 Chenyu Gao
Chenyu Gao Ting Huang†
Ting Huang†- Department of Neurology, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
Around 15% of patients with Parkinson's disease (PD) have a family history, and 5–10% have confirmed genetic causes. PRKN is the most common gene responsible for early-onset Parkinson's disease (EOPD), while rare variants of PLA2G6 likely raise PD susceptibility in the Chinese population. We investigated the genetic information of 13 members of a Han Chinese family with known EOPD by whole-exome sequencing and Sanger sequencing, and analyzed the clinical history, physical examination, blood laboratory test, and brain imaging data of the patients. Two members, including the proband, were suspected of having EOPD. A novel homozygous frameshift mutation, c.856delT, and a compound heterozygous mutation, c.1321T>C/c.856delT of PRKN, were identified, as well as two single nucleotide variants of PLA2G6 and TENM4. The proband exhibited a rare symmetrical resting tremor limited to her lower limbs and never exhibited signs of rigidity. 18F-DOPA PET/CT scan indicated a symmetrical reduced signaling in the striatum. The novel frameshift mutation and compound heterozygous mutation of PRKN are likely to be the genetic causes of EOPD in this family.
Introduction
Parkinson's disease (PD), the second most common neurodegenerative disorder after Alzheimer's disease (1), affects 1% of persons over the age of 65 and 3% of those older than 80 (2). PD is a type of alpha synucleinopathy with aggregation of abnormally folded α-synuclein proteins, which form intracellular inclusions within Lewy bodies and Lewy neurites, while the specific subnuclei of the substantia nigra pars compacta (SNpc) are damaged, causing the loss of dopaminergic neurons (3). In this way, PD generates a series of clinical symptoms, classified as motor symptoms and nonmotor symptoms. Resting tremor, bradykinesia, and rigidity are defined as classical parkinsonian motor symptoms, which usually develop unilaterally and later spread to the contralateral side, but ordinarily remain asymmetrical throughout the disease course (4, 5). Nonmotor symptoms include hyposmia, cognitive disorder, sleep disorder, autonomic dysfunction, and psychiatric symptoms (4, 5). To diagnose PD, not only are the mandatory criteria needed but also diagnostic exclusion criteria and red flags for PD diagnosis should be considered (4, 6).
Although PD was categorized as an idiopathic disorder due to its unknown etiology, around 15% of the patients have a family history of the disease, and 5–10% have a monogenic form of the disease with Mendelian inheritance (7, 8). At least 23 loci and 19 genes (10 autosomal dominant genes and 9 autosomal recessive genes) for parkinsonism have been identified as pathogenic up to 2018, 10 genes of which are proven to be associated with early-onset Parkinson's disease (EOPD) (9). PRKN is the most common gene responsible for EOPD, causing nearly 49% of EOPD cases with autosomal recessive inheritance and 19% of sporadic EOPD cases (10). PLA2G6 mutations were first reported to be related to adult-onset levodopa-responsive dystonia-parkinsonism in 2009 (11), and more than 18 PLA2G6 variants have been confirmed to be associated with familial dystonia-parkinsonism and PD thus far (9). It has been suggested that rare variants of PLA2G6 may raise PD susceptibility in the Chinese population (12).
In this study, we discuss a Chinese family carrying a novel frameshift mutation and a compound heterozygous mutation of PRKN, as well as two single nucleotide variants (SNVs) of PLA2G6 and TENM4. We collected the clinical data of the proband as well as the other affected member of the family, and analyzed the family pedigree by investigating the DNA sequencing results of 13 members from four generations. The proband exhibited a rare symmetrical resting tremor limited to her lower limbs, without the appearance of rigidity, which is incompatible with the MDS clinical diagnostic criteria.
Materials and Methods
Subjects
We investigated the genetic information of 13 members in a family with EOPD from the Jiangxi Province of Southern China. Two female members appeared to have symptoms of PD, while other family members did not exhibit related clinical manifestations. The two affected family members were examined by three experienced neurologists from Nanjing First Hospital, Nanjing Medical University, and data collected included clinical history review, physical examinations, and blood laboratory tests. All participants, or their legal guardians, provided informed consent. The study was approved by the ethics committee of Nanjing First Hospital, Nanjing Medical University. All procedures were conducted strictly according to the Administrative Regulations on Medical Institutions, issued by the State Council of the P.R. China.
