases, including HD. Thus far, a small pilot (155) and one case study (156) have reported that Memantine improves motor, but not cognitive deficits, in HD patients. However, it may be premature to conclude from these small-scale studies, especially given the fact that both trials were based on the selection of patients in advanced stages of disease that may have further tainted the true potential of Memantine. Hence, Memantine is currently under investigation in the clinical trial MITIGATE-HD, which recruited a large number of pre-manifest HD, early HD and control individuals providing a better opportunity to investigate this compound as a treatment for cognitive impairments. However, the secondary outcome measures of the MITIGATE-HD study do not include CSF t-tau or p-tau nor tau PET imaging evaluation and it will therefore not be possible to determine whether the improvement in cognition, if any, is due to the ability of Memantine to reduce tau hyperphosphorylation.
Views on Targeting Pathological Forms of P-Tau
Although targeting the mechanisms of phosphorylation/dephosphorylation of tau is a seemingly logical strategy, several limitations should be considered. Based on the outcomes of AD clinical trials, the major drawback is the dysregulation of essential kinases and phosphatases responsible for the post-translational modifications of unrelated tau substrates. For instance, GSK-3β phosphorylates more than 100 substrates, while PP2A has more than 300 known targets. Furthermore, both enzymes are themselves regulated by a number of signaling pathways (157, 158). Pharmacological alterations of these complex signaling pathways may lead to activation of compensatory mechanisms, which would in turn generate unpredictable outcomes (158). For example, pharmacological activation of the PP2B phosphatase could promote dephosphorylation of mHTT at serine 421, leading to the disruption of axonal transport and cellular distribution of important neurotrophic factors (159). Small molecules aimed at specifically targeting the multi-substrate enzymes involved in tau phosphorylation are accompanied by decisive limitations, which constrain their use in the treatment of HD and other tauopathies. However, the promising clinical results obtained with lithium and Memantine do warrant further investigation in the context of HD.
Inhibiting the Formation of Tau Aggregates
When hyperphosphorylated tau detaches from the microtubules, it relocates to the somatodendritic compartment and self-assembles into increasingly complex aggregate species. This process begins with soluble oligomers which grow into pre-fibrils and ultimately form insoluble NFTs (66) [reviewed in (160)]. These NFTs accumulate with disease progression in AD (34, 161, 162) as well as in HD (44–46, 99) (Table 1), and therapeutic strategies to prevent tau aggregation have therefore become a major focus of research (Figure 1). However, the perspective that accumulation of NFTs causes neurodegeneration has been more recently challenged (163, 164) [reviewed in (165)]. Indeed, mislocalized, soluble misfolded, soluble hyperphosphorylated forms of tau and tau oligomers are emerging as candidate neurotoxic entities (166–168). For example, the mislocalization of tau to dendritic spines has been established in AD and seemingly induces synaptic impairments in rTg4510 and P301S tau mouse models (167, 169–171) as well as the loss of dendritic spines in AD patients (172). Spinal alterations have been observed in R6/2 mice (173), but the relationship between tau and spine instability in HD remains to be elucidated. Additionally, as observed in AD and HD, pre-tangles and NFTs have been identified in several brain regions, predominantly in the putamen, cortex and hippocampus (22, 27, 43, 44, 46, 98–100). It is therefore reasonable to anticipate that inhibiting tau aggregation at early and late stages of fibrillization may improve HD-related neurotoxicity and, on a larger scale, cognitive deficits.
The design of inhibitors of tau aggregation has already generated a significant number of small molecules for drug screening. Among 3,000 anti-tau aggregation molecules studied, fibrillization inhibitors have been identified as the most effective in preventing the formation of NFTs in Tg animals expressing mutated human tau [reviewed in (174)]. For example, Methylene Blue and its derivatives have been shown to stabilize tau in a conformation that prevents its fibrillization (175), but other neurotoxic forms of tau, such as oligomers, do not appear to be affected (164, 176). Nonetheless, treatment of JNPL3 Tg mice expressing the P301L tau mutation results in the amelioration of cognitive deficits (177) and Methylene Blue is the only tau-targeting treatment that has reached phase III in AD. Importantly, this drug can inhibit the aggregation of other self-assembling proteins including mHTT (178), decrease the formation of mHTT inclusion bodies in primary mouse neurons and R6/2 mice, and improve motor deficits in these animals (178).
Views on Inhibiting the Formation of Tau Aggregates
The current state of the field offers histological evidence of tau aggregation in post-mortem HD brain tissue, but available experimental animal models do not recapitulate pathological tau inclusions. Establishing alternative models that reproduce key features and molecular dysfunctions of HD is a prerequisite to evaluate the contributions of tau aggregates to pathology as well as the potential of anti-tau aggregation therapies. Additionally, it is becoming increasingly clear that pathological forms do not solely consist of NFTs, but include aggregate intermediates and soluble post-translationally modified tau. Considering the similarities among tauopathies, an efficient therapeutic approach for HD should therefore target multiple tau species, including oligomers and hyperphosphorylated forms of the protein (27).
Tau Immunotherapies
Tau-based immunotherapies refer to the neutralization and clearance of the tau protein by host-generated antibodies (active immunization) or by the administration of tau-specific antibodies (passive immunization). Immunotherapies can be designed to target a variety of tau species, including t-tau, hyperphosphorylated tau, extracellular tau, oligomeric tau or tau fragments (Figure 1) (165, 179–181). This approach provides greater precision and flexibility to therapeutic designs, but a successful clinical outcome relies on the identification of the exact pathological forms of tau responsible for a specific phenotype. Multiple forms of tau are potential candidates for immunotherapies in HD, including tau aggregates (27), p-tau (27, 32), tau oligomers (27) and caspase-2 cleaved Δtau314 (84). Each form appears to be associated with distinct disease phenotypes/stages, and tau hyperphosphorylation at S396, S404, T205, and S199 epitopes has been observed in stage 4 HD patients (32). The presence of tau oligomers (T22 and TOMA positive staining) has also been detected in the putamen of stage 4 HD patients (27). Caspase-2 cleaved Δtau314 protein is a form of tau associated with dementia in Lewy body disease (182) and is found in greater concentrations in the caudate nucleus and prefrontal cortex of HD subjects when compared to healthy controls (84). The following paragraphs will survey the available literature to propose additional therapeutic frameworks for the next generation of HD drugs.
Active Immunization
Active immunization by exposure of the host immune system to pathological forms of tau has also been considered to induce a long-lasting anti-tau immunity. Several peptides have been designed using tau pathological forms such as p-tau (181, 183), oligomers (180, 184) and truncated tau (185). Immunization of animals recapitulating AD features can result in pathophysiological and behavioral improvements (181), with the host-generated antibodies depict specific recognition of p-tau (181). Based on these findings, two anti-tau vaccines have been tested in AD clinical trials; one targeting S396 and S404 phosphorylated tau (ACI-35) (186) and the other targeting the pathological N-truncated form of tau (AADvac-1 or Axon peptide 108 conjugated to keyhole limpet hemocyanin) (187). Importantly, the ACI-35 vaccine was designed against pathological forms of tau also found in HD patients and further investigations could establish its potential as a candidate immunotherapy to ameliorate cognitive deficits in this patient population (27, 99).
Passive Immunization
Passive immunization is achieved by administering pre-formed antibodies to recognize a specific antigen and constitutes an alternative immunization approach that does not solely rely on the immune system of the host. Antibodies can further be engineered to express desirable properties and bind tau within intracellular and/or extracellular compartments. Several antibodies have been designed to target various forms of tau and the promising antibodies that have reached clinical trials are discussed below.
Passive immunization to target intracellular tau
Following peripheral administration, anti-tau antibodies are able to cross the blood brain barrier to reach neuronal and non-neuronal elements. The antibodies are internalized by neurons via receptor-mediated endocytosis (188), bind tau and the emerging tau-antibody complex is subsequently degraded through the proteasomal system (189). A significant number of tau passive immunotherapies have been designed to target p-tau, and the anti-tau pS202 monoclonal antibody (named CP13) has thus far demonstrated a superior efficacy compared to other anti-p-tau antibodies (190). Indeed, CP13 reduces soluble and insoluble total and p-tau levels in the cortex and hindbrain of a Tg mouse model expressing the human P301L mutation and which is characterized by a severe tauopathy (190). In HD, increased pS202 levels correlate with alterations in tau phosphorylation in both R6/2 and zQ175 mouse models (56, 64), as well as in patients (27, 42, 43). In addition to p-tau, antibodies have been designed to target other pathological forms of the protein. The humanized antibody LY3303560 (also referred to as Zagotenemab) is derived from a monoclonal antibody used in histology (MC1) to identify pathological conformations of tau (191) in both AD (191) and HD human brain tissue (33). It has also demonstrated greater affinity toward soluble tau aggregates compared to tau monomers in vitro (179). This immunotherapy is safe and well-tolerated in humans, as established by the successful completion of a phase I and an ongoing phase II clinical trial evaluating its efficacy in AD (192). CP13 and LY3303560 are therefore attractive immunotherapy candidates that could be tested for efficacy in preclinical models of HD. Animal models such as zQ175 or KI140 mice recapitulate a slow disease progression and the gradual acquisition of molecular and behavioral HD phenotypes, and could therefore provide a suitable experimental system to evaluate the benefits of immunotherapies on HD-related cognitive as well as motor deficits (116, 144).
