Abstract
Purpose: Concerns of drug–drug interactions (DDIs) between anti-seizure medications (ASMs) and non-vitamin K oral anticoagulants (NOACs) have emerged in recent case reports and guidelines. Theoretically, the induction of hepatic cytochrome P450 3A4 (CYP3A4) enzyme and permeability glycoprotein (P-GP) efflux transporter protein systems may reduce the effect of NOACs. We aimed to investigate whether such DDIs are clinically relevant in a real-world situation.
Methods: We retrospectively reviewed 320 ischemic stroke patients with atrial fibrillation (Af) and grouped them according to different potential interactions with CYP3A4 and P-GP. Ischemic stroke events, transient ischemic attack (TIA) events, follow-up duration, baseline characteristics, concomitant ASMs, and stroke risk factors were collected. Statistical analysis included Kaplan–Meier survival curves and the log-rank test.
Results: Overall, 320 ischemic stroke with Af patients received NOACs. Among the NOAC users, 75 also took ASMs, including 56 that have potential DDIs: 43 (13.4%) were categorized as potential CYP and P-GP DDIs and 13 (4.1%) as P-GP-only DDIs. The remaining 264 (82.5%) patients were used as controls including 19 exposed to nonsignificant DDI ASMs and 245 patients without ASM exposure. The incidence rates of recurrent stroke/TIA events in both CYP3A4 and P-GP DDIs, P-GP DDIs only, and no DDIs were 7.5, 2.1, and 8.4/100 person-years, respectively. Kaplan–Meier survival curves and the log-rank test did not show significant differences among the groups.
Conclusions: The recurrent stroke rate of NOAC users with potential DDIs was not higher than in those without potential DDIs in this single-institute study. Our results suggest that theoretical interactions between ASMs and NOACs may not be as severe as previously thought in a real-world situation.
Introduction
Atrial fibrillation (Af) is a major risk factor for cardioembolic stroke and requires oral anticoagulant (OAC) therapy. Post-stroke epilepsy has been reported to occur in 5–17% of ischemic stroke patients, which is even higher (31%) in patients with cardioembolic stroke (1). Long-term anti-seizure medication (ASM) treatment is necessary for seizure control in these patients. Recently, non-vitamin K oral anticoagulants (NOACs) have become the mainstream treatment for non-valvular Af patients due to their efficacy and safety profile (2). Real-world studies have reported lower incidence rates of stroke with better adherence to guidelines (3, 4). With the increasing use of NOACs, the issue of potential drug–drug interactions (DDIs) in polypharmacy was raised in the 2018 European Heart Rhythm Association Practical Guide on the use of NOACs in patients with Af, including the possible interactions between ASMs and NOACs (5).
Theoretically, DDIs between NOACs and ASMs mainly focus on the effect of permeability glycoprotein (P-GP) and cytochrome P450 3A4 enzyme (CYP3A4) systems (5). Induction of CYP3A4 may increase hepatic clearance of drugs, and induction of P-GP may increase the re-secretion of medications in the gut and clearance in the kidneys (6, 7). Many ASMs are known to affect these systems. Oxcarbazepine and topiramate can induce CYP3A4 enzyme (8, 9), while levetiracetam can induce P-GP (10). In addition, carbamazepine, phenobarbital, phenytoin, and valproic acid have been shown to be able to induce both CYP3A4 and P-GP, although the effect varies in different models (11–18). Critical DDIs may be associated with failure of the anticoagulation effect and expose the patients to the risk of recurrent stroke and systemic embolism (19, 20). On the other hand, switching ASMs to avoid DDIs has been associated with a 16.7–21.7% increased risk in 6-month seizure recurrence rate (21, 22). Therefore, selecting the most appropriate ASMs for patients with post-stroke epilepsy and Af needs to consider the DDIs between ASMs and NOACs. Currently, the guideline recommendations for DDIs are mainly based on cell/animal models or case reports (8, 10). Due to the lack of strong clinical evidence, this study aimed to investigate whether theoretical DDIs between ASMs and NOACs have an impact on stroke prevention in a real-world setting.
Materials and Methods
Study Design
We retrospectively reviewed patients admitted for ischemic stroke due to Af at the Department of Neurology of Kaohsiung Chang Gung Memorial Hospital from January 2013 to December 2017. This study was approved by the local Institutional Review Board of Kaohsiung Chang Gung Memorial Hospital (IRB No. 201901169B0).
Definitions and Criteria
Ischemic stroke was diagnosed by an episode of neurologic dysfunction caused by vascular stenosis or occlusion leading to focal cerebral, spinal cord, or retinal infarction within a specific vascular territory. Cardioembolic stroke was defined as arterial occlusion presumably due to an embolus arising in the heart (23). Transient ischemic attack (TIA) was diagnosed by neurologic symptoms of ischemic origin that lasted for least than 24 h (24).
Clinical Assessment
We included ischemic stroke patients aged ≥18 years who used NOACs for Af and had at least 3 months of follow-up data. Patients who received other medications that potentially interact with CYP3A4 or P-GP (either induction or inhibition) were excluded: verapamil, amiodarone, quinidine, human immunodeficiency virus (HIV) medications, rifampicin, ketoconazole, cyclosporine, and tacrolimus (5, 25, 26).