Genetic Analysis
Peripheral blood samples were collected from 13 family participants for DNA sequencing, including two patients and 11 persons who appeared healthy. Genomic DNA was extracted following the standard protocol of the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Exomes of gDNA isolated from the two patients were captured by liquid chip technology. Samples of the two patients underwent paired-end sequencing on the Illumina HiSeq X-ten platform (Illumina Inc., San Diego, CA, USA). Sanger sequencing was performed to validate candidate variants of 13 family participants using an ABI 3730 XL genetic analyzer (Applied Biosystems Inc., Foster City, CA, USA). All potential variants were filtered according to the Single Nucleotide Polymorphism Database, the 1,000 Genomes Project (1000G), and the Genome Aggregation Database (gnomAD). Sorting Intolerant from Tolerant (SIFT) and Polymorphism Phenotyping v2 (Polyphen-2) were used to predict the impact of an amino acid substitution caused by missense mutation on the protein's structure and function. The impact of synonymous variants, short InDel variants, and variants across the intron–exon border was predicted by MutationTaster.
Neuroimaging Analysis
Computed tomography (CT) and 3.0T magnetic resonance imaging (MRI) scans were performed on the proband's head. She also underwent positron emission computed tomography (PET/CT) scan of the brain, and to eliminate interference, she was injected with imaging agent 18F-FDG 24 h after the 18F-DOPA injection after a 6-h fast. After 90 min, a static brain scan was performed in accordance with the standard operating procedures of Nanjing First Hospital, and the images were reconstructed with CT-based attenuation correction. The other patient claimed to have difficulty traveling, and thus she was unable to finish the imaging examination.
Results
Family Pedigree Map
A novel frameshift mutation and a compound heterozygous mutation of PRKN were detected in a Han Chinese family with a history of EOPD, whose members also carried other SNVs of PLA2G6 and TENM4. The family pedigree was established based on genetic analysis of 13 members from four generations, including 11 unaffected members (Figure 1A, II-1,2,5,6,7, III-2,3,5,6,9, and IV-1) and two with EOPD (Figure 1A, III-4,8). Three members of the family (Figure 1A, I-1, II-4, and IV-2) refused to participate in the study, and another five family members (Figure 1A, II-3, III-1,7,10,11) were not able to take part in the research. There is no consanguineous marriage in the family according to the proband's statement.
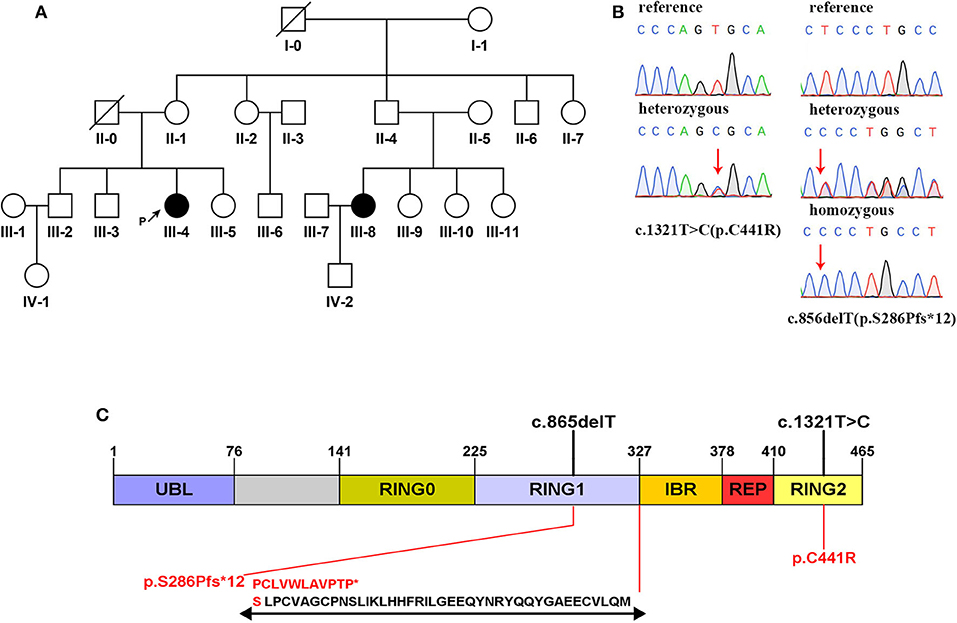
Figure 1. (A) Family pedigree map. Circle: female; square: male; black: the affected; white: the healthy; arrow with letter P on the side: the proband; slash: the deceased. (B) Sanger sequencing analysis of the mutation c.1321C>T and the mutation c.856delT. (C) Domain structure of the parkin protein (13). Two mutations detected in this study are marked in black above the icon. The amino acid changes are marked in red below the icon. Specific amino acid sequence changes caused by the frameshift mutation are shown at the bottom.