Passive immunization to target extracellular tau
In addition to neurotoxic effects resulting from post-translational modifications, tau has been suggested to adopt prion-like properties that result in its transcellular propagation through the extracellular milieu, to ultimately seed pathology in healthy recipient cells (193). Evidence from cellular (194, 195) and animal models (39–41), as well as post-mortem tissue (36, 161, 162), support this hypothesis (Table 1). For example, a single injection of AD brain homogenate in the cortex or hippocampus of a naïve mouse induces endogenous tau misfolding and aggregation with detrimental consequences on physiological functions and behavior (41). Extracellular tau is particularly efficient in corrupting functional endogenous tau and initiating pathological aggregates that lead to the formation of NFTs (196). A number of mechanisms of tau spreading have been identified and include cytoplasmic exchange by tunneling nanotubes (197), as well as transsynaptic (198) or transcellular propagation via exocytosis and endocytosis (194, 195, 199). Several antibodies targeting extracellular tau have been designed to prevent transcellular propagation and seeding of tau-related pathology. Antibodies are now available to recognize a diversity of extracellular tau isoforms and fragments. The antibody C2N-8E12 (also referred to as ABBV-8E12) recognizes the isoform tau-F (441 aa) (200), while the antibody RO7105705 (also referred to as Semorinemab) binds the N-terminus of all six tau isoforms (201). Gosuranemab (also referred to as BMS-986168) was engineered using a tau fragment released into the conditioned media prepared from AD patient-derived cortical neurons (202, 203). Despite differences in the targeted epitopes, all of these antibodies have been shown to ameliorate tau pathology and behavioral deficits in mouse models that express MAPT mutations (JNPL3, P301L and P301S) (203–206). These observations suggest that reducing extracellular levels of tau is a promising strategy to improve AD pathology. Favorable to their use is the fact that antibodies tested in phase I clinical trial have met safety and tolerability criteria (207) [reviewed in (208)]. Similar routes of tau propagation have been suggested to occur in HD patients who received fetal grafts to replace cell loss generated by the disease process (42). However, these observations originate from post-mortem analyses on a few rare cases and more definitive evidence is needed to conclude that tau can indeed propagate in the HD brain. Furthermore, HD patients have lower concentrations of t-tau in the CSF in comparison to AD affected individuals, (96, 97, 209–212), and whether CSF t-tau levels truly reflect extracellular tau load in the central nervous system (CNS) of HD patients remains to be elucidated.
Views on Tau Immunotherapies
Research in the field of immunotherapy has made significant progress in the development of anti-tau treatments for AD and is gaining momentum in HD [reviewed in (101, 213)]. Antibody-based therapies have the distinct advantage of being highly specific to the selected epitopes and can target intracellular or extracellular tau both in the periphery and in the CNS. Antibodies have a low molecular weight and are therefore suitable for nanocarrier-based delivery approaches to reduce their degradation, mitigate the host immune response and control the rate of antibody release (214). Furthermore, administration of tau-targeting antibodies by injection is a straightforward and safe procedure (207). Based on the concept that mHTT can propagate between cells and template pathology in a prion-like fashion, active and passive immunization strategies targeting extracellular mHTT are attracting interest (215–222) [extensively reviewed in (213)], and could be tested in parallel to a tau, or mHTT/tau combined immunization. Furthermore, halting accumulation and propagation of pathogenic proteins in HD patients at early stages of the disease, before they are afflicted by neurodegeneration and cognitive dysfunction, may provide the most effective protection.
However, a major limitation of current studies is that the majority of tau-related dysregulations have not been reported in premanifest or early-stage diseased patients. As a result, the association of pathological forms of tau with late stage disease could cast doubts on the validity of targeting tau in HD (32). A broad investigation on a larger population of HD patients, matched for age, sex and CAG repeat length, would provide more accurate information on the progression of tau pathology. In particular, future studies could take advantage of the technological advances achieved with PET scans and specific anti-tau ligands (223), to enable the analysis of pathological tau in the brain of living patients with a degree of specificity that is not possible with post-mortem analyses. By doing so, tau abnormalities could be directly compared to cognitive performance and importantly, changes over time. For instance, the PET ligand [18F]MK-6240, which has already been validated in AD, is a new generation tau ligand with high sensitivity for NFTs and negligible off-target binding (224), which makes it ideal for defining the extent of tau pathology in HD brains and investigating its relationship with cognitive profile.
Mitigation of Tau-Induced Cellular Alterations by Stabilizing Microtubules
Structural and functional damage to the neuronal cytoskeleton constitutes a key event in the pathogenesis of tauopathies (225–227). Therefore, microtubule-stabilizing small molecules have been developed and studied as a therapeutic approach to treat tauopathies (Figure 1). Alterations of microtubule-dependent axonal transport is a characteristic of HD pathology (78, 79) and could result from the independent, but coordinated effects of mHTT and tau (Table 1). mHTT has been shown to interact with and destabilize microtubules, resulting in alterations of axonal transport and loss of neuronal viability (78, 79, 228). Tau has further been found to recruit mHTT to the microtubule network (56), which could precipitate the destabilization of microtubules. For example, microtubule stabilization using the small molecule Taxol, a semisynthetic taxane derivative, inhibits the entry of mHTT into the nucleus and increases neuronal survival in HD primary striatal and cortical neurons (78). These observations suggest that tau and mHTT can both induce microtubule destabilization, which support the restoration of microtubule functions as a therapeutic approach for HD.
The cytoskeleton stabilizing agents Epothilone and Taxane derivatives have been found to interact with tubulin, restore rapid axonal transport, and alleviate cognitive and motor impairments in mouse models of tauopathies (T44 tau Tg mice; PS19 tau Tg mice) (71, 229). However, the Epothilone D phase I clinical trial was discontinued due to toxicity and severe side-effects were reported for taxoid TPI 287 (abeotaxane) (230). On the other hand, the microtubule-stabilizing peptide Davunetide (NAP) (231, 232) was well-tolerated in preclinical toxicology and clinical safety studies (233). NAP demonstrated highly potent neuroprotective properties by reducing the activity of the microtubule-severing protein katanin (234), inhibiting programmed cell death and restoring mitochondrial function in the PD-related A53T α-synuclein SH-SY5Y cell culture model (235). In a Tg schizophrenia mouse model (activity-dependent neuroprotective protein (ADNP)+/− Tg mice), intranasal administration of NAP decreased brain levels of p-tau and improved cognition (236). The treatment of patients suffering from mild cognitive impairments with NAP showed improved attention and working memory (233). However, no improvement of cognitive deficits was observed in Progressive Supranuclear Palsy (PSP) patients treated with NAP, which could be partly explained by an ineffective dosage (237).
Views on Stabilizing Microtubules
The regulation and restoration of microtubule function is an attractive strategy substantiated by promising preclinical results. Epothilone and Taxane derivatives were approved by the FDA in the early 1990s [history reviewed in (238)] as chemotherapies to treat aggressive cancers and despite significant side-effects, they are among the most relied upon therapies to treat solid tumors (239, 240). In the case of neurodegenerative diseases, the toxicity of microtubule-stabilizing agents is a major drawback that may limit their clinical use, altogether. However, new microtubules stabilizing agents such as Davunetide and others [reviewed in (241)] exhibit promising disease-mitigating outcomes in animal models, with more manageable side-effects in clinical trials, and could therefore be the new frontier in the search for microtubules stabilizing strategies to ameliorate tau pathology in neurodegenerative diseases, including HD.
Modulation of MAPT Gene Expression
Mutations in the MAPT gene have been associated with neurodegenerative diseases such as Frontotemporal dementia (20, 242–245), PSP (245) and AD (17–21); diseases characterized by abnormal accumulation of pathological forms of tau and that further correlate with cognitive deficits. Impaired alternative splicing of exon 10 of the MAPT gene leads to an imbalance in the amount of 3R and 4R tau isoforms within cells, and a change in 3R to 4R ratio has been associated with specific tauopathies. For instance, NFTs are found to be immunopositive for both 4R and 3R in AD (28), while patients with Pick's disease predominantly express the 3R tau isoform (246). As a result, temporarily altering MAPT expression or splicing using antisense oligonucleotides (ASO) has been considered (Figure 1) (247). To our knowledge, no MAPT mutations have been associated with HD, but patients express lower 3R and increased 4R tau mRNA and protein levels (32) and the 4R isoform is enriched in tau nuclear rods found in the striatum (31). Therefore, selectively reducing the levels of 4R tau without altering the overall expression of tau protein in HD could restore protein homeostasis (31, 32).
Tau is involved in essential cellular functions (248) and as a result, tau silencing could raise safety concerns that would limit its translation to the clinic. Studies in tau knock-out animal models have demonstrated that loss of Mapt causes cognitive and motor deficits in an age- and strain-dependent manner (249). A marked reduction in the number of dopaminergic neurons was observed in the substantia nigra pars compacta of Mapt −/− mice, which correlated with PD-like motor deficits in 12-month old animals (249, 250). However, the partial or total downregulation of the Mapt gene in the R6/1 Tg (115 CAG repeats) HD mouse model improves motor behavior and does not induce significant side-effects (31). Furthermore, the reduction of tau protein levels using the ASO BIIB080 prevents NFTs deposition and clears pre-existing tau aggregates, mitigates neuronal loss, ameliorates behavior (nesting) and extends the survival of PS19 Tg mice expressing the disease-associated P301S tau mutation (251). In non-human primates, ASO BIIB080 reduces tau mRNA and protein levels in the brain, spinal cord and CSF (251). These promising results enabled the advancement of ASO BIIB080 to phase I clinical trial that is currently ongoing (252).
Views on Modulating MAPT Gene Expression
Modulation of gene expression is a novel therapeutic avenue to treat tauopathies and mRNA-targeting strategies using ASO have recently been proposed as a potential treatment for HD (253). The preliminary results of a clinical trial on IONIS-HTTRx, an ASO drug that targets HTT, reported a sharp decrease in mHTT CSF levels with no serious adverse events (254, 255). To our knowledge, silencing more than one gene via ASOs has never been tested, but lowering both HTT and tau expression levels in HD patients has recently been proposed (256). However, gene silencing and modification of gene expression are novel technologies with only a few treatments approved for clinical use. For instance Fomivirsen, an antisense antiviral drug for the treatment of retinitis cytomegalovirus, was the first antisense drug approved by the FDA in 1998 (257), and the second ASO therapy to reach the market (Mipomersen, to treat familial hypercholesterolemia) was FDA approved only 15 years later. As of today, little is known about the possible long-term consequences of modulating protein levels with ASOs, and it is essential to extensively investigate whether modulation of both MAPT and HTT expression could ameliorate disease outcome in HD patients without inducing long-lasting side-effects.