The patients' age, sex, underlying medical condition, follow-up duration, stroke risk factors, and medication history were recorded. The NOAC regimen was evaluated according to the 2014 American Heart Association (AHA)/American College of Cardiology (ACC)/Heart Rhythm Society (HRS) guidelines for Af management (2). The primary endpoint was any ischemic stroke or TIA event during follow-up.
Carbamazepine, oxcarbazepine, phenobarbital, phenytoin, topiramate, and valproic acid were defined as being CYP3A4 inducers, and levetiracetam, carbamazepine, phenobarbital, phenytoin, and valproic acid were defined as being P-GP inducers. The patients were divided into groups according to the ASMs as: “both CYP3A4 and P-GP inducers,” “CYP3A4 inducers,” “P-GP inducers,” and “control group” (Figure 1). The control group was made up of patients without ASM exposure and patients receiving ASMs that have no interaction with CYP3A4 and P-GP.
Figure 1
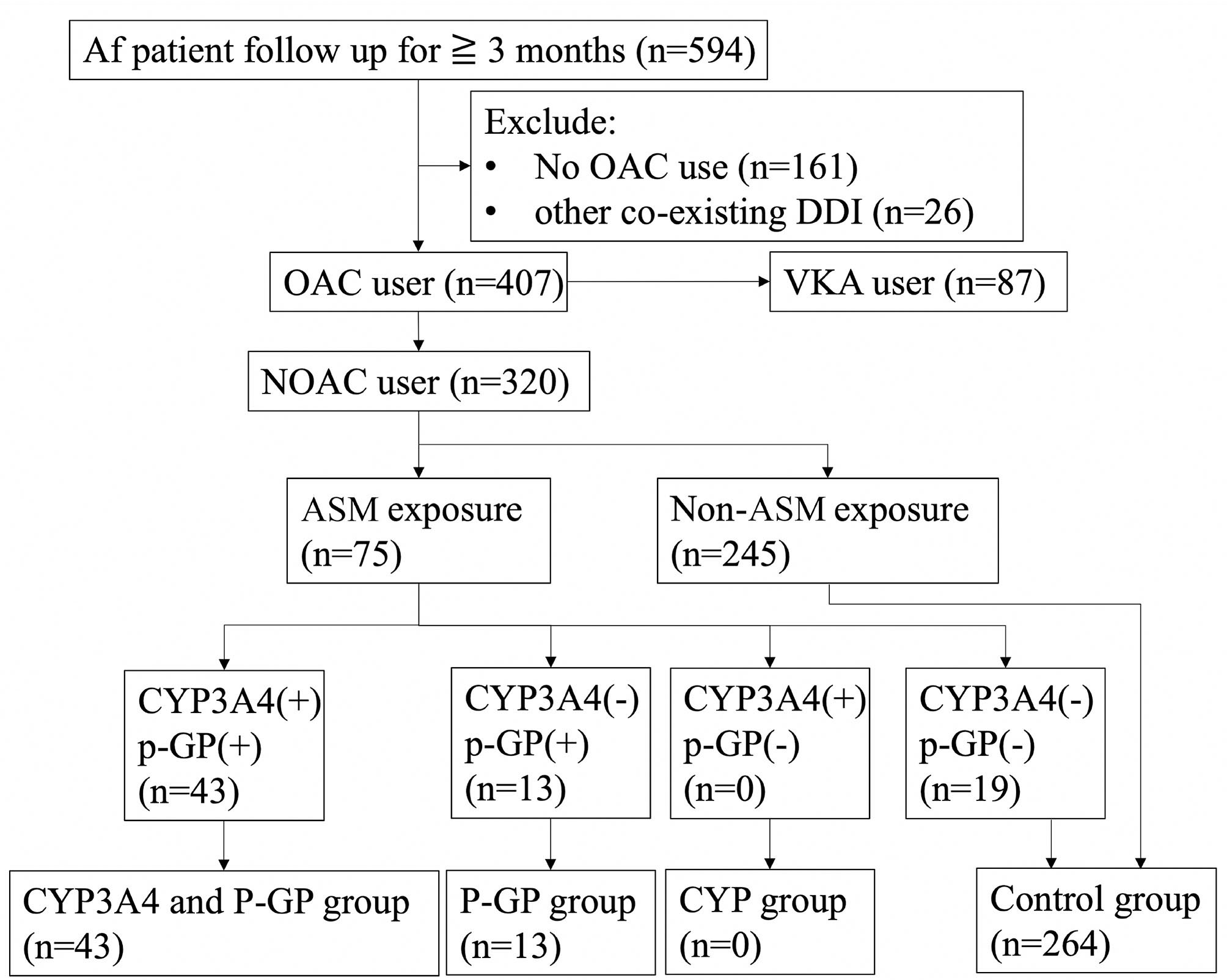
Flow diagramof patient inclusion and exclusion. Af, atrial fibrillation; OAC, oral anticoagulant; VKA, vitamin K antagonist; DDI, drug–drug interaction; ASM, anti-seizure medication; CYP3A4, cytochrome P450 3A4; P-GP, permeability glycoprotein; n, number.
Because ASMs may have varying effects on CYP3A4, we further divided the ASMs into strong CYP3A4 inducer group (carbamazepine, phenobarbital, phenytoin) and weak CYP3A4 inducer group (topiramate, valproic acid, oxcarbazepine) (8, 12, 15, 18, 27).