Clinical Features
The proband (III-4) is a 45-year-old woman with an 11-year history of PD-like tremor in both lower limbs. She was found to have resting tremor in two legs at the age of 34 with fatigue complaints. The symptoms gradually worsened in the following years, but still progressed at a slow rate. In 2017, a large-scale polyclinic suspected her of having PD, and she began oral medication (Madopar 125 mg tid) then. When the prescription worked well, the patient reduced the drug dose, which made the symptoms worse again. Routine and biochemical blood examination showed no significant abnormalities. Blood copper, ceruloplasmin, and liver ultrasound were normal, and no K–F rings were observed. Nevertheless, her younger cousin (III-8), another affected member of the family, appears to have typical PD symptoms, including resting tremor, rigidity, and bradykinesia. She first noticed weakness in her left leg and difficulty moving in 2005, without tremor signs at that time. In her left limb, however, she gradually developed tremor, rigidity, and bradykinesia. The symptoms peaked in 2015. She had great difficulty in drinking and dressing, and started to take Madopar at that time. Although the medicine was effective, the patient took it irregularly (125 mg tid/bid). The clinical features of these two patients are summarized in Table 1. One (III-6) of the proband's cousins has a history of psychosis.
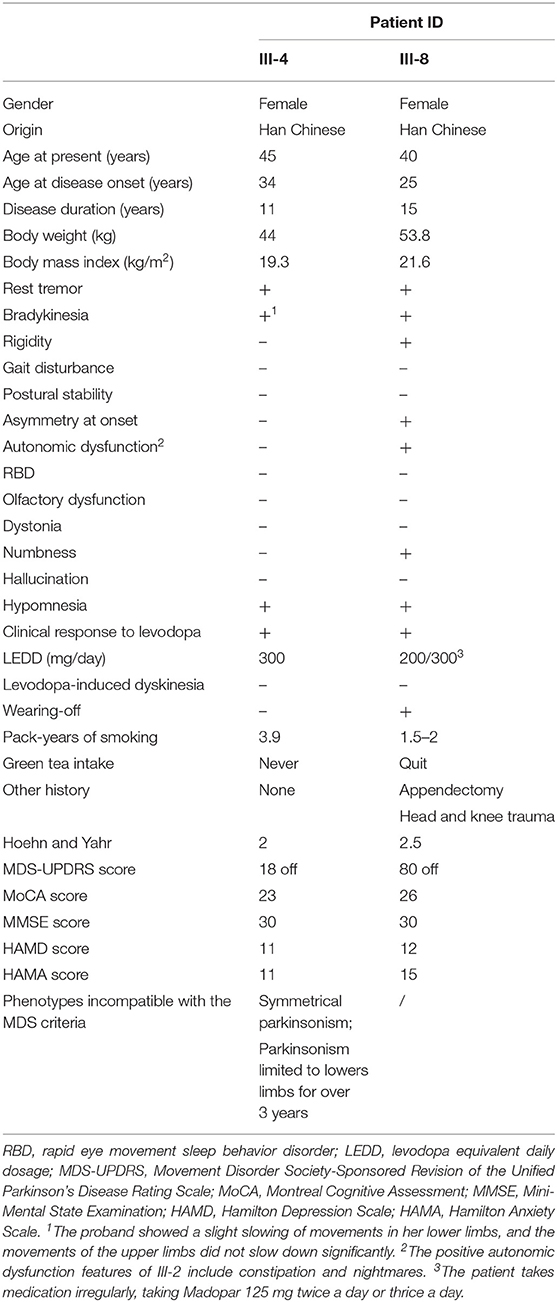
Table 1. Clinical features of the two patients in this family.