Additional Tau-Targeting Approaches
In addition to phosphorylation, tau can undergo a number of post-translational modifications such as glycosylation (9), acetylation (7), truncation (258), glycation (259), nitration (260) and ubiquitination (261) [reviewed in (262)]. These modifications have been associated with tauopathies and mainly investigated in AD brains, with observation of increases in acetylated (7), caspase-truncated (263) and N-glycosylated forms of tau (9). Several molecules have been tested in preclinical and clinical studies to target these different disease-associated post-translational modifications. For example, Salsalate is a nonsteroidal anti-inflammatory drug that decreases t-tau levels and acetylated tau at the K174 residue, ameliorates hippocampal atrophy and memory deficits in PS19 Tg mice (8), and has consequently advanced to phase 1 PSP clinical trial (264). It remains undetermined whether tau post-translational modifications, such as acetylation or glycosylation, occur in HD and only a broader understanding of all forms of tau alterations in the disease would support the exploration of these additional tau-targeting approaches.
Another aspect of pathology is the impairment of protein degradation via alterations of the UPS, which has been observed and associated with tauopathies, and proposed to mediate the accumulation of tau within cells (265, 266) [reviewed in (267)]. The search for drugs that increase the degradation of tau by activating the UPS led to the identification of Rolipram (268). Rolipram has been found to activate the UPS by stimulating the protein kinase A (PKA)/cAMP pathway in ex vivo cortical brain slices of rTg4510 mice and to reduce both t-tau and insoluble tau levels (266). Rolipram also decreases t-tau and p-tau levels in a Tg mouse model of early-stage tauopathy expressing the proteasome 26S-targeted fragment (rTg4510:Ub-G76V-GFP) and improves reversal learning behavior in experimental animals (266).
Disease-associated post-translational modifications of tau, as well as dysfunction of the UPS, are compelling alternatives to explore for the treatment of tauopathies. In contrast to the therapeutic strategies discussed in sections Therapeutic Strategies to Target Pathological Forms of Tau, Mitigation of tau-induced cellular alterations by stabilizing microtubules, Modulation of MAPT Gene Expression of this review, the relevance of these pathways remains to be demonstrated in models of HD, and the functional and mechanistic contributions of UPS dysfunction to HD pathology are unclear (269). HD-related alterations of the UPS have been hinted at, with early studies showing sequestration of UPS components within mHTT inclusion bodies (87, 270) in Tg HD mouse models (R6/1, R6/2 and R6/5 Tg - 130-155 CAG repeats) (87) as well as in human brain tissue (270). However, further characterization suggested that UPS impairments could be a transient phenotype, as global UPS activity was not significantly impaired in the R6/2 model and in the HD94 mouse model that expresses mHTT fragments under a tetracycline-responsive system (271, 272). An explanation for these seemingly contradictory findings was proposed by Schipper-Krom et al. who suggested that proteasome recruitment into inclusion bodies is a dynamic and reversible process, that does not inhibit the catalytic activity of the sequestered proteasome units (273), thus supporting observations that the UPS activity may not be impaired in HD mouse models. Nonetheless, pharmacological and genetic modulation of the proteasome activity suggest its implication in mHTT aggregation and cell survival, as a reduced activity is associated with increased aggregation (88, 274) and increased activity promotes the survival of HD striatal neurons (275).
Conclusion
HD patients and their families carry a heavy emotional and financial burden as they face the challenges of a very complex disease, which manifests with a blend of motor, neuropsychiatric and cognitive deficits. Efficient symptomatic treatments, and more importantly disease-modifying therapies, are urgently needed. The diversity of symptoms and difficulty to target the root of the disease, a mutation in the HTT gene, suggests that combinatorial therapies that attenuate multiple dysfunctional proteins and pathways may ultimately be the best strategy to tackle the complex portrait of HD (Table 1). Here, we have reviewed compelling evidence suggesting that HD patients develop features of tauopathies and tau-related dysfunctions. These include the (i) altered exon 10 splicing of MAPT (31, 32), (ii) correlations between H2 MAPT haplotype and severity of cognitive decline (27), (iii) dysfunction of tau-targeting kinases and phosphatases (55, 56, 64, 83), (iv) increased soluble hyperphosphorylated tau (32), (v) altered levels of t-tau in the brain and CSF (32, 96, 97, 209), and (vi) presence of brain NFTs (27, 98, 99). The accumulating evidence of tau pathology encompasses broad dysregulations at the genetic and molecular levels, in CNS tissue and associated biological fluids, and therefore argue in favor of the classification of HD as a secondary tauopathy. In-depth studies are now needed to determine if tau dysfunction directly causes some of the features associated with HD, and in particular cognitive deficits.
Therapeutic approaches targeting tau pathology have been initially designed to treat AD (Figure 1) (101) or primary tauopathies such as PSP (276), based on a large body of evidence suggesting a strong connection between alterations of tau function and cognitive decline [extensively reviewed in (277)]. HD pathology is also associated with both pathological forms of tau and cognitive impairments, thus tau-targeting therapies may offer a completely new angle to treat HD-associated cognitive dysfunction. A diversity of tau-targeting methodologies has been developed, from small molecules to immunotherapies and modulation of gene expression using ASO. These therapies are at various stages of drug development, providing exciting and hopeful prospects toward the betterment of the HD condition.
Author Contributions
MM and SS contributed equally to reviewing the literature and writing the first draft of the manuscript. ARJ, MA, and FC contributed to discussions regarding the manuscript, revised, and finalized the review. All authors contributed to the article and approved the submitted version.
Funding
FC is a recipient of a Researcher Chair from the Fonds de Recherche du Québec en Santé (FRQS, 35059) providing salary support and operating funds, and receives funding from the Canadian Institutes of Health Research (CIHR, PJT-162164 and PJT-168865) to conduct her HD-related research. At the time of the study, MM was supported by a Desjardins bourse de prestige Didier Mouginot from the Fondation du CHU de Québec and a bourse d'excellence du Centre Thématique de Recherche en Neurosciences also from CHU de Québec. SS holds a Bourse de formation Desjardins pour la recherche et innovation from CHU de Québec, and MA is a recipient of a postdoctoral fellowship from FRQS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. (1994) 369:488–91. doi: 10.1038/369488a0
2. Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic tau mutant. J Neurosci. (2011) 31:2511–25. doi: 10.1523/JNEUROSCI.5245-10.2011
3. Wang J-Z, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. (2008) 85:148–75. doi: 10.1016/j.pneurobio.2008.03.002
4. Alonso AD, Cohen LS, Corbo C, Morozova V, ElIdrissi A, Phillips G, et al. Hyperphosphorylation of tau associates with changes in its function beyond microtubule stability. Front Cell Neurosci. (2018) 12:338. doi: 10.3389/fncel.2018.00338
5. Jameson L, Frey T, Zeeberg B, Dalldorf F, Caplow M. Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry. (1980) 19:2472–9. doi: 10.1021/bi00552a027
6. Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. (1984) 259:5301–5.