For patient follow-up, patients were followed up regularly in our clinic every 1–3 months according to their clinical condition. Only the regular follow-up periods in our clinic were included for analysis. As for the ASM co-administered groups, only the times when NOAC and ASMs were both given were counted as the duration of follow-up.
Statistical Analysis
All statistical analyses were performed using SPSS v.24.0 for Mac (SPSS Inc., Chicago, IL, USA). Descriptive summaries were reported as the mean ± standard deviation for continuous variables and as number (percentage) for categorical variables. For nonparametric variables, we used the median value with the interquartile range (IQR) and the Mann–Whitney U test with two-sided test. Incidence rates were calculated as the number of ischemic events divided by the time at risk and presented per 100 person-years. The cumulative incidence of ischemic events was evaluated using Kaplan–Meier survival curves, which were visually inspected to check the proportional hazards assumption. Differences between groups were evaluated using the log-rank test. A p value of ≤ 0.05 was considered to be statistically significant.
Results
During the study period, 594 patients with a diagnosis of Af and ischemic stroke were reviewed. Twenty-six patients were excluded because they received other co-medications with potential CYP3A4 or P-GP interaction besides ASMs. Eighty-seven patients receiving vitamin K antagonist (VKA) and another 161 patients who did not receive OAC treatment were excluded.
A total of 320 stroke with Af patients received NOAC treatment, including 75 patients who received ASMs. Among them, 56 patients were exposed to CYP3A4 or P-GP inducing ASMs and 19 exposed to ASMs without significant DDIs. Forty-three of them (13.4%) were classified into the CYP3A4 plus P-GP group and 13 (4.1%) were classified as P-GP-only group. The remaining 264 (82.5%) were used as controls, including 19 patients with nonsignificant DDI ASM exposure and 245 patients who did not take ASMs. No patient was classified as CYP3A4-only group. No significant differences were noted between the CYP3A4 and P-GP group and control group except that the CYP3A4 and P-GP group had significantly fewer male patients and dyslipidemia compared with the control group. Similarly, no significant differences were noted between the P-GP group and control group except that the P-GP group had significantly fewer male patients and a longer follow-up duration than those of the control group (Table 1).
Table 1
| Total N | CYP and P-GP | P-GP | Control | p value | P-GP vs. Control | CYP and P-GP vs. Control |
|---|---|---|---|---|---|---|
| 43 | 13 | 264 | CYP and P-GP vs. P-GP | |||
| Age (year, median, IQR) | 71 (67.3–79.3) | 72.2 (68–77.1) | 75.3 (69–81.2) | 0.741 | 0.147 | 0.179 |
| Male sex (N, %) | 18 (41.9) | 4 (30.8) | 164 (60) | 0.477 | 0.031 | 0.019 |
| Follow-up (months, median, IQR) | 12.3 (4.8–28.8) | 30 (15.6–38.4) | 20.4 (10.8–40.8) | 0.003 | 0.027 | 0.067 |
| CHA2DS2-VASc (median, IQR) | 3 (2-4) | 3 (3-4) | 3 (2-4) | 0.364 | 0.669 | 0.272 |
| CHF (N, %) | 7 (16.3) | 42 (15.9) | 2 (15.4) | 0.939 | 0.96 | 0.951 |
| Hypertension (N, %) | 19 (44.2) | 152 (57.6) | 6 (46.2) | 0.901 | 0.418 | 0.102 |
| Vascular disease history (N, %) | 6 (14) | 35 (13.3) | 2 (15.4) | 0.898 | 0.826 | 0.901 |
| DM (N, %) | 6 (14) | 65 (24.6) | 3 (23.1) | 0.437 | 0.9 | 0.125 |
| Prior stroke/TIA history (N, %) | 3 (7) | 34 (12.9) | 2 (15.4) | 0.356 | 0.793 | 0.271 |
| Creatinine (mg/dl) (median, IQR) | 0.8 (0.77–0.89) | 0.86 (0.72–1.11) | 0.96 (0.77–1.14) | 0.467 | 0.053 | 0.241 |
| Dyslipidemia (N, %) | 10 (23.3) | 5 (38.5) | 125 (47.3) | 0.282 | 0.532 | 0.003 |
| BMI (median, IQR) | 25.8 (24.7–28.5) | 27.2 (25–28.8) | 24 (21.3–27.2) | 0.069 | 0.091 | 0.82 |
| Smoking (N, %) | 7 (16.3) | 1 (7.7) | 50 (18.9) | 0.442 | 0.308 | 0.678 |
| Nonstandard dose reduction (N, %) | 26 (72.9) | 10 (76.9) | 164 (64.1) | 0.282 | 0.282 | 0.836 |
| New-onset heart failure (N, %) | 1 (2.3) | 0 | 3 (1.1) | 0.454 | ||
| Mortality (N, %) | 8 (18) | 2 (15) | 32 (12) | 1 | 0.665 | 0.23 |
| Recurrent admission (100 p-yr, N) | 61 (11) | 35 (3) | 50 (81) | 0.705 | 0.584 | 0.295 |
| Embolic event IR (100 p-yr, N) | 7.3 (2) | 0 (0) | 7.4 (42) | 0.809 | ||
| Hemorrhagic event IR (100 p-yr, N) | 2.9 (2) | 5.4 (2) | 3.8 (22) | 0.628 | 0.708 | 0.843 |
| Total event IR (100 p-yr, N) | 10.2 (7) | 5.4 (2) | 11.2 (64) | 0.486 | 0.374 | 0.662 |
The DDIs of ASMs in patients taking NOACs.