Neuroimaging
CT and 3.0T MRI scans of the proband were negative, and no obvious brainstem or cerebellar atrophy was observed (Figures 2A–D). The brain PET/CT scan showed some abnormalities. Nothing abnormal was found in her 18F-FDG PET/CT imaging, as no reduced signaling was detected in the lenticular nucleus and thalamus (Figures 2E,F). However, 18F-DOPA PET/CT scan indicated reduced signaling in the striatum, which was mostly symmetrical (Figures 2G,H).
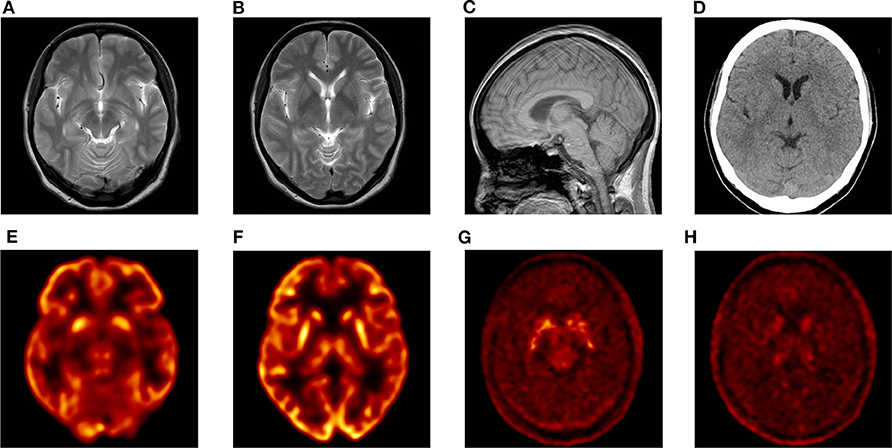
Figure 2. Neuroimaging images of the proband. (A–C) 3.0T MRI images: no obvious abnormalities. (D) CT image: no obvious abnormalities. (E,F) 18F-FDG PET/CT image: no obvious abnormalities. (G,H) 18F-DOPA PET/CT image: bilateral striatum dopamine neurons damaged.
Molecular Genetic Findings
Mutations in exon 7 and exon 12 of PRKN were detected in the patients by whole-exome sequencing, which reached an average coverage of >100 × and a minimal coverage of 20 × in 97% of the target regions. A compound heterozygous mutation, c.1321T>C (p.C441R)/c.856delT (p.S286Pfs*12), was detected in the proband (III-4), and a homozygous mutation, c.856delT (p.S286Pfs*12), was detected in her younger cousin (III-8). The two candidate mutations were validated by Sanger sequencing in all participants (Figure 1B). The proband's mother (II-1) and her affected cousin's mother (II-5) both carried a heterozygous PRKN mutation, c.856delT, and most of her relatives (II-2,6, III-5,9) carried the same heterozygous mutation. Her older brother (III-2) was found to be heterozygous for the mutation, c.1321T>C. The minor allele frequency (MAF) of c.1321T>C was recorded to be 0.00004 in gnomAD. It was classified as a disease-causing mutation (DM) in the Human Gene Mutation Database (HGMD, ID CM033800) and predicted to be “probably damaging” by Polyphen-2 and to “affect protein function” by SIFT. No allele frequency of c.856delT was recorded in gnomAD. Furthermore, it was neither recorded in HGMD nor reported by any other researchers. The prediction of MutationTaster on this mutation is that it is “disease causing.” The specific data are shown in Table 2.
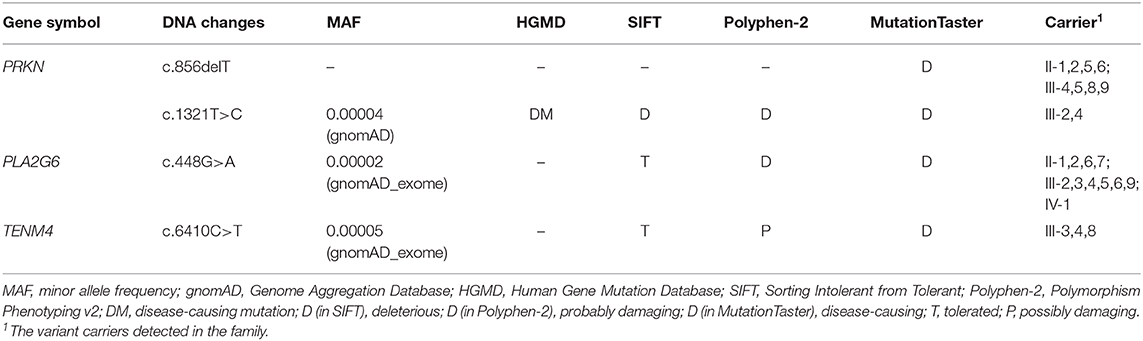
Table 2. DNA sequencing results of the family.