7. Min S-W, Cho S-H, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. (2010) 67:953–66. doi: 10.1016/j.neuron.2010.08.044
8. Min S-W, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. (2015) 21:1154–62. doi: 10.1038/nm.3951
9. Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med. (1996) 2:871–5. doi: 10.1038/nm0896-871
10. Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem. (1996) 271:28741–4. doi: 10.1074/jbc.271.46.28741
11. Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C, et al. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol. (2003) 163:1021–31. doi: 10.1016/S0002-9440(10)63462-1
12. Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J Biol Chem. (2006) 281:9919–24. doi: 10.1074/jbc.M510127200
13. Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. EMBO J. (1993) 12:365–70. doi: 10.1002/j.1460-2075.1993.tb05665.x
14. Mudher A, Brion J-P, Avila J, Medina M, Buée L. EuroTau: towing scientists to tau without tautology. Acta Neuropathol Commun. (2017) 5:90. doi: 10.1186/s40478-017-0491-z
15. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
16. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. (1998) 95:7737–41. doi: 10.1073/pnas.95.13.7737
17. Sala Frigerio C, Lau P, Troakes C, Deramecourt V, Gele P, Van Loo P, et al. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer's disease patients. Alzheimers Dement. (2015) 11:1265–76. doi: 10.1016/j.jalz.2015.02.007
18. Jin SC, Pastor P, Cooper B, Cervantes S, Benitez BA, Razquin C, et al. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer's disease Ibero-American cohort. Alzheimers Res Ther. (2012) 4:34. doi: 10.1186/alzrt137
19. Momeni P, Pittman A, Lashley T, Vandrovcova J, Malzer E, Luk C, et al. Clinical and pathological features of an Alzheimer's disease patient with the MAPT Delta K280 mutation. Neurobiol Aging. (2009) 30:388–93. doi: 10.1016/j.neurobiolaging.2007.07.013
20. Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum Mol Genet. (2012) 21:3500–12. doi: 10.1093/hmg/dds161
21. Schulte EC, Fukumori A, Mollenhauer B, Hor H, Arzberger T, Perneczky R, et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson's disease. Eur J Hum Genet. (2015) 23:1328–33. doi: 10.1038/ejhg.2014.300
22. Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC, et al. The H1c haplotype at the MAPT locus is associated with Alzheimer's disease. Hum Mol Genet. (2005) 14:2399–404. doi: 10.1093/hmg/ddi241
23. Zhang C-C, Zhu J-X, Wan Y, Tan L, Wang H-F, Yu J-T, et al. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. (2017) 8:44994–5007. doi: 10.18632/oncotarget.16690
24. Pastor P, Moreno F, Clarimón J, Ruiz A, Combarros O, Calero M, et al. MAPT H1 haplotype is associated with late-onset Alzheimer's Disease risk in APOEε4 noncarriers: results from the dementia genetics Spanish consortium. J Alzheimers Dis. (2016) 49:343–52. doi: 10.3233/JAD-150555
25. Sánchez-Juan P, Moreno S, de Rojas I, Hernández I, Valero S, Alegret M, et al. The MAPT H1 haplotype is a risk factor for Alzheimer's Disease in APOE ε4 non-carriers. Front Aging Neurosci. (2019) 11:327. doi: 10.3389/fnagi.2019.00327/full
26. Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, et al. Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol. (2017) 16:701–11. doi: 10.1016/S1474-4422(17)30161-8
27. Vuono R, Winder-Rhodes S, de Silva R, Cisbani G, Drouin-Ouellet J, REGISTRY Investigators of the European Huntington's Disease Network, et al. The role of tau in the pathological process and clinical expression of Huntington's disease. Brain. (2015) 138(Pt 7):1907–18. doi: 10.1093/brain/awv107
28. Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. (1992) 8:159–68. doi: 10.1016/0896-6273(92)90117-V
29. Uchihara T, Hara M, Nakamura A, Hirokawa K. Tangle evolution linked to differential 3- and 4-repeat tau isoform deposition: a double immunofluorolabeling study using two monoclonal antibodies. Histochem Cell Biol. (2012) 137:261–7. doi: 10.1007/s00418-011-0891-2
30. Uematsu M, Nakamura A, Ebashi M, Hirokawa K, Takahashi R, Uchihara T. Brainstem tau pathology in Alzheimer's disease is characterized by increase of three repeat tau and independent of amyloid β. Acta Neuropathol Commun. (2018) 6:1. doi: 10.1186/s40478-017-0501-1
31. Fernández-Nogales M, Cabrera JR, Santos-Galindo M, Hoozemans JJM, Ferrer I, Rozemuller AJM, et al. Huntington's disease is a four-repeat tauopathy with tau nuclear rods. Nat Med. (2014) 20:881–5. doi: 10.1038/nm.3617
32. St-Amour I, Turgeon A, Goupil C, Planel E, Hébert SS. Co-occurrence of mixed proteinopathies in late-stage Huntington's disease. Acta Neuropathol. (2018) 135:249–65. doi: 10.1007/s00401-017-1786-7
33. Gratuze M, Cisbani G, Cicchetti F, Planel E. Is Huntington's disease a tauopathy? Brain. (2016) 139(Pt 4):1014–25. doi: 10.1093/brain/aww021
34. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. (1992) 42(3 Pt 1):631–9. doi: 10.1212/WNL.42.3.631
35. Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. (2016) 139(Pt 5):1551–67. doi: 10.1093/brain/aww027
36. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
37. Hallinan GI, Vargas-Caballero M, West J, Deinhardt K. Tau misfolding efficiently propagates between individual intact hippocampal neurons. J Neurosci. (2019) 39:9623–32. doi: 10.1523/JNEUROSCI.1590-19.2019
38. Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. (2016) 19:1085–92. doi: 10.1038/nn.4328
39. de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. (2012) 73:685–97. doi: 10.1016/j.neuron.2011.11.033
40. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. (2009) 11:909–13. doi: 10.1038/ncb1901
41. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA. (2013) 110:9535–40. doi: 10.1073/pnas.1301175110
42. Cisbani G, Maxan A, Kordower JH, Planel E, Freeman TB, Cicchetti F. Presence of tau pathology within foetal neural allografts in patients with Huntington's and Parkinson's disease. Brain. (2017) 140:2982–92. doi: 10.1093/brain/awx255
43. Jellinger KA. Alzheimer-type lesions in Huntington's disease. J Neural Transm. (1998) 105:787–99. doi: 10.1007/s007020050095
44. McIntosh GC, Jameson HD, Markesbery WR. Huntington disease associated with Alzheimer disease. Ann Neurol. (1978) 3:545–8. doi: 10.1002/ana.410030616
45. Myers RH, Sax DS, Schoenfeld M, Bird ED, Wolf PA, Vonsattel JP, et al. Late onset of Huntington's disease. J Neurol Neurosurg Psychiatry. (1985) 48:530–4. doi: 10.1136/jnnp.48.6.530
46. Reyes MG, Gibbons S. Dementia of the Alzheimer's type and Huntington's disease. Neurology. (1985) 35:273–7. doi: 10.1212/WNL.35.2.273
47. Hye A, Kerr F, Archer N, Foy C, Poppe M, Brown R, et al. Glycogen synthase kinase-3 is increased in white cells early in Alzheimer's disease. Neurosci Lett. (2005) 373:1–4. doi: 10.1016/j.neulet.2004.10.031
48. Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. (1997) 56:70–8. doi: 10.1097/00005072-199701000-00007
49. Swatton JE, Sellers LA, Faull RLM, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer's and Down syndrome but not in schizophrenia human brain. Eur J Neurosci. (2004) 19:2711–9. doi: 10.1111/j.0953-816X.2004.03365.x
50. Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. (1999) 402:615–22. doi: 10.1038/45159
51. Yasojima K, Kuret J, DeMaggio AJ, McGeer E, McGeer PL. Casein kinase 1 delta mRNA is upregulated in Alzheimer disease brain. Brain Res. (2000) 865:116–20. doi: 10.1016/S0006-8993(00)02200-9
52. Kimura R, Kamino K, Yamamoto M, Nuripa A, Kida T, Kazui H, et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum Mol Genet. (2007) 16:15–23. doi: 10.1093/hmg/ddl437
53. Ryoo S-R, Cho H-J, Lee H-W, Jeong HK, Radnaabazar C, Kim Y-S, et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between down syndrome and Alzheimer's disease. J Neurochem. (2008) 104:1333–44. doi: 10.1111/j.1471-4159.2007.05075.x
54. Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin M-L, Yardin C, et al. Tau protein kinases: involvement in Alzheimer's disease. Ageing Res Rev. (2013) 12:289–309. doi: 10.1016/j.arr.2012.06.003
55. L'Episcopo F, Drouin-Ouellet J, Tirolo C, Pulvirenti A, Giugno R, Testa N, et al. GSK-3β-induced Tau pathology drives hippocampal neuronal cell death in Huntington's disease: involvement of astrocyte-neuron interactions. Cell Death Dis. (2016) 7:e2206. doi: 10.1038/cddis.2016.104
56. Blum D, Herrera F, Francelle L, Mendes T, Basquin M, Obriot H, et al. Mutant huntingtin alters tau phosphorylation and subcellular distribution. Hum Mol Genet. (2015) 24:76–85. doi: 10.1093/hmg/ddu421
57. Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. (1993) 61:921–7. doi: 10.1111/j.1471-4159.1993.tb03603.x
58. Liu F, Grundke-Iqbal I, Iqbal K, Gong C-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. (2005) 22:1942–50. doi: 10.1111/j.1460-9568.2005.04391.x
59. Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin M-L, Terro F. Tau protein phosphatases in Alzheimer's disease: the leading role of PP2A. Ageing Res Rev. (2013) 12:39–49. doi: 10.1016/j.arr.2012.06.008
60. Rahman A, Grundke-Iqbal I, Iqbal K. Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem Res. (2005) 30:277–87. doi: 10.1007/s11064-005-2483-9
61. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. (2009) 3:112–9. doi: 10.1016/j.molmed.2009.01.003
62. Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. (2007) 32:23654. doi: 10.1074/jbc.m703269200
63. Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, et al. Phosphorylation of different tau sites during progression of Alzheimer's disease. Acta Neuropathol Commun. (2018) 6:52. doi: 10.1186/s40478-018-0557-6
64. Gratuze M, Noël A, Julien C, Cisbani G, Milot-Rousseau P, Morin F, et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington's disease. Hum Mol Genet. (2015) 24:86–99. doi: 10.1093/hmg/ddu456
65. Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. PNAS. (1994) 12:5562–6. doi: 10.1073/pnas.91.12.5562
66. Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. (1996) 2:783–7. doi: 10.1038/nm0796-783
67. Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. PNAS. (1997) 1:298–303. doi: 10.1073/pnas.94.1.298
68. Merrick SE, Trojanowski JQ, Lee VM-Y. Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons (NT2N Cells). J Neurosci. (1997) 15:5726–37. doi: 10.1523/JNEUROSCI.17-15-05726.1997
69. Sherman MA, LaCroix M, Amar F, Larson ME, Forster C, Aguzzi A, et al. Soluble conformers of Aβ and tau alter selective proteins governing axonal transport. J Neurosci. (2016) 37:9647–58. doi: 10.1523/JNEUROSCI.1899-16.2016
70. Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. (1999) 3:751—62. doi: 10.1016/s0896-6273(00)81127-7
71. Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan A-ML, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. (2010) 30:13861–6. doi: 10.1523/JNEUROSCI.3059-10.2010
72. Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, et al. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. (2003) 5:1623–7. doi: 10.1016/s0002-9440(10)64296-4
73. Barten DM, Fanara P, Andorfer C, Hoque N, Wong PYA, Husted KH, et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J Neurosci. (2012) 21:7137–45. doi: 10.1523/JNEUROSCI.0188-12.2012
74. King ME, Kan H-M, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J Cell Biol. (2006) 4:541–6. doi: 10.1083/jcb.200605187
75. Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. PNAS. (2002) 9:6364–9. doi: 10.1073/pnas.092136199
76. Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, et al. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. (2013) 22:2920–37. doi: 10.1038/emboj.2013.207
77. Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci. (2010) 36:11938–50. doi: 10.1523/JNEUROSCI.2357-10.2010
78. Trushina E, Heldebrant MP, Perez-Terzic CM, Bortolon R, Kovtun IV, Badger JD, et al. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington's disease. Proc Natl Acad Sci USA. (2003) 100:12171–6. doi: 10.1073/pnas.2034961100
79. Trushina E, Dyer RB, Badger JD, Ure D, Eide L, Tran DD, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. (2004) 24:8195–209. doi: 10.1128/MCB.24.18.8195-8209.2004
80. Chang DTW, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. (2006) 2:388–400. doi: 10.1016/j.nbd.2005.12.007
81. Orr AL, Li S, Wang C-E, Li H, Wang J, Rong J, et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. (2008) 11:2783–92. doi: 10.1523/JNEUROSCI.0106-08.2008
82. Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. (2004) 1:127–38. doi: 10.1016/j.cell.2004.06.018
83. Fernández-Nogales M, Hernández F, Miguez A, Alberch J, Ginés S, Pérez-Navarro E, et al. Decreased glycogen synthase kinase-3 levels and activity contribute to Huntington's disease. Hum Mol Genet. (2015) 24:5040–52. doi: 10.1093/hmg/ddv224
84. Liu P, Smith BR, Huang ES, Mahesh A, Vonsattel JPG, Petersen AJ, et al. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington's disease patients. Acta Neuropathol Commun. (2019) 7:111. doi: 10.1186/s40478-019-0764-9
85. Maxan A, Cicchetti F. Tau: a common denominator and therapeutic target for neurodegenerative disorders. J Exp Neurosci. (2018) 12:1179069518772380. doi: 10.1177/1179069518772380
86. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's disease collaborative research group. Cell. (1993) 72:971–83. doi: 10.1016/0092-8674(93)90585-E
87. Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. (1997) 90:537–48. doi: 10.1016/S0092-8674(00)80513-9
88. Jana NR, Zemskov EA, Wang GH, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. (2001) 10:1049–59. doi: 10.1093/hmg/10.10.1049
89. Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell. (2004) 14:95–104. doi: 10.1016/S1097-2765(04)00151-0
90. Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. (2001) 293:493–8. doi: 10.1126/science.1059581
91. Ha AD, Fung VSC. Huntington's disease. Curr Opin Neurol. (2012) 25:491–8. doi: 10.1097/WCO.0b013e3283550c97
92. Tang C, Feigin A. Monitoring Huntington's disease progression through preclinical and early stages. Neurodegener Dis Manag. (2012) 2:421–435. doi: 10.2217/nmt.12.34
93. Winder-Rhodes SE, Hampshire A, Rowe JB, Peelle JE, Robbins TW, Owen AM, et al. Association between MAPT haplotype and memory function in patients with Parkinson's disease and healthy aging individuals. Neurobiol Aging. (2015) 36:1519–28. doi: 10.1016/j.neurobiolaging.2014.12.006
94. Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. (2016) 8:338ra66. doi: 10.1126/scitranslmed.aaf2362
95. Koychev I, Gunn RN, Firouzian A, Lawson J, Zamboni G, Ridha B, et al. PET tau and amyloid-β burden in mild Alzheimer's Disease: divergent relationship with age, cognition, and cerebrospinal fluid biomarkers. J Alzheimers Dis. (2017) 60:283–93. doi: 10.3233/JAD-170129
96. Rodrigues FB, Byrne L, McColgan P, Robertson N, Tabrizi SJ, Leavitt BR, et al. Cerebrospinal fluid total tau concentration predicts clinical phenotype in Huntington's disease. J Neurochem. (2016) 139:22–5. doi: 10.1111/jnc.13719
97. Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, Miller JRC, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest. (2015) 125:1979–86. doi: 10.1172/JCI80743
98. Caparros-Lefebvre D, Kerdraon O, Devos D, Dhaenens CM, Blum D, Maurage CA, et al. Association of corticobasal degeneration and Huntington's disease: can tau aggregates protect huntingtin toxicity? Mov Disord. (2009) 24:1089–90. doi: 10.1002/mds.22204
99. Davis MY, Keene CD, Jayadev S, Bird T. The co-occurrence of Alzheimer's disease and Huntington's disease: a neuropathological study of 15 elderly Huntington's disease subjects. J Huntingtons Dis. (2014) 3:209–17. doi: 10.3233/JHD-140111
100. Moss RJ, Mastri AR, Schut LJ. The coexistence and differentiation of late onset Huntington's Disease and Alzheimer's Disease: a case report and review of the literature. J Am Geriatr Soc. (1988) 36:237–42. doi: 10.1111/j.1532-5415.1988.tb01807.x
101. Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. (2018) 14:399–415. doi: 10.1038/s41582-018-0013-z
102. Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer's disease. Lancet. (1986) 2:421–6. doi: 10.1016/S0140-6736(86)92134-3
103. Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. (2000) 33:95–130. doi: 10.1016/S0165-0173(00)00019-9
104. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. (1986) 83:4913–7. doi: 10.1073/pnas.83.13.4913
105. Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J Biol Chem. (2012) 287:893–904. doi: 10.1074/jbc.M111.306472
106. Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ, et al. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. (2003) 348:1333–41. doi: 10.1056/NEJMoa013128
107. Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease. Arch Neurol. (2006) 63:49–54. doi: 10.1001/archneur.63.1.49
108. Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, et al. A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. (2009) 35:359–67. doi: 10.1016/j.nbd.2009.05.025
109. Noble W, Hanger DP, Miller CCJ, Lovestone S. The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol. (2013) 4:83. doi: 10.3389/fneur.2013.00083
110. Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res. (1999) 34:21–9. doi: 10.1016/S0168-0102(99)00026-7
111. Leroy K, Yilmaz Z, Brion J-P. Increased level of active GSK-3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. (2007) 33:43–55. doi: 10.1111/j.1365-2990.2006.00795.x
112. Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. (2001) 20:27–39. doi: 10.1093/emboj/20.1.27
113. Spittaels K, Haute CV den, Dorpe JV, Geerts H, Mercken M, Bruynseels K, et al. Glycogen synthase kinase-3β phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. (2000) 275:41340–9. doi: 10.1074/jbc.M006219200
114. Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, et al. Amyloid activates GSK-3β to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. (2008) 172:786–98. doi: 10.2353/ajpath.2008.070904
115. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) 87:493–506. doi: 10.1016/S0092-8674(00)81369-0
116. Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet M-F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. (2003) 465:11–26. doi: 10.1002/cne.10776
117. Leggio GM, Catania MV, Puzzo D, Spatuzza M, Pellitteri R, Gulisano W, et al. The antineoplastic drug flavopiridol reverses memory impairment induced by Amyloid-ß1-42 oligomers in mice. Pharmacol Res. (2016) 106:10–20. doi: 10.1016/j.phrs.2016.02.007
118. Zhang M, Li J, Chakrabarty P, Bu B, Vincent I. Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann-Pick type C mice. Am J Clin Pathol. (2004) 165:843–53. doi: 10.1016/S0002-9440(10)63347-0
119. Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann–Pick Type C murine model. J Neurosci. (2002) 22:6515–25. doi: 10.1523/JNEUROSCI.22-15-06515.2002
120. Engel T, Goñi-Oliver P, Lucas J, Avila J, Hernández F. Chronic lithium administration to FTDP-17 tau and GSK-3β overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem. (2006) 99:1445–55. doi: 10.1111/j.1471-4159.2006.04139.x
121. Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis. (2003) 5:301–8. doi: 10.3233/JAD-2003-5405
122. Caccamo A, Oddo S, Tran LX, LaFerla FM. Lithium reduces tau phosphorylation but not Aβ or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. (2007) 170:1669–75. doi: 10.2353/ajpath.2007.061178
123. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. (2005) 102:6990–5. doi: 10.1073/pnas.0500466102
124. Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz H-J, et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis. (2015) 45:75–88. doi: 10.3233/JAD-141959
125. Matsunaga S, Kishi T, Annas P, Basun H, Hampel H, Iwata N. Lithium as a Treatment for Alzheimer's Disease: a systematic review and meta-analysis. J Alzheimers Dis. (2015) 48:403–10. doi: 10.3233/JAD-150437
126. Andén NE, Dalén P, Johansson B. Baclofen and lithium in Huntington's chorea. Lancet. (1973) 2:93. doi: 10.1016/S0140-6736(73)93285-6
127. Vestergaard P, Baastrup PC, Petersson H. Lithium treatment of Huntington's chorea. A placebo-controlled clinical trial. Acta Psychiatr Scand. (1977) 56:183–8. doi: 10.1111/j.1600-0447.1977.tb03561.x
128. Wood NI, Morton AJ. Chronic lithium chloride treatment has variable effects on motor behaviour and survival of mice transgenic for the Huntington's disease mutation. Brain Res Bull. (2003) 61:375–83. doi: 10.1016/S0361-9230(03)00141-2
129. Scheuing L, Chiu C-T, Liao H-M, Linares GR, Chuang D-M. Preclinical and clinical investigations of mood stabilizers for Huntington's disease: what have we learned? Int J Biol Sci. (2014) 10:1024–38. doi: 10.7150/ijbs.9898
130. Ishii T, Hashimoto E, Ukai W, Tateno M, Yoshinaga T, Saito S, et al. Lithium-induced suppression of transcription repressor NRSF/REST: effects on the dysfunction of neuronal differentiation by ethanol. Eur J Pharmacol. (2008) 593:36–43. doi: 10.1016/j.ejphar.2008.07.021