NOAC, non-vitamin K oral anticoagulant; CYP, cytochrome P450 3A4; P-GP, permeability glycoprotein; TIA, transient ischemic attack; CHF, congestive heart failure; DM, diabetes mellitus; IQR, interquartile range; N, number; %, percentage; 100 p-yr, 100 persons per year; BMI, body mass index; DDI, drug–drug interaction; ASM, anti-seizure medication. Bold values indicate statistically significant.
Among patients taking ASMs with NOACs (N = 56), the average number of ASMs used was 1.4 (range 1–5). Thirty-seven used one ASM, 16 used two ASMs, two used three ASMs, and one used five ASMs. All patients in the P-GP group used one ASM (N = 13), which was levetiracetam. For patients in the CYP and P-GP group, they used levetiracetam, valproic acid, phenytoin, topiramate, lamotrigine, carbamazepine, or zonisamide (Table 2).
Table 2
| CYP and P-GP group | P-GP group | Control group | ||
|---|---|---|---|---|
| Total N | 43 | 13 | 264 | |
| ASM daily dose (median, N, IQR) | LEV | 1,000 [15 (35%), 1,000–1,975] | 1, 000 [13 (100%), 1,000–1,600] | |
| VPA | 1,300 [26 (60%), 1,000–1,500] | |||
| PHT | 300 [17 (40%), 300–300] | |||
| LMT | 275 [3 (7%), 212.5–337.5] | |||
| TPM | 600 [1 (2%)] | |||
| CBZ | 800 [2 (5%), 800–800] | |||
| ZNS | 500 [2 (5%), 500–500] | |||
| PER | 2 [6 (14%), 2–4] | 2 [1 (8%)] | 2 [9 (3%), 2–4] | |
| LCS | 300 [4 (9%), 200–400] | 200 [2 (15%), 200–200] | 200 [10 (4%), 200–200] |
ASM doses among groups.
ASM, anti-seizure medication; IQR, interquartile range; N, number; NOAC, non-vitamin K oral anticoagulant; CYP, cytochrome P450 3A4; P-GP, permeability glycoprotein; LEV, levetiracetam; VPA, valproic acid; PHT, phenytoin; TPM, topiramate; LMT, lamotrigine; CBZ, carbamazepine; ZNS, zonisamide; PER, perampanel; LCS, lacosamide.
There was no significant difference in the incidence of recurrent stroke/TIA events between the CYP3A4 and P-GP group and the control group (7.3 vs. 7.4/100 person-years, p = 0.809). Although the P-GP group had no recurrent stroke/TIA events, it also had the smallest number of patients (n = 20). During the follow-up period, no systemic embolic events were observed. Among them, five (12%) of the CYP3A4 and P-GP group, two (15%) of the P-GP group, and 43 (16%) of the controls were lost to follow-up due to failed to return to the clinics or moved to other institutes for follow-up.
Because the effect of CYP3A4 may be varied between ASMs, we further divided the CYP3A4 inducer category into strong and weak groups. The incidence of ischemic events for the strong CYP3A4 inducer group is 3.3 (100 person-years, total/event N = 17/1), weak CYP3A4 inducer group is 10.6 (100 person-years, total/event N = 26/4), and control group is 7.4 (100 person-years, total/event N = 264/42). No statistically significant difference was observed between groups (Table 3).
Table 3
| Strong inducer | Weak Inducer | Control | p value | |||
|---|---|---|---|---|---|---|
| ASM | Carbamazepine phenobarbital phenytoin | Topiramate valproic acid oxcarbazepine | Strong vs. control | Weak vs. control | Strong vs. weak | |
| Total N | 17 | 26 | 264 | |||
| Age (median, IQR) | 77.6 (66–78.7) | 72 (67–83) | 75.3 (69–81.2) | 0.388 | 0.362 | 0.886 |
| Male Gender (N, %) | 3 (17.6) | 16 (62) | 164 (60) | <0.001 | 0.834 | 0.003 |
| Follow-up (months, median, IQR) | 20.4 (8.4–32.4) | 13.2 (4.8–26.4) | 20.4 (10.8–40.8) | 0.45 | 0.041 | 0.583 |
| CHA2DS2-VASc (median, IQR) | 4 (2.25–4.75) | 3 (2-3) | 3 (2-4) | 0.68 | 0.112 | 0.22 |
| Dyslipidemia (N, %) | 5 (29.4) | 7 (27) | 125 (47.3) | 0.108 | 0.057 | 0.951 |
| Smoking (N, %) | 1 (5.9) | 5 (19) | 50 (18.9) | 0.154 | 0.961 | 0.199 |
| Creatinine (mg/dl, median, IQR) | 0.84 (0.65–0.95) | 0.95 (0.78–1.38) | 0.96 (0.77–1.14) | 0.014 | 0.593 | 0.068 |
| Nonstandard dose reduction (N, %) | 10 (58.8) | 18 (69.2) | 164 (64.1) | 0.58 | 0.846 | 0.359 |
| Embolic event IR (N) (100 p-yr) | 3.3 (1) | 10.6 (4) | 7.4 (42) | 0.477 | 0.344 | 0.512 |
DDIs of strong and weak CYP3A4 inducers on NOAC user.