Heterozygous variants c.448G>A (p.A2137V) of PLA2G6 and c.6410C>T (p.E150K) of TENM4 were detected in the proband, while c.6410C>T of TENM4 was also found in the other affected family member. Details are shown in Table 2.
Discussion
PRKN, the parkin RBR E3 ubiquitin protein ligase gene located in chromosome 6q25.2–q27.7 with 12 exons, was first reported as the genetic cause of autosomal recessive juvenile parkinsonism (ARJP) in 1998 in unrelated Japanese families (14, 15). Mutations of PRKN, mainly including exon rearrangements, deletions, or missense mutations, are also common in patients with EOPD in China (13, 16). Our team identified a novel heterozygous deletion and duplication of PRKN in a Han Chinese family with EOPD recently (17). American experts first detected compound heterozygous mutation Ex5 del/c.1321T>C of PRKN in patients with PD in 2002, and missense mutation c.1321T>C has been found in Asians with EOPD in recent years (18–21). However, the frameshift mutation c.856delT found in this study has never been reported before, nor has the compound heterozygous mutation c.1321T>C/c.856delT. Therefore, we conducted further protein function analysis in silico to predict the pathogenicity of these mutations.
The mutation c.856delT detected in this family results in amino acid alternation, in which the amino acid 286 is changed from serine to proline, which generates a new reading frame, stopping prematurely at codon 12 downstream (Figure 1C). The parkin protein is an E3 ubiquitin ligase implicated in mitophagy with 465 amino acids that functions in the ubiquitin–proteasome system, and the ubiquitin-like domain interacts with the RING1 domain in negatively regulating protein activity (9, 13, 22). Meanwhile, approximately 25% of all reported PRKN mutations have been found in the RING1 domain, as well as the mutation c.856delT found in our study (13). According to the prediction of MutationTaster, the novel frameshift mutation leads to amino acid sequence and splice site changes, and the protein features might be affected, which may ultimately result in the expression of truncated protein or becoming a target for nonsense-mediated mRNA decay. Accordingly, we infer that the mutation c.856delT of PRKN is deleterious. Furthermore, the missense mutation c.1321T>C is predicted to be deleterious by SIFT and Polyphen-2 and reported to be expected to affect ubiquitination (20). Therefore, it is reasonable to consider the mutation c.1321T>C as pathogenic, too.
For further cosegregation analysis, we performed direct sequencing of candidate genes on 13 members of this family. Mutation c.856delT was found in eight family members, while mutation c.1321T>C was found in two of them. All of the unaffected members carried a single heterozygous type. It can easily be determined that the mutation c.856delT is inherited from the proband's mother. Thus, we infer that the proband's father could be heterozygous for c.1321T>C, and her affected cousin's father (II-4), just like his siblings, carried a single heterozygous mutation, c.856delT. From the analysis above, these two candidate mutations are cosegregated from the disease phenotype in the family, and we believe that they are likely to be the genetic cause of PD in this family.
However, there were differences in the onset and manifestation of PD in the two patients of the family. The clinical features of PRKN-related PD were notable for the following points: early onset, mostly before 45 years old, responsive to levodopa, progressing slowly, commonly exhibiting dystonia, nearly half exhibiting hyperreflexia, no observed olfactory dysfunction, and limited suffering from dementia (9, 23–28). Patient III-8 appears to have typical symptoms of PRKN-related PD, such as early onset, slow progression, and no olfactory or cognitive dysfunction. In contrast, III-4 has a symmetrical tremor only in her legs, which has never extended to her upper limbs in the past 11 years. Moreover, she never exhibits rigidity and is more flexible than her affected cousin. According to the MDS clinical diagnostic criteria for PD mentioned previously (6), III-4 has one red flag (symmetrical parkinsonism, offset by one supported criteria) and one absolute exclusion criteria (parkinsonism limited to lower limbs for over 3 years). Principally, she would not be clinically diagnosed with PD for appearing incompatible with the diagnostic criteria. However, III-8 could be clinically diagnosed according to the criteria mentioned before, and she carries a homozygous frameshift mutation of PRKN, which is carried as heterozygous type by III-4. Based on our inferences above, the two affected members of the family could both be genetically diagnosed with PD.