131. Kopnisky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang D-M. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. (2003) 116:425–35. doi: 10.1016/S0306-4522(02)00573-0
132. Nonaka S, Hough CJ, Chuang D-M. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-d-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci USA. (1998) 95:2642–7. doi: 10.1073/pnas.95.5.2642
133. Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacol. (2001) 158:100–6. doi: 10.1007/s002130100871
134. Hellweg R, Lang UE, Nagel M, Baumgartner A. Subchronic treatment with lithium increases nerve growth factor content in distinct brain regions of adult rats. Mol Psychiatry. (2002) 7:604–8. doi: 10.1038/sj.mp.4001042
135. Jornada LK, Moretti M, Valvassori SS, Ferreira CL, Padilha PT, Arent CO, et al. Effects of mood stabilizers on hippocampus and amygdala BDNF levels in an animal model of mania induced by ouabain. J Psychiatr Res. (2010) 44:506–10. doi: 10.1016/j.jpsychires.2009.11.002
136. Yasuda S, Liang M-H, Marinova Z, Yahyavi A, Chuang D-M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. (2009) 14:51–9. doi: 10.1038/sj.mp.4002099
137. Akkouh IA, Skrede S, Holmgren A, Ersland KM, Hansson L, Bahrami S, et al. Exploring lithium's transcriptional mechanisms of action in bipolar disorder: a multi-step study. Neuropsychopharmacology. (2020) 45:947–55. doi: 10.1038/s41386-019-0556-8
138. Farah R, Khamisy-Farah R, Amit T, Youdim MBH, Arraf Z. Lithium's gene expression profile, relevance to neuroprotection a cDNA microarray study. Cell Mol Neurobiol. (2013) 33:411–20. doi: 10.1007/s10571-013-9907-x
139. Danivas V, Moily NS, Thimmaiah R, Muralidharan K, Purushotham M, Muthane U, et al. Off label use of lithium in the treatment of Huntington's disease: a case series. Indian J Psychiatry. (2013) 55:81–3. doi: 10.4103/0019-5545.105522
140. Leonard DP, Kidson MA, Brown JG, Shannon PJ, Taryan S. A double blind trial of lithium carbonate and haloperidol in Huntington's chorea. Aust N Z J Psychiatry. (1975) 9:115–8. doi: 10.3109/00048677509159834
141. Lim NKH, Hung LW, Pang TY, Mclean CA, Liddell JR, Hilton JB, et al. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington's disease affected brain. Hum Mol Genet. (2014) 23:4051–63. doi: 10.1093/hmg/ddu119
142. Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. (2005) 166:1761–71. doi: 10.1016/S0002-9440(10)62486-8
143. Xifró X, Giralt A, Saavedra A, García-Martínez JM, Díaz-Hernández M, Lucas JJ, et al. Reduced calcineurin protein levels and activity in exon-1 mouse models of Huntington's disease: Role in excitotoxicity. Neurobiol Dis. (2009) 36:461–9. doi: 10.1016/j.nbd.2009.08.012
144. Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS ONE. (2012) 7:e49838. doi: 10.1371/journal.pone.0049838
145. Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. (1999) 24:186–91. doi: 10.1016/S0968-0004(99)01375-4
146. Kins S, Crameri A, Evans DR, Hemmings BA, Nitsch RM, Gotz J. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem. (2001) 276:38193–200. doi: 10.1074/jbc.M102621200
147. Rui M, Ng KS, Tang Q, Bu S, Yu F. Protein phosphatase PP2A regulates microtubule orientation and dendrite pruning in Drosophila. EMBO Rep. (2020) 21:e48843. doi: 10.15252/embr.201948843
148. Viquez NM, Li CR, Wairkar YP, DiAntonio A. The B′ protein phosphatase 2A regulatory subunit well-rounded regulates synaptic growth and cytoskeletal stability at the drosophila neuromuscular junction. J Neurosci. (2006) 26:9293–303. doi: 10.1523/JNEUROSCI.1740-06.2006
149. Wang J, Xie R, Kou X, Liu Y, Qi C, Liu R, et al. A protein phosphatase 2A deficit in the hippocampal CA1 area impairs memory extinction. Mol Brain. (2019) 12:51. doi: 10.1186/s13041-019-0469-9
150. Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA. (2010) 107:21830–5. doi: 10.1073/pnas.0912793107
151. Wei H, Zhang H, Wang X, Xie J, An D, Wan L, et al. Direct activation of protein phosphatase 2A (PP2A) by tricyclic sulfonamides ameliorates Alzheimer's disease pathogenesis in cell and animal models. Neurotherapeutics. (2020) doi: 10.1007/s13311-020-00841-6
152. Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. (2004) 566:261–9. doi: 10.1016/j.febslet.2004.04.047
153. Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. (2010) 65:178–90. doi: 10.1016/j.neuron.2010.01.008
154. Wu H-M, Tzeng N-S, Qian L, Wei S-J, Hu X, Chen S-H, et al. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. (2009) 34:2344–57. doi: 10.1038/npp.2009.64
155. Beister A, Kraus P, Kuhn W, Dose M, Weindl A, Gerlach M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington's disease. J Neural Transm Suppl. (2004) 68:117–22. doi: 10.1007/978-3-7091-0579-5_14
156. Cankurtaran ES, Ozalp E, Soygur H, Cakir A. Clinical experience with risperidone and memantine in the treatment of Huntington's disease. J Natl Med Assoc. (2006) 98:1353–5.
157. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. (2015) 148:114–31. doi: 10.1016/j.pharmthera.2014.11.016
158. Wlodarchak N, Xing Y. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol. (2016) 51:162–84. doi: 10.3109/10409238.2016.1143913
159. Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain. (2009) 2:33. doi: 10.1186/1756-6606-2-33
160. Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol. (2013) 4:114. doi: 10.3389/fneur.2013.00114
161. Braak H, Braak E. Staging of alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. (1995) 16:271–8. doi: 10.1016/0197-4580(95)00021-6
162. Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer's disease. In: Jellinger K, Fazekas F, Windisch M, editors. Ageing and Dementia. Vienna: Springer. (1998). p. 127–40. doi: 10.1007/978-3-7091-6467-9_11
163. Iqbal K, Alonso A del C, Grundke-Iqbal I. Cytosolic abnormally hyperphosphorylated tau but not paired helical filaments sequester normal MAPs and inhibit microtubule assembly. J Alzheimers Dis. (2008) 14:365–70. doi: 10.3233/JAD-2008-14402
164. Takashima A, Soeda Y. Granular tau oligomer as toxic tau aggregation form in AD. Alzheimers Dement. (2014) 10:P161. doi: 10.1016/j.jalz.2014.04.150
165. Kopeikina KJ, Hyman BT, Spires-Jones TL. Soluble forms of tau are toxic in Alzheimer's disease. Transl Neurosci. (2012) 3:223–33. doi: 10.2478/s13380-012-0032-y
166. Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. (2007) 27:3650–62. doi: 10.1523/JNEUROSCI.0587-07.2007
167. Hoffmann NA, Dorostkar MM, Blumenstock S, Goedert M, Herms J. Impaired plasticity of cortical dendritic spines in P301S tau transgenic mice. Acta Neuropathol Commun. (2013) 1:82. doi: 10.1186/2051-5960-1-82
168. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. (2011) 6:39. doi: 10.1186/1750-1326-6-39
169. Kopeikina KJ, Polydoro M, Tai H-C, Yaeger E, Carlson GA, Pitstick R, et al. Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. (2013) 521:1334–53. doi: 10.1002/cne.23234
170. Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, et al. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. (2010) 223:385–93. doi: 10.1016/j.expneurol.2009.07.029
171. Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. (2007) 53:337–51. doi: 10.1016/j.neuron.2007.01.010
172. Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rábano A, Avila J, et al. The influence of phospho-τ on dendritic spines of cortical pyramidal neurons in patients with Alzheimer's disease. Brain. (2013) 136(Pt 6):1913–28. doi: 10.1093/brain/awt088
173. Murmu RP, Li W, Holtmaat A, Li J-Y. Dendritic spine instability leads to progressive neocortical spine loss in a mouse model of Huntington's Disease. J Neurosci. (2013) 33:12997–3009. doi: 10.1523/JNEUROSCI.5284-12.2013
174. Bulic B, Pickhardt M, Mandelkow E-M, Mandelkow E. Tau protein and tau aggregation inhibitors. Neuropharmacology. (2010) 59:276–89. doi: 10.1016/j.neuropharm.2010.01.016
175. Crowe A, James MJ, Lee VM-Y, Smith AB, Trojanowski JQ, Ballatore C, et al. Aminothienopyridazines and methylene blue affect tau fibrillization via cysteine oxidation. J Biol Chem. (2013) 288:11024–37. doi: 10.1074/jbc.M112.436006
176. Soeda Y, Saito M, Maeda S, Ishida K, Nakamura A, Kojima S, et al. Methylene blue inhibits formation of tau fibrils but not of granular tau oligomers: a plausible key to understanding failure of a clinical trial for Alzheimer's disease. J Alzheimers Dis. (2019) 68:1677–86. doi: 10.3233/JAD-181001
177. Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, et al. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS ONE. (2012) 7:e52389. doi: 10.1371/journal.pone.0052389
178. Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, et al. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington's disease models. J Neurosci. (2012) 32:11109–19. doi: 10.1523/JNEUROSCI.0895-12.2012
179. Alam R, Driver D, Wu S, Lozano E, Key SL, Hole JT, et al. Preclinical characterization of an antibody [LY3303560] targeting aggregated tau. Alzheimers Dement. (2017) 13:P592–3. doi: 10.1016/j.jalz.2017.07.227
180. Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. (2014) 40(Suppl. 1):S97–111. doi: 10.3233/JAD-132477
181. Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, López-Deber MP, Reis P, et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS ONE. (2013) 8:e72301. doi: 10.1371/journal.pone.0072301
182. Smith BR, Nelson KM, Kemper LJ, Leinonen-Wright K, Petersen A, Keene CD, et al. A soluble tau fragment generated by caspase-2 is associated with dementia in Lewy body disease. Acta Neuropathol Commun. (2019) 7:124. doi: 10.1186/s40478-019-0765-8
183. Selenica M-LB, Davtyan H, Housley SB, Blair LJ, Gillies A, Nordhues BA, et al. Epitope analysis following active immunization with tau proteins reveals immunogens implicated in tau pathogenesis. J Neuroinflammation. (2014) 11:152. doi: 10.1186/s12974-014-0152-0
184. Castillo-Carranza DL, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, et al. Passive immunization with tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci. (2014) 34:4260–72. doi: 10.1523/JNEUROSCI.3192-13.2014
185. Paholikova K, Salingova B, Opattova A, Skrabana R, Majerova P, Zilka N, et al. N-terminal truncation of microtubule associated protein tau dysregulates its cellular localization. J Alzheimers Dis. (2015) 43:915–26. doi: 10.3233/JAD-140996
186. AC Immune SA. A Study to Evaluate the Safety, Tolerability and Immunogenicity of Tau Targeted Vaccines in Participants With Early Alzheimer's Disease. Report No. NCT04445831 (2020). Available online at: www.clinicaltrials.gov (accessed October 7, 2020).