NOAC, non-vitamin K oral anticoagulant; CYP, cytochrome P450 3A4; IQR, interquartile range; N, number; %, percentage; CHF, chronic heart failure; DM, diabetes mellitus; TIA, transient ischemic attack; IR, incidence rate; 100 p-yr, 100 persons per year; DDI, drug–drug interaction. Bold values indicate statistically significant.
Because an insufficient dose of NOACs may affect the risk of recurrent stroke, we further analyzed this confounding factor. Nonstandard dose reductions were noted in 200 (62.5%) patients. The incidence rates of standard dose and nonstandard dose reductions were 6.2 (100 person-years) and 7.3 (100 person-years) (p = 0.815), respectively. We performed a subanalysis of the patients who received an adequate NOAC dose and NOAC underdose groups. For adequate NOAC dose, there was no significant difference in the incidence rate among the CYP3A4 and P-GP, P-GP, and control groups [4.1, 0, and 6.7 per 100 person-years (p = 0.941); event-free ratio, 88.4, 100, and 84.1%, respectively]. Similarly, for NOAC underdose group, no significant difference in the incidence rate among the CYP3A4 and P-GP, P-GP, and control groups [9.3, 0, and 7.7 per 100 person-years (p = 0.802); event-free ratio, 84.6, 100, and 83.5%, respectively].
Kaplan–Meier survival curves of recurrent stroke/TIA events with different DDIs are illustrated in Figure 2. The results showed no significant difference between different DDIs and control groups (log-rank test, p = 0.809).
Figure 2
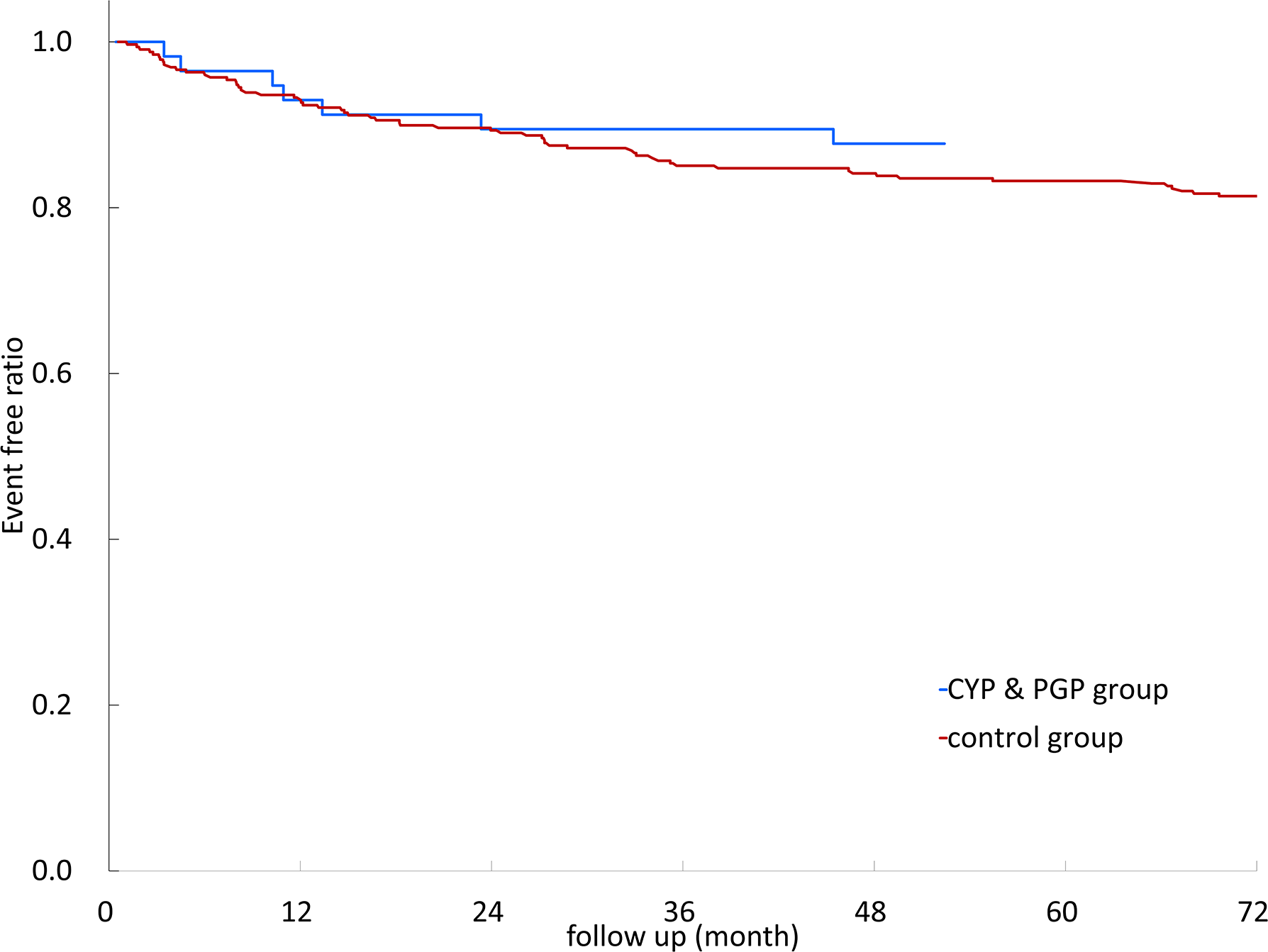
Kaplan–Meier survival curves of non-vitamin K oral anticoagulants (NOACs) with different anti-seizure medication (ASM) drug–drug interactions. CYP3A4, cytochrome P450 3A4; P-GP, permeability glycoprotein; CYP3A4&P-GP, CYP3A4 and P-GP.