Furthermore, 18F-DOPA PET/CT scan of III-4 showed reduced signaling in the bilateral striatum. Combining her family history, genetic examination results, and PET/CT imaging, she could be considered to have PD. However, the symmetrical reduced signaling in the 18F-DOPA PET/CT scan seems atypical among the images of patients with PD. Although there is a report suggesting that patients with DJ1-related PD exhibit early-onset levodopa-responsive symmetrical parkinsonism (29), to the best of our knowledge, there is no report of another patient with PD with an 11-year duration who exhibits symmetrical tremor exclusively limited to the lower limbs and no manifestation of rigidity. The patient appears incompatible with the MDS clinical diagnostic criteria, and although it may be an individual case, we, nonetheless, think it helps to enrich our understanding of PD diagnosis. Furthermore, the cause of the particular phenotype may also be genetic.
Additionally, we found that heterozygous variants c.448G>A (p.A2137V) of PLA2G6 and c.6410C>T (p.E150K) of TENM4 in the proband were predicted as “tolerated” by SIFT. Predictions on Polyphen reveal that the variant-locating sequences are relatively conserved, and the MAFs recorded in gnomAD are both lower than 0.01. Moreover, the variant c.6410C>T is also identified as a heterozygous type in the other affected family member. Studies show that some homozygous and compound heterozygous mutations of PLA2G6 were found to be associated with EOPD in the Asian population (30–32). A homozygous missense mutation of PLA2G6 was confirmed to contribute to autosomal recessive early-onset parkinsonism (AREP) in Han Chinese families without dystonia (33). It is also reported that a Chinese EOPD patient carrying a heterozygous mutation c.1321T>C of PRKN appeared to have symmetrical parkinsonism at onset (19). We boldly speculate that the special phenotype of the proband may be related to the two variants she carries, even though the other affected family member does not carry these mutations. However, whether or not these variants contribute to the symmetrical symptoms still requires further study. Some missense mutations of TENM4 are confirmed to cause essential tremor in Caucasian populations; however, the relationship between TENM4 and essential tremor in the Han Chinese population still needs further clarification (34–36). A recent study identifies TENM4 as a novel candidate gene for a Han Chinese family with schizophrenia (37). The effect of the TENM4 variant found in this study on the disease phenotype of the family still remains unclear.
Nevertheless, there are still some drawbacks in our research. We failed to obtain brain imaging data from the other patient of the family due to her trouble traveling, which might have been helpful in analyzing the cause of the difference in clinical features between the two patients. Furthermore, some members, including a patient's father, refused to participate in this study, which has an impact on pedigree analysis.
In conclusion, a novel homozygous frameshift mutation and a compound heterozygous mutation of PRKN were found in this EOPD family. The mutations c.856delT and c.1321T> C/c.856delT are considered pathogenic. Moreover, the proband's symptoms appeared incompatible with the MDS clinical diagnostic criteria. The differences in clinical phenotypes of the two patients in this family might be related to the slight difference in their genotypes. Further studies are required to clarify the pathogenicity of these mutations and to definitely understand their functional effects. In addition, we believe that clinical doctors need to consider whether it is necessary to improve the diagnostic criteria for PD and draw up targeted diagnostic criteria for familial hereditary PD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by ethics committee of Nanjing First Hospital, Nanjing Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YZ and YT conceptualized the study. CG and TH were responsible for the diagnosis and clinical evaluation. CG and ZY were in charge of the genetic analysis. CG, TH, and YT did the investigation. RC was responsible for the neuroimaging data and video collection. CG wrote the manuscript—original draft preparation. ZY, YT, and YZ wrote, reviewed, and edited the manuscript. CG and TH contributed equally to the study and should be considered as co-first authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Major Scientific and Technological Items of Clinical Medicine, Jiangsu Province (BL2014014) and the Medical Innovation Team of Jiangsu Province (CXTDA2017030).