187. Axon Neuroscience SE. Safety Study of AADvac1, a Tau Peptide-KLH-Conjugate Active Vaccine to Treat Alzheimer's Disease. Report No. NCT01850238 (2013). Available online at: www.clinicaltrials.gov (accessed October 7, 2020).
188. Fuller JP, Stavenhagen JB, Teeling JL. New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer's Disease. Front Neurosci. (2014) 8:235. doi: 10.3389/fnins.2014.00235
189. McEwan WA, Falcon B, Vaysburd M, Clift D, Oblak AL, Ghetti B, et al. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc Natl Acad Sci USA. (2017) 114:574–9. doi: 10.1073/pnas.1607215114
190. d'Abramo C, Acker CM, Jimenez H, Davies P. Passive immunization in JNPL3 transgenic mice using an array of phospho-tau specific antibodies. PLoS ONE. (2015) 10:e0135774. doi: 10.1371/journal.pone.0135774
191. Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. (1997) 48:128–32. doi: 10.1002/(SICI)1097-4547(19970415)48:2<128::AID-JNR5>3.0.CO;2-E
192. Eli Lilly and Company. Assessment of Safety, Tolerability, and Efficacy of LY3303560 in Early Symptomatic Alzheimer's Disease. Report No. NCT03518073. (2020). Available online at: clinicaltrials.gov (accessed Jun 23, 2020).
193. Evans LD, Wassmer T, Fraser G, Smith J, Perkinton M, Billinton A, et al. Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. (2018) 22:3612–24. doi: 10.1016/j.celrep.2018.03.021
194. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. (2009) 284:12845–52. doi: 10.1074/jbc.M808759200
195. Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem. (2015) 290:14893–903. doi: 10.1074/jbc.M115.652693
196. Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer's disease and PART. Acta Neuropathol. (2018) 136:57–67. doi: 10.1007/s00401-018-1855-6
197. Abounit S, Wu JW, Duff K, Victoria GS, Zurzolo C. Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion. (2016) 10:344–51. doi: 10.1080/19336896.2016.1223003
198. Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, de Strooper B, et al. Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. (2015) 11:1176–83. doi: 10.1016/j.celrep.2015.04.043
199. Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. (2013) 288:1856–70. doi: 10.1074/jbc.M112.394528
200. Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem. (2012) 287:19440–51. doi: 10.1074/jbc.M112.346072
201. Plotkin SS, Cashman NR. Passive immunotherapies targeting Aβ and tau in Alzheimer's disease. Neurobiol Dis. (2020) 144:e105010. doi: 10.1016/j.nbd.2020.105010
202. Mullard A. Stem-cell discovery platforms yield first clinical candidates. Nat Rev Drug Disc. (2015) 14:589–91. doi: 10.1038/nrd4708
203. Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging. (2015) 36:693–709. doi: 10.1016/j.neurobiolaging.2014.09.007
204. Lee S-H, Le Pichon CE, Adolfsson O, Gafner V, Pihlgren M, Lin H, et al. Antibody-mediated targeting of tau in vivo does not require effector function and microglial engagement. Cell Rep. (2016) 16:1690–700. doi: 10.1016/j.celrep.2016.06.099
205. Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. (2013) 80:402–14. doi: 10.1016/j.neuron.2013.07.046
206. Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, et al. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol. (2015) 2:278–88. doi: 10.1002/acn3.176
207. Boxer AL, Qureshi I, Ahlijanian M, Grundman M, Golbe LI, Litvan I, et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol. (2019) 18:549–58. doi: 10.1016/S1474-4422(19)30139-5
208. Jadhav S, Avila J, Schöll M, Kovacs GG, Kövari E, Skrabana R, et al. A walk through tau therapeutic strategies. Acta Neuropathol Commun. (2019) 7:22. doi: 10.1186/s40478-019-0664-z
209. Constantinescu R, Romer M, Zetterberg H, Rosengren L, Kieburtz K. Increased levels of total tau protein in the cerebrospinal fluid in Huntington's disease. Parkinsonism Relat Disord. (2011) 17:714–5. doi: 10.1016/j.parkreldis.2011.06.010
210. Niemelä V, Landtblom A-M, Blennow K, Sundblom J. Tau or neurofilament light-Which is the more suitable biomarker for Huntington's disease? PLoS ONE. (2017) 12:e0172762. doi: 10.1371/journal.pone.0172762
211. Rodrigues FB, Byrne LM, McColgan P, Robertson N, Tabrizi SJ, Zetterberg H, et al. Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington's disease. PLoS ONE. (2016) 11:e0163479. doi: 10.1371/journal.pone.0163479
212. Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. (2016) 15:673–84. doi: 10.1016/S1474-4422(16)00070-3
213. Denis HL, David LS, Cicchetti F. Antibody-based therapies for Huntington's disease: current status and future directions. Neurobiol Dis. (2019) 132:104569. doi: 10.1016/j.nbd.2019.104569
214. Wagh A, Law B. Methods for conjugating antibodies to nanocarriers. Methods Mol Biol. (2013) 1045:249–66. doi: 10.1007/978-1-62703-541-5_15
215. Bartl S, Oueslati A, Southwell AL, Siddu A, Parth M, David LS, et al. Inhibiting cellular uptake of mutant huntingtin using a monoclonal antibody: implications for the treatment of Huntington's disease. Neurobiol Dis. (2020) 141:104943. doi: 10.1016/j.nbd.2020.104943
216. Cicchetti F, Lacroix S, Cisbani G, Vallières N, Saint-Pierre M, St-Amour I, et al. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol. (2014) 76:31–42. doi: 10.1002/ana.24174
217. Gosset P, Maxan A, Alpaugh M, Breger L, Dehay B, Tao Z, et al. Evidence for the spread of human-derived mutant huntingtin protein in mice and non-human primates. Neurobiol Dis. (2020) 141:104941. doi: 10.1016/j.nbd.2020.104941
218. Jeon I, Cicchetti F, Cisbani G, Lee S, Li E, Bae J, et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. (2016) 132:577–92. doi: 10.1007/s00401-016-1582-9
219. Masnata M, Cicchetti F. The evidence for the spread and seeding capacities of the mutant huntingtin protein in in vitro systems and their therapeutic implications. Front Neurosci. (2017) 11:647. doi: 10.3389/fnins.2017.00647
220. Masnata M, Sciacca G, Maxan A, Bousset L, Denis HL, Lauruol F, et al. Demonstration of prion-like properties of mutant huntingtin fibrils in both in vitro and in vivo paradigms. Acta Neuropathol. (2019) 137:981–1001. doi: 10.1007/s00401-019-01973-6
221. Maxan A, Sciacca G, Alpaugh M, Tao Z, Breger L, Dehay B, et al. Use of adeno-associated virus-mediated delivery of mutant huntingtin to study the spreading capacity of the protein in mice and non-human primates. Neurobiol Dis. (2020) 141:104951. doi: 10.1016/j.nbd.2020.104951
222. Rieux M, Alpaugh M, Sciacca G, Saint-Pierre M, Masnata M, Denis HL, et al. Shedding a new light on Huntington's disease: how blood can both propagate and ameliorate disease pathology. Mol Psychiatry. (2020) doi: 10.1038/s41380-020-0787-4
223. Giehl K, Reetz K, Dogan I, Werner CJ, Schulz JB, Hammes J, et al. Tau pathology in Huntington's disease: a brief in vivo PET-imaging report. Basal Ganglia. (2017) 8:13. doi: 10.1016/j.baga.2017.02.038
224. Pascoal TA, Shin M, Kang MS, Chamoun M, Chartrand D, Mathotaarachchi S, et al. In vivo quantification of neurofibrillary tangles with [18F]MK-6240. Alzheimer's Res Ther. (2018) 10:74. doi: 10.1186/s13195-018-0402-y
225. Gong N-J, Chan C-C, Leung L-M, Wong C-S, Dibb R, Liu C. Differential microstructural and morphological abnormalities in mild cognitive impairment and Alzheimer's disease: evidence from cortical and deep gray matter. Hum Brain Mapp. (2017) 38:2495–508. doi: 10.1002/hbm.23535
226. Praprotnik D, Smith MA, Richey PL, Vinters HV, Perry G. Filament heterogeneity within the dystrophic neurites of senile plaques suggests blockage of fast axonal transport in Alzheimer's disease. Acta Neuropathol. (1996) 91:226–35. doi: 10.1007/s004010050420
227. Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. (1990) 11:29–37. doi: 10.1016/0197-4580(90)90059-9
228. Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. (2003) 40:41–52. doi: 10.1016/S0896-6273(03)00569-5
229. Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA. (2005) 102:227–31. doi: 10.1073/pnas.0406361102
230. Tsai RM, Miller Z, Koestler M, Rojas JC, Ljubenkov PA, Rosen HJ, et al. Reactions to multiple ascending doses of the microtubule stabilizer TPI-287 in patients with Alzheimer disease, progressive supranuclear palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol. (2019) 77:215–24. doi: 10.1001/jamaneurol.2019.3812
231. Gozes I, Divinski I. NAP, a neuroprotective drug candidate in clinical trials, stimulates microtubule assembly in the living cell. Curr Alzheimer Res. (2007) 4:507–9. doi: 10.2174/156720507783018208
232. Sari Y. Activity-dependent neuroprotective protein-derived peptide, NAP, preventing alcohol-induced apoptosis in fetal brain of C57BL/6 mouse. Neuroscience. (2009) 158:1426–35. doi: 10.1016/j.neuroscience.2008.11.021
233. Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M, et al. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord. (2013) 35:325–36. doi: 10.1159/000348347
234. Sudo H, Baas PW. Strategies for diminishing katanin-based loss of microtubules in tauopathic neurodegenerative diseases. Hum Mol Genet. (2011) 20:763–78. doi: 10.1093/hmg/ddq521
235. Melo TQ, van Zomeren KC, Ferrari MFR, Boddeke HWGM, Copray JCVM. Impairment of mitochondria dynamics by human A53T α-synuclein and rescue by NAP (davunetide) in a cell model for Parkinson's disease. Exp Brain Res. (2017) 235:731–42. doi: 10.1007/s00221-016-4836-9
236. Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, et al. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. (2007) 323:438–49. doi: 10.1124/jpet.107.129551
237. Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. (2014) 13:676–85. doi: 10.1016/S1474-4422(14)70088-2
238. Weaver BA. How taxol/paclitaxel kills cancer cells. Mol Biol Cell. (2014) 25:2677–81. doi: 10.1091/mbc.e14-04-0916
239. Caparica R, Lambertini M, Azambuja E de. How I treat metastatic triple-negative breast cancer. ESMO Open. (2019) 4(Suppl. 2):e000504. doi: 10.1136/esmoopen-2019-000504