Discussion
In this retrospective cohort study, we reported real-world data on DDIs between ASMs and NOACs. Surprisingly, we found that the use of CYP3A4 and/or P-GP inducers did not significantly alter the incidence of recurrent stroke/TIA even in patients who received underdosed NOACs despite the limited cohort size collected from a single institute.
Previous studies have suggested that several ASMs may affect the CYP3A4 and P-GP system. Carbamazepine and phenytoin could induce CYP3A4 and P-GP in healthy volunteers; however, the potency and efficacy were low compared with strong inducers such as rifampin (11, 12). Phenobarbital and valproic acid have also been reported to induce CYP3A4 and P-GP based on mouse/rat models and human cell line studies (13–18). NOACs are mainly metabolized by CYP3A4 and secreted into the gut by the P-GP system. With the increasing use of NOACs, DDIs have become an important issue, especially as elderly patients often have multiple medical conditions. The 2018 European Heart Rhythm Association Practical Guide reviewed potential DDIs for NOACs and highlighted that carbamazepine, levetiracetam, oxcarbazepine, phenobarbital, phenytoin, topiramate, and valproic acid should not be used or used with caution or avoided in patients taking NOACs (5). Although theoretical DDIs have been observed in animal and cell line models, this may not always reflect the same degree of interaction in humans. This is demonstrated in our study where we did not observe an increase in cardioembolic stroke in patients receiving potential interacting ASMs, both in the standard NOACs dose or underdose groups.
There are several possible explanations for this discrepancy between theoretical interactions and real-world data. First, the dose of ASMs may be relevant. For example, the enzyme-inducing effect is known to occur at a higher dose of many ASMs (≥1,200 mg/day for oxcarbazepine, ≥800 mg/day for carbamazepine, and ≥400 mg/day for topiramate) (9, 28). Patients in our study usually received lower ASM doses that may not induce liver enzymes. Second, the P-GP system is known to have a species-specific effect so that it can be induced by phenytoin and levetiracetam in mice but not in humans (29, 30). The phase 1 study of levetiracetam on healthy volunteers did not have significant DDIs on P-GP (29).
In this retrospective study, we found that co-medication of potential interacting ASMs to NOACs did not increase the risk of recurrent ischemic events in post-stroke epilepsy patients. This suggests that DDIs involving CYP3A4 and/or P-GP in ASMs may not be a major clinical concern. However, this finding should be interpreted with caution. The study sample size was relatively small (N = 320), included only Asian population, and retrospective with limited follow-up duration [median 20.2 (IQR: 13.2–30) months], or lost to follow-up, which may underestimate the risk of recurrent stroke. In addition, the high prevalence of underdosing NOACs in this study group may affect the outcomes. The use of lower-dose NOAC in the Asian population is relatively common (15–59%) because of the concerns of higher bleeding tendencies to VKA and lower body mass (31, 32). Underdosing of NOACs may exacerbate the risk of DDIs on recurrent stroke due to a lower blood concentration; however, we did not observe a significant increase in embolic events in the NOAC underdose group. We also did not observe a significant increase of embolic stroke when comparing patients with underdosed NOAC vs. standard NOAC dose. This is in accordance with the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation II study (ORBIT-AF II) where there is no difference between standard and underdosing groups (33). Another limitation was that direct NOAC serum concentration monitoring was not routinely performed in our clinical practice. Lastly, similar to warfarin, it is possible that genetic factors that influence the metabolism or transportation of the NOACs may have an effect on the DDIs, which was not evaluated in this study.
In conclusion, our single-institute study suggests that DDIs between ASMs and NOACs in Asian post-stroke epilepsy patients may not be as severe as previously thought in a single institutional real-world situation. Further international studies with a larger sample size, different ethnicities, direct monitoring of NOAC serum concentration, and longer follow-up duration or meta-analysis are warranted to clarify the clinical significance of such DDIs.
Statements
Data availability statement
The datasets presented in this article are not readily available because the dataset generated and analyzed during the current study is not publicly available. The consent form participants did not cover data sharing but are available from the corresponding author on reasonable request. Requests to access the datasets should be directed to menghan@cgmh.org.tw.
Ethics statement
The studies involving human participants were reviewed and approved by Chang Gung Medical Foundation Institutional Reviewer Board 201901169B0. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
C-JH, S-HC, C-HL, Y-TL, C-WH, and M-HT contributed to the acquisition and interpretation of the data and revising the manuscript for intellectual content. C-JH, S-HC, and M-HT contributed to the design and conceptualization of the study; analysis and interpretation of the data, drafting, revising, and final approval of the manuscript for intellectual content. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by grants from the Chang Gung Medical Foundation (CMRPG8I0411) to C-JH and the Ministry of Science and Technology, Taiwan (MOST107-2314-B-182A-057-MY3), and Chang Gung Medical Foundation (CMRPG8J1161) to M-HT.