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the voluntary participants and investigators for their cooperation and efforts in collecting the genetic information, clinical data, and DNA samples. We also thank the Hangzhou Xiangyin medical laboratory for the DNA sequencing. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.582323/full#supplementary-material
References
1. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. (2014) 311:1670–83. doi: 10.1001/jama.2014.3654
2. Pringsheim T, Jette N, Frolkis A, Steeves TDL. The prevalence of Parkinson's disease: a systematic review and meta-analysis. Mov Disord. (2014) 29:1583–90. doi: 10.1002/mds.25945
3. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
4. Homayoun H. Parkinson disease. Ann Intern Med. (2018) 169:ITC33–ITC48. doi: 10.7326/AITC201809040
5. Kalia LV, Lang AE. Parkinson's disease. Lancet (London, England). (2015) 386:896–912. doi: 10.1016/S0140-6736(14)61393-3
6. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. (2015) 30:1591–601. doi: 10.1002/mds.26424
7. Kalinderi K, Bostantjopoulou S, Fidani L. The genetic background of Parkinson's disease: current progress and future prospects. Acta Neurol Scand. (2016) 134:314–26. doi: 10.1111/ane.12563
8. Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. (2009) 18:R48–59. doi: 10.1093/hmg/ddp012
9. Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. (2018) 42:72–85. doi: 10.1016/j.arr.2017.12.007
10. Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. (2000) 342:1560–7. doi: 10.1056/NEJM200005253422103
11. Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. (2009) 65:19–23. doi: 10.1002/ana.21415
12. Shen T, Pu J, Lai HY, Xu L, Si X, Yan Y, et al. Genetic analysis of ATP13A2, PLA2G6 and FBXO7 in a cohort of Chinese patients with early-onset Parkinson's disease. Sci Rep. (2018) 8:14028. doi: 10.1038/s41598-018-32217-4
13. Fan K, Hu P, Song C, Deng X, Wen J, Liu Y, et al. Novel compound heterozygous PRKN variants in a Han-Chinese family with early-onset Parkinson's disease. Parkinsons Dis. (2019) 2019:9024894. doi: 10.1155/2019/9024894
14. Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa-Hattori Y, et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am J Hum Genet. (1997) 60:588–96.
15. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. (1998) 392:605–8. doi: 10.1038/33416
16. Li H, Yusufujiang A, Naser S, Zhu Y, Maimaiti M, He X, et al. Mutation analysis of PARK2 in a Uyghur family with early-onset Parkinson's disease in Xinjiang, China. J Neurol Sci. (2014) 342:21–4. doi: 10.1016/j.jns.2014.03.044
17. Huang T, Gao C-Y, Wu L, Gong P-Y, Wang J-Z, Tian Y-Y, et al. Han Chinese family with early-onset Parkinson's disease carries novel compound heterozygous mutations in the PARK2 gene. Brain Behav. (2019) 9:e01372. doi: 10.1002/brb3.1372
18. West A, Periquet M, Lincoln S, Lücking CB, Nicholl D, Bonifati V, et al. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet. (2002) 114:584–91. doi: 10.1002/ajmg.10525
19. Chan DK, Mok V, Ng PW, Yeung J, Kwok JB, Fang ZM, et al. PARK2 mutations and clinical features in a Chinese population with early-onset Parkinson's disease. J Neural Transm (Vienna). (2008) 115:715–9. doi: 10.1007/s00702-007-0011-6
20. Ghazavi F, Fazlali Z, Banihosseini SS, Hosseini S-R, Kazemi MH, Shojaee S, et al. PRKN, DJ-1, and PINK1 screening identifies novel splice site mutation in PRKN and two novel DJ-1 mutations. Mov Disord. (2011) 26:80–9. doi: 10.1002/mds.23417
21. Zhao Y, Qin L, Pan H, Liu Z, Jiang L, He Y, et al. The role of genetics in Parkinson's disease: a large cohort study in Chinese mainland population. Brain J Neurol. (2020) 143:2220–34. doi: 10.1093/brain/awaa167