240. Quick QA. Epothilone B induces glioblastoma cell death via survivin down-regulation. Exp Oncol. (2008) 30:195–201.
241. Varidaki A, Hong Y, Coffey ET. Repositioning microtubule stabilizing drugs for brain disorders. Front Cell Neurosci. (2018) 12:226. doi: 10.3389/fncel.2018.00226
242. Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S, et al. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol. (2002) 51:525–30. doi: 10.1002/ana.10163
243. Iyer A, Lapointe NE, Zielke K, Berdynski M, Guzman E, Barczak A, et al. A novel MAPT mutation, G55R, in a frontotemporal dementia patient leads to altered tau function. PLoS ONE. (2013) 8:e76409. doi: 10.1371/journal.pone.0076409
244. Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, et al. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol. (2000) 48:939–43. doi: 10.1002/1531-8249(200012)48:6<939::AID-ANA17>3.0.CO;2-1
245. Stanford PM, Brooks WS, Teber ET, Hallupp M, McLean C, Halliday GM, et al. Frequency of tau mutations in familial and sporadic frontotemporal dementia and other tauopathies. J Neurol. (2004) 251:1098–104. doi: 10.1007/s00415-004-0489-x
246. Caffrey TM, Wade-Martins R. Functional MAPT haplotypes: Bridging the gap between genotype and neuropathology. Neurobiol Dis. (2007) 27:1–10. doi: 10.1016/j.nbd.2007.04.006
247. Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, et al. Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron. (2016) 90:941–7. doi: 10.1016/j.neuron.2016.04.042
248. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. (2017) 133:665–704. doi: 10.1007/s00401-017-1707-9
249. Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. (2012) 18:291–5. doi: 10.1038/nm.2613
250. Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett. (2000) 279:129–32. doi: 10.1016/S0304-3940(99)00964-7
251. DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. (2017) 9:eaag0481. doi: 10.1126/scitranslmed.aag0481
252. Ionis Pharmaceuticals, Inc. A Randomized, Double-Blind, Placebo-Controlled Study, Followed by an Open-Label Extension, to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Multiple Ascending Doses of Intrathecally Administered ISIS 814907 in Patients With Mild Alzheimer's Disease. Report No.: NCT03186989 (2020). Available online at: clinicaltrials.gov (accessed June 23, 2020).
253. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting huntingtin expression in patients with Huntington's Disease. N Engl J Med. (2019) 380:2307–16. doi: 10.1056/NEJMoa1900907
254. Mullard A. Pioneering antisense drug heads into pivotal trials for Huntington disease. Nat Rev Drug Discov. (2019) 18:161–3. doi: 10.1038/d41573-019-00018-7
255. Smith AV, Tabrizi SJ. Therapeutic antisense targeting of huntingtin. DNA Cell Biol. (2019) 39:154–8. doi: 10.1089/dna.2019.5188
256. Fernández-Nogales M, Lucas JJ. Altered levels and isoforms of tau and nuclear membrane invaginations in Huntington's disease. Front Cell Neurosci. (2019) 13:574. doi: 10.3389/fncel.2019.00574
257. Oberemok VV, Laikova KV, Repetskaya AI, Kenyo IM, Gorlov MV, Kasich IN, et al. A Half-century history of applications of antisense oligonucleotides in medicine, agriculture and forestry: we should continue the journey. Molecules. (2018) 23:1302. doi: 10.3390/molecules23061302
258. Mena R, Edwards PC, Harrington CR, Mukaetova-Ladinska EB, Wischik CM. Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer's disease. Acta Neuropathol. (1996) 91:633–41. doi: 10.1007/s004010050477
259. Nacharaju P, Ko L, Yen SH. Characterization of in vitro glycation sites of tau. J Neurochem. (1997) 69:1709–19. doi: 10.1046/j.1471-4159.1997.69041709.x
260. Reynolds MR, Berry RW, Binder LI. Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: implications for Alzheimer's disease. Biochemistry. (2005) 44:1690–700. doi: 10.1021/bi047982v
261. Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. (2006) 281:10825–38. doi: 10.1074/jbc.M512786200
262. Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: Implications for Alzheimer's disease. Neurochem Int. (2011) 58:458–71. doi: 10.1016/j.neuint.2010.12.023
263. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. (2004) 114:121–30. doi: 10.1172/JCI200420640
264. Boxer A. A 6 Month, Open-Label, Pilot Futility Clinical Trial of Oral Salsalate for Progressive Supranuclear Palsy. Report No.: NCT02422485 (2018). Available online at: clinicaltrials.gov (accessed July 1, 2020).
265. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. (2000) 75:436–9. doi: 10.1046/j.1471-4159.2000.0750436.x
266. Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. (2016) 22:46–53. doi: 10.1038/nm.4011
267. van Leeuwen FW, Hol EM, Fischer DF. Frameshift proteins in Alzheimer's disease and in other conformational disorders: time for the ubiquitin-proteasome system. J Alzheimers Dis. (2006) 9(3 Suppl):319–25. doi: 10.3233/JAD-2006-9S336
268. Lokireddy S, Kukushkin NV, Goldberg AL. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci USA. (2015) 112:E7176–85. doi: 10.1073/pnas.1522332112
269. Ortega Z, Lucas JJ. Ubiquitin–proteasome system involvement in Huntington's disease. Front Mol Neurosci. (2014) 7:77. doi: 10.3389/fnmol.2014.00077
270. DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. (1997) 277:1990–3. doi: 10.1126/science.277.5334.1990
271. Bett JS, Cook C, Petrucelli L, Bates GP. The ubiquitin-proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's Disease. PLoS ONE. (2009) 4:e5128. doi: 10.1371/journal.pone.0005128
272. Ortega Z, Díaz-Hernández M, Maynard CJ, Hernández F, Dantuma NP, Lucas JJ. Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J Neurosci. (2010) 30:3675–88. doi: 10.1523/JNEUROSCI.5673-09.2010
273. Schipper-Krom S, Juenemann K, Jansen AH, Wiemhoefer A, Nieuwendijk R van den, Smith DL, et al. Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. (2014) 588:151–9. doi: 10.1016/j.febslet.2013.11.023
274. Nekrasov ED, Vigont VA, Klyushnikov SA, Lebedeva OS, Vassina EM, Bogomazova AN, et al. Manifestation of Huntington's disease pathology in human induced pluripotent stem cell-derived neurons. Mol Neurodegener. (2016) 11:27. doi: 10.1186/s13024-016-0092-5
275. Seo H, Sonntag K-C, Kim W, Cattaneo E, Isacson O. Proteasome activator enhances survival of Huntington's disease neuronal model cells. PLoS ONE. (2007) 2:e238. doi: 10.1371/journal.pone.0000238
276. Boxer AL, Yu J-T, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. (2017) 16:552–63. doi: 10.1016/S1474-4422(17)30157-6
Keywords: tauopathy, tau hyperphosphorylation, cognitive deficits, tau-targeting treatments, tau aggregation inhibitors, tau immunotherapy, microtubule stabilizers, gene silencing
Citation: Masnata M, Salem S, de Rus Jacquet A, Anwer M and Cicchetti F (2020) Targeting Tau to Treat Clinical Features of Huntington's Disease. Front. Neurol. 11:580732. doi: 10.3389/fneur.2020.580732
Received: 06 July 2020; Accepted: 17 September 2020;
Published: 19 November 2020.
Edited by:
Sonia Do Carmo, McGill University, CanadaReviewed by:
Rocio Gomez-Pastor, University of Minnesota Twin Cities, United StatesJose Lucas, Consejo Superior de Investigaciones Científicas (CSIC), Spain
Copyright © 2020 Masnata, Salem, de Rus Jacquet, Anwer and Cicchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Cicchetti, ZnJhbmNlc2NhLmNpY2NoZXR0aUBjcmNodWRlcXVlYmVjLnVsYXZhbC5jYQ==
†These authors have contributed equally to this work