Acknowledgments
The authors wish to thank the Stroke Center of Kaohsiung Chang Gung Memorial Hospital.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Galovic M Dohler N Erdelyi-Canavese B Felbecker A Siebel P Conrad J et al . Prediction of late seizures after ischaemic stroke with a novel prognostic model (the SeLECT score): a multivariable prediction model development and validation study. Lancet Neurol. (2018) 17:143–52. 10.1016/S1474-4422(17)30404-0
2.
January CT Wann LS Alpert JS Calkins H Cigarroa JE Cleveland JC Jr et al . 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. (2014) 64:e1–76. 10.1161/CIR.0000000000000041
3.
Chan YH See LC Tu HT Yeh YH Chang SH Wu LS et al . Efficacy and safety of apixaban, dabigatran, rivaroxaban, and warfarin in Asians with nonvalvular atrial fibrillation. J Am Heart Assoc. (2018) 7:e008150. 10.1161/JAHA.117.008150
4.
Lip GYH Keshishian A Li X Hamilton M Masseria C Gupta K et al . Effectiveness and safety of oral anticoagulants among nonvalvular atrial fibrillation patients. Stroke. (2018) 49:2933–44. 10.1161/STROKEAHA.118.020232
5.
Steffel J Verhamme P Potpara TS Albaladejo P Antz M Desteghe L et al . The 2018 European heart rhythm association practical guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur Heart J. (2018) 39:1330–93. 10.1093/eurheartj/ehy136
6.
Gnoth MJ Buetehorn U Muenster U Schwarz T Sandmann S . In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Ther. (2011) 338:372–80. 10.1124/jpet.111.180240
7.
Hartter S Koenen-Bergmann M Sharma A Nehmiz G Lemke U Timmer W et al . Decrease in the oral bioavailability of dabigatran etexilate after co-medication with rifampicin. Br J Clin Pharmacol. (2012) 74:490–500. 10.1111/j.1365-2125.2012.04218.x
8.
Nallani SC Glauser TA Hariparsad N Setchell K Buckley DJ Buckley AR et al . Dose-dependent induction of cytochrome P450 (CYP) 3A4 and activation of pregnane X receptor by topiramate. Epilepsia. (2003) 44:1521–8. 10.1111/j.0013-9580.2003.06203.x
9.
Andreasen AH Brosen K Damkier P . A comparative pharmacokinetic study in healthy volunteers of the effect of carbamazepine and oxcarbazepine on cyp3a4. Epilepsia. (2007) 48:490–6. 10.1111/j.1528-1167.2007.00924.x
10.
Moerman L Wyffels L Slaets D Raedt R Boon P De Vos F . Antiepileptic drugs modulate P-glycoproteins in the brain: a mice study with (11)C-desmethylloperamide. Epilepsy Res. (2011) 94:18–25. 10.1016/j.eplepsyres.2010.12.013
11.
Giessmann T May K Modess C Wegner D Hecker U Zschiesche M et al . Carbamazepine regulates intestinal P-glycoprotein and multidrug resistance protein MRP2 and influences disposition of talinolol in humans. Clin Pharmacol Ther. (2004) 76:192–200. 10.1016/j.clpt.2004.04.011
12.
Lim ML Min SS Eron JJ Bertz RJ Robinson M Gaedigk A et al . Coadministration of lopinavir/ritonavir and phenytoin results in two-way drug interaction through cytochrome P-450 induction. J Acquir Immune Defic Syndr. (2004) 36:1034–40. 10.1097/00126334-200408150-00006
13.
Eyal S Lamb JG Smith-Yockman M Yagen B Fibach E Altschuler Y et al . The antiepileptic and anticancer agent, valproic acid, induces P-glycoprotein in human tumour cell lines and in rat liver. Br J Pharmacol. (2006) 149:250–60. 10.1038/sj.bjp.0706830
14.
Cerveny L Svecova L Anzenbacherova E Vrzal R Staud F Dvorak Z et al . Valproic acid induces CYP3A4 and MDR1 gene expression by activation of constitutive androstane receptor and pregnane X receptor pathways. Drug Metab Dispos. (2007) 35:1032–41. 10.1124/dmd.106.014456
15.
Ohno M Motojima K Okano T Taniguchi A . Induction of drug-metabolizing enzymes by phenobarbital in layered co-culture of a human liver cell line and endothelial cells. Biol Pharm Bull. (2009) 32:813–7. 10.1248/bpb.32.813
16.
Jing X Liu X Wen T Xie S Yao D Liu X et al . Combined effects of epileptic seizure and phenobarbital induced overexpression of P-glycoprotein in brain of chemically kindled rats. Br J Pharmacol. (2010) 159:1511–22. 10.1111/j.1476-5381.2009.00634.x
17.
Puranik YG Birnbaum AK Marino SE Ahmed G Cloyd JC Remmel RP et al . Association of carbamazepine major metabolism and transport pathway gene polymorphisms and pharmacokinetics in patients with epilepsy. Pharmacogenomics. (2013) 14:35–45. 10.2217/pgs.12.180
18.
Alvariza S Fagiolino P Vazquez M Feria-Romero I Orozco-Suarez S . Chronic administration of phenytoin induces efflux transporter overexpression in rats. Pharmacol Rep. (2014) 66:946–51. 10.1016/j.pharep.2014.06.007
19.
Eriksson BI Dahl OE Ahnfelt L Kalebo P Stangier J Nehmiz G et al . Dose escalating safety study of a new oral direct thrombin inhibitor, dabigatran etexilate, in patients undergoing total hip replacement: BISTRO I. J Thromb Haemost. (2004) 2:1573–80. 10.1111/j.1538-7836.2004.00890.x
20.
Rankin J Nagar M Crosby J Toomari N Pietras R Ben-Zur UM . Possible failure of novel direct-acting oral anticoagulants in management of pulmonary embolism: a case report. J Med Case Rep. (2016) 10:346. 10.1186/s13256-016-1135-9
21.
Wang SP Mintzer S Skidmore CT Zhan T Stuckert E Nei M et al . Seizure recurrence and remission after switching antiepileptic drugs. Epilepsia. (2013) 54:187–93. 10.1111/j.1528-1167.2012.03652.x
22.
Finamore JM Sperling MR Zhan T Nei M Skidmore CT Mintzer S . Seizure outcome after switching antiepileptic drugs: a matched, prospective study. Epilepsia. (2016) 57:1294–300. 10.1111/epi.13435
23.
Adams HP Jr Bendixen BH Kappelle LJ Biller J Love BB Gordon DL Marsh EE 3rd . Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in acute stroke treatment. Stroke. (1993) 24:35–41. 10.1161/01.STR.24.1.35
24.
Albers GW Caplan LR Easton JD Fayad PB Mohr JP Saver JL et al . Transient ischemic attack–proposal for a new definition. N Engl J Med. (2002) 347:1713–6. 10.1056/NEJMsb020987
25.
Wang L Zhang D Raghavan N Yao M Ma L Frost CE et al . In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. (2010) 38:448–58. 10.1124/dmd.109.029694
26.
Mueck W Kubitza D Becka M . Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. (2013) 76:455–66. 10.1111/bcp.12075
27.
Sugiyama I Murayama N Kuroki A Kota J Iwano S Yamazaki H et al . Evaluation of cytochrome P450 inductions by anti-epileptic drug oxcarbazepine, 10-hydroxyoxcarbazepine, and carbamazepine using human hepatocytes and HepaRG cells. Xenobiotica. (2016) 46:765–74. 10.3109/00498254.2015.1118774
28.
Rosenfeld WE Doose DR Walker SA Nayak RK . Effect of topiramate on the pharmacokinetics of an oral contraceptive containing norethindrone and ethinyl estradiol in patients with epilepsy. Epilepsia. (1997) 38:317–23. 10.1111/j.1528-1157.1997.tb01123.x
29.
Levy RH Ragueneau-Majlessi I Baltes E . Repeated administration of the novel antiepileptic agent levetiracetam does not alter digoxin pharmacokinetics and pharmacodynamics in healthy volunteers. Epilepsy Res. (2001) 46:93–9. 10.1016/S0920-1211(01)00253-4
30.
Baltes S Gastens AM Fedrowitz M Potschka H Kaever V Loscher W . Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. (2007) 52:333–46. 10.1016/j.neuropharm.2006.07.038
31.
Yamashita Y Uozumi R Hamatani Y Esato M Chun YH Tsuji H et al . Current status and outcomes of direct oral anticoagulant use in real-world atrial fibrillation patients- Fushimi AF Registry. Circ J. (2017) 81:1278–85. 10.1253/circj.CJ-16-1337
32.
Ono T Ikemura N Kimura T Ueda I Tanaka H Tokuda H et al . Contemporary trend of reduced-dose non-vitamin K anticoagulants in Japanese patients with atrial fibrillation: a cross-sectional analysis of a multicenter outpatient registry. J Cardiol. (2019) 73:14–21. 10.1016/j.jjcc.2018.09.003
33.
Steinberg BA Shrader P Pieper K Thomas L Allen LA Ansell J et al . Frequency and outcomes of reduced dose non-vitamin K antagonist anticoagulants: results from ORBIT-AF II (the outcomes registry for better informed treatment of atrial fibrillation II). J Am Heart Assoc. (2018) 7:e007633. 10.1161/JAHA.117.007633
Summary
Keywords
epilepsy, ASM, drug–drug interaction, stroke, NOAC
Citation
Ho C-J, Chen S-H, Lin C-H, Lu Y-T, Hsu C-W and Tsai M-H (2021) Non-vitamin K Oral Anticoagulants and Anti-seizure Medications: A Retrospective Cohort Study. Front. Neurol. 11:588053. doi: 10.3389/fneur.2020.588053
Received
28 July 2020
Accepted
28 December 2020
Published
26 February 2021
Volume
11 - 2020
Edited by
Edoardo Ferlazzo, University of Catanzaro, Italy
Reviewed by
Christian Brandt, Mara Hospital, Germany; Kusum Sinha, Penn State College of Medicine, United States; Luigi Francesco Iannone, University of Catanzaro, Italy
Updates
Copyright
© 2021 Ho, Chen, Lin, Lu, Hsu and Tsai.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng-Han Tsai menghan@cgmh.org.tw
This article was submitted to Epilepsy, a section of the journal Frontiers in Neurology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.