22. Shimura H, Hattori N, Kubo Si, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. (2000) 25:302–5. doi: 10.1038/77060
23. Lohmann E, Periquet M, Bonifati V, Wood NW, De Michele G, Bonnet A-M, et al. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol. (2003) 54:176–85. doi: 10.1002/ana.10613
24. Kim HJ, Kim H-J, Lee J-Y, Yun JY, Kim SY, Park SS, et al. Phenotype analysis in patients with early onset Parkinson's disease with and without parkin mutations. J Neurol. (2011) 258:2260–7. doi: 10.1007/s00415-011-6110-1
25. Elia AE, Del Sorbo F, Romito LM, Barzaghi C, Garavaglia B, Albanese A. Isolated limb dystonia as presenting feature of Parkin disease. J Neurol Neurosurg Psychiatry. (2014) 85:827–8. doi: 10.1136/jnnp-2013-307294
26. Tassin J, Dürr A, Bonnet AM, Gil R, Vidailhet M, Lücking CB, et al. Levodopa-responsive dystonia. GTP cyclohydrolase I or parkin mutations? Brain J Neurol. (2000) 123:1112–21. doi: 10.1093/brain/123.6.1112
27. Khan NL, Katzenschlager R, Watt H, Bhatia KP, Wood NW, Quinn N, et al. Olfaction differentiates parkin disease from early-onset parkinsonism and Parkinson disease. Neurology. (2004) 62:1224–6. doi: 10.1212/01.WNL.0000118281.66802.81
28. Alcalay RN, Caccappolo E, Mejia-Santana H, Tang MX, Rosado L, Orbe Reilly M, et al. Cognitive and motor function in long-duration PARKIN-associated Parkinson disease. JAMA neurology. (2014) 71:62–7. doi: 10.1001/jamaneurol.2013.4498
29. Abbas MM, Govindappa ST, Sudhaman S, Thelma BK, Juyal RC, Behari M, et al. Early Onset Parkinson's disease due to DJ1 mutations: an Indian study. Parkinsonism Related Disord. (2016) 32:20–4. doi: 10.1016/j.parkreldis.2016.04.024
30. Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. (2010) 75:1356–61. doi: 10.1212/WNL.0b013e3181f73649
31. Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, et al. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. (2012) 159B:183–91. doi: 10.1002/ajmg.b.32012
32. Shi Ch, Tang Bs, Wang L, Lv Zy, Wang J, Luo Lz, et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology. (2011) 77:75–81. doi: 10.1212/WNL.0b013e318221acd3
33. Xie F, Cen Z, Ouyang Z, Wu S, Xiao J, Luo W. Homozygous p.D331Y mutation in PLA2G6 in two patients with pure autosomal-recessive early-onset parkinsonism: further evidence of a fourth phenotype of PLA2G6-associated neurodegeneration. Parkinsonism Related Disord. (2015) 21:420–2. doi: 10.1016/j.parkreldis.2015.01.012
34. Hor H, Francescatto L, Bartesaghi L, Ortega-Cubero S, Kousi M, Lorenzo-Betancor O, et al. Missense mutations in TENM4, a regulator of axon guidance and central myelination, cause essential tremor. Hum Mol Genet. (2015) 24:5677–86. doi: 10.1093/hmg/ddv281
35. Yan Y-P, Xu C-Y, Gu L-Y, Zhang B, Shen T, Gao T, et al. Genetic testing of FUS, HTRA2, and TENM4 genes in Chinese patients with essential tremor. CNS Neurosci Ther. (2020) 26:837–41. doi: 10.1111/cns.13305
36. Chao YX, Lin Ng EY, Tio M, Kumar P, Tan L, Au WL, et al. Essential tremor linked TENM4 mutation found in healthy Chinese individuals. Parkinsonism Related Disord. (2016) 31:139–40. doi: 10.1016/j.parkreldis.2016.05.003
Keywords: Parkinson's disease, early-onset, PRKN mutations, genetic analysis, whole-exome sequence
Citation: Gao C, Huang T, Chen R, Yuan Z, Tian Y and Zhang Y (2020) A Han Chinese Family With Early-Onset Parkinson's Disease Carrying Novel Frameshift Mutation and Compound Heterozygous Mutation of PRKN Appearing Incompatible With MDS Clinical Diagnostic Criteria. Front. Neurol. 11:582323. doi: 10.3389/fneur.2020.582323
Received: 13 July 2020; Accepted: 28 August 2020;
Published: 09 October 2020.
Edited by:
Chin-Hsien Lin, National Taiwan University Hospital, TaiwanReviewed by:
Manabu Funayama, Juntendo University, JapanJifeng Guo, Central South University, China
Copyright © 2020 Gao, Huang, Chen, Yuan, Tian and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youyong Tian, dHl5OTk1NkBuam11LmVkdS5jbg==; Yingdong Zhang, Wmhhbmd5ZDYyQGFsaXl1bi5jb20=
†These authors have contributed equally